
Abstract
Extracellular vesicles (EVs), such as exosomes and microvesicles, encapsulate proteins and microRNAs (miRNAs) as new modulators of both intercellular crosstalk and disease pathogenesis. The composition of EVs is modified by various triggers to maintain physiological homeostasis. In response to cigarette smoke exposure, the lungs develop emphysema, myofibroblast accumulation and airway remodelling, which contribute to chronic obstructive pulmonary disease (COPD). However, the lung disease pathogenesis through modified EVs in stress physiology is not understood. Here, we investigated an EV-mediated intercellular communication mechanism between primary human bronchial epithelial cells (HBECs) and lung fibroblasts (LFs) and discovered that cigarette smoke extract (CSE)-induced HBEC-derived EVs promote myofibroblast differentiation in LFs. Thorough evaluations of the modified EVs and COPD lung samples showed that cigarette smoke induced relative upregulation of cellular and EV miR-210 expression of bronchial epithelial cells. Using co-culture assays, we showed that HBEC-derived EV miR-210 promotes myofibroblast differentiation in LFs. Surprisingly, we found that miR-210 directly regulates autophagy processes via targeting ATG7, and expression levels of miR-210 are inversely correlated with ATG7 expression in LFs. Importantly, autophagy induction was significantly decreased in LFs from COPD patients, and silencing ATG7 in LFs led to myofibroblast differentiation. These findings demonstrate that CSE triggers the modification of EV components and identify bronchial epithelial cell-derived miR-210 as a paracrine autophagy mediator of myofibroblast differentiation that has potential as a therapeutic target for COPD. Our findings show that stressor exposure changes EV compositions as emerging factors, potentially controlling pathological disorders such as airway remodelling in COPD.
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Chronic obstructive pulmonary disease (COPD) is characterized by emphysema and progressive airflow limitation. Airway remodelling, in which the differentiation of fibroblasts to myofibroblasts plays a pivotal role, is a direct result of the inflammatory response due to exposure to inhaled cigarette smoke and leads to narrowing of the small airways (Citation1, Citation2). Increasing evidence has demonstrated that the processes of remodelling the small airways are attributed to excessive production of various triggers by bronchial epithelial cells (Citation3). Bronchial epithelial cells act as a source of various cytokines/chemokines, adhesion molecules and growth factors that modulate other elements of the airway wall and immune cells against cigarette smoke (Citation4). Therefore, alterations in bronchial epithelial cells have the potential to change the finely balanced reciprocal interaction between the bronchial epithelial and mesenchymal cell types that influences cellular differentiation, known as the epithelial–mesenchymal trophic unit (Citation5, Citation6). Airway remodelling in response to pathologic stimuli may change normal homeostatic autocrine and paracrine interactions between the epithelium and the surrounding fibroblast sheath; however, the detailed molecular mechanisms underlying the development of airway remodelling are not yet fully understood. Currently, autophagy, an intracellular degradation system through an autophagosomal–lysosomal pathway, is increasingly recognized as significant in various human diseases. Autophagy is a dynamic process that can rapidly change its status in response to disease activity and environmental stressors (Citation7). Autophagy-related (Atg) proteins identified in yeast include core Atg proteins that are required for autophagosome formation and are highly conserved in mammals (Citation8). Among the core Atg proteins, the physiological and pathological roles of Atg5 and Atg7 in autophagy machinery have been widely investigated in murine models (Citation9, Citation10). Remarkably, recent studies have shown that autophagy regulation plays an important role as a novel mechanism possibly underlying lung disease pathogenesis, including cellular senescence and lung fibrosis (Citation11, Citation12).
Extracellular vesicles (EVs), such as exosomes and microvesicles, are released by a variety of cells into their environment enclosed by a phospholipid bilayer. EVs have been recognized as novel modulators of cell-to-cell communication and orchestrators of health and disease (Citation13, Citation14). EVs can act as autocrine/paracrine effectors, based on evidence that they are able to transport a characteristic composition of proteins and nucleic acids, such as messenger RNA (mRNA) and microRNA (miRNA) (Citation15). Importantly, exposure to various stressors can modify the composition of EVs to change the surrounding microenvironment through EV cell-to-cell communication (Citation16). In normal cell physiology, EV secretion is a protective process to eliminate harmful components during adverse conditions, such as cigarette smoke exposure (Citation17). Currently, miRNAs, one composition of EVs, are known to play a regulatory role in the processes of cell differentiation and cell phenotypic alteration. The dysregulated expression of miRNAs has been linked to lung disease conditions, such as pulmonary fibrosis and inflammatory respiratory diseases (Citation18, Citation19). Furthermore, distinct miRNA profiles in extracellular spaces have also been related to disease pathologies, leading to interest in the use of EV miRNAs as disease biomarkers (Citation20). Such new mechanisms of intercellular communication raise the possibility that the miRNA cargo via EVs might modulate disease phenotypes and functions of recipient cells. However, the cell types in the lungs that were related to EV-mediated crosstalk and their paracrine effects remain unclear. Therefore, we hypothesized that EVs modified by cigarette smoke exposure may regulate lung disease phenotypes as novel paracrine signalling modulators during airway remodelling in COPD pathology.
Here, we demonstrate a novel pathogenic mechanism of COPD through modified EVs in cigarette stress physiology within the airway microenvironment. We report EV miR-210-mediated intercellular crosstalk between human bronchial epithelial cells (HBECs) and lung fibroblasts (LFs), which contributes to the development of myofibroblast differentiation via autophagy suppression, which may be associated with airway remodelling in COPD pathogenesis. miR-210 has already been reported to directly regulate multiple transcripts associated with diverse cellular functions that are known to influence normal developmental physiology as well as hypoxia-dependent disease states (Citation21). We found that miR-210 also directly targets ATG7 in LFs, which results in the modulation of the autophagy process. Therefore, our findings showed for the first time that autophagy processes are regulated by the EV cargo to cause myofibroblast differentiation in the lungs.
Methods
Cell culture and clinical samples
Normal airways were collected from 1st through 4th order bronchi from pneumonectomy and lobectomy specimens. Informed consent was obtained from all surgical participants as part of the approved on-going research protocol by the Ethics Committee of Jikei University School of Medicine. HBECs were isolated using protease treatment and characterized as previously described (Citation22). Freshly isolated HBECs were plated onto Rat Tail and Collagen type I-coated dishes and incubated overnight; the medium was then changed to Bronchial Epithelial Growth Medium (BEGM, Lonza, Tokyo, Japan). HBECs were serially passaged and used for experiments until passage 4. LFs were cultured from lung tissues using the explant technique. Briefly, fibroblasts outgrown from lung fragments were cultured in Dulbecco's modified Eagle's medium with 10% foetal calf serum and an antibiotic–antimycotic (Invitrogen, Grand Island, NY). LFs were serially passaged and used for experiments until passage 4. Human lung tissue samples were derived from the resected lungs of lung cancer patients who had recurrence after pulmonary resection at the National Cancer Center Hospital. The study protocol was approved by the Institutional Review Board of the National Cancer Center. All materials were obtained with written informed consent and were provided by the National Cancer Center Biobank.
EV purification and analysis
Before collection of the culture medium, the cells (HBECs or BEAS-2B) were washed with PBS, and the medium was switched to fresh BEGM (for HBECs) or Advanced RPMI containing an antibiotic–antimycotic and 2 mM l-glutamine (for BEAS-2B). After incubation for 48 h, the conditioned medium was collected and centrifuged at 2,000×g for 10 min at 4°C to thoroughly remove the cellular debris. The supernatant was filtered through a 0.22-µm filter (Millipore, Billerica, MA) to expectedly remove the microvesicles. The conditioned medium was then used for EV isolation (mainly exosomes). For EV preparation, the conditioned medium was ultracentrifuged in Beckman SW41Ti rotor at 35,000 rpm for 70 min at 4°C. The pellets were washed with 11 ml of PBS by ultracentrifugation at 35,000 rpm for 70 min at 4°C and resuspended in PBS. The putative EV fraction was measured for its protein content using a Quant-iT™ Protein Assay with Qubit®2.0 Fluorometer (Invitrogen). Nanoparticle tracking analysis was performed using the NanoSight LM10HS with a blue laser system (NanoSight, Amesbury, UK) on isolated EVs diluted 500-fold with PBS for analysis.
Results
HBEC-derived EVs are transported to primary LFs
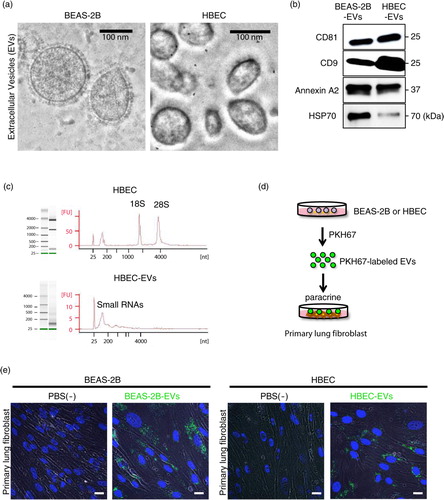
We first isolated EVs by ultracentrifugation from conditioned medium collected from immortalized bronchial epithelial cells (BEAS-2B cell line) or primary HBECs and characterized them extensively. Electron microscopic analysis revealed typical bilayer membrane vesicles that were heterogeneous in size, ranging from approximately 50 to 150 nm (a). Western blotting analysis confirmed the presence of EV marker proteins CD81, CD9, annexin A2 and heat shock 70 kDa protein 4 (HSP70) in the BEAS-2B- and HBEC-derived EVs (b). In addition, cytochrome c was detectable in the whole cell lysates, but it was absent in the BEAS-2B- and HBEC-derived EVs (Supplementary Fig. 1). When we estimated the isolated RNAs from HBECs and HBEC-derived EVs, we found that the RNA profile of HBEC-derived EVs contained only minute amounts of ribosomal RNA (18S and 28S), as compared with that of HBECs, but large amounts of small RNAs, such as miRNAs (c). These data suggest that we have successfully isolated EVs from the conditioned medium. To investigate whether bronchial epithelial cell-derived EVs are crucially involved in the paracrine actions of subepithelial fibroblasts, we labelled secreted BEAS-2B- or HBEC-derived EVs with a green fluorescent marker, that is, PKH67; labelled EVs were incubated with cultured primary LFs (d). Analysis of EV uptake performed in primary LFs by confocal microscopy revealed uptake of labelled EVs (e). Therefore, we hypothesize that bronchial epithelial cell-derived EVs function locally in paracrine signals during homeostasis and disease progression in lungs.
Fig. 1. Human bronchial epithelial cell-derived EVs are transported to primary lung fibroblasts (LFs). (a) Electron microscopy images of BEAS-2B- and HBEC-derived EVs, showing a size of approximately 50–150 nm in diameter. (b) Western blot of these cell-derived EVs for CD81, CD9, annexin A2 and HSP70. (c) HBECs (upper panel) and HBEC-derived EVs (lower panel) were analysed with a bioanalyser. Gels and electropherograms are shown. The left gel lane is the ladder standard, and the right lane is the total RNA from HBECs and HBEC-derived EVs. The y-axis of the electropherogram shows the signal intensities in arbitrary fluorescence units (FU), and the x-axis shows the size of the RNA in nucleotides (nt). (d) Schematic representation of the EV uptake experiment. BEAS-2B- and HBEC-derived EVs were labelled with PKH67 and incubated with primary LFs. (e) Purified BEAS-2B- and HBEC-derived EVs or vehicle PBS(-) as a control was labelled with PKH67 (green) and incubated with primary LFs. Nuclei were counterstained with DAPI (blue). Scale bar: 20 µm.
Cigarette smoke extract-induced HBEC-derived EVs promote lung myofibroblast differentiation
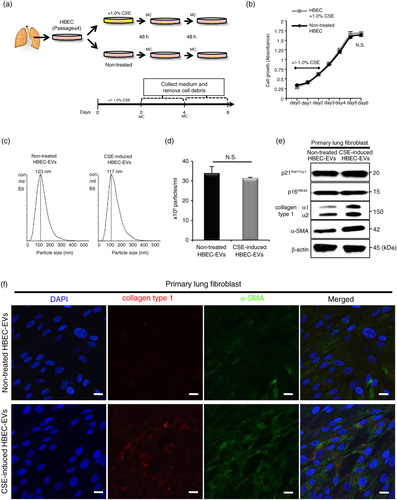
Bronchial epithelial cells have first-line contact with harmful substances during cigarette smoke exposure, resulting in stress-induced phenotypic alterations, which might be involved in the pathogenesis of airway remodelling in COPD. Moreover, EV composition and function are changeable in response to a variety of physiological stressors (Citation16). Based on the labelling experiments, we assumed that EVs modified by cigarette smoke can change the paracrine interactions between the epithelium and the surrounding fibroblast. To clarify the EV-mediated interaction between bronchial epithelial cells and LFs during cigarette smoke exposure, we analysed the role of cigarette smoke extract (CSE)-induced HBEC-derived EVs in the microenvironment of the airway. After HBECs were incubated in the non-FBS-containing medium with or without a low concentration of CSE (1.0%) for 2 days, we collected conditioned medium (on days 4 and 6) and isolated EVs by ultracentrifugation (a). Cell proliferation analysis showed that a low concentration of CSE did not affect HBEC growth (b). These data suggest that a low concentration of CSE did not induce cellular senescence or apoptosis. Furthermore, the particle sizes and counts of the EV preparations measured by the NanoSight particle tracking system showed no significant differences between CSE-induced HBEC-derived EVs and non-treated HBEC-derived EVs (c and d). To investigate the possible paracrine effects of CSE-induced HBEC-derived EVs, LFs were cultured with the EVs (5 µg/ml). Western blotting (e and Supplementary Fig. 2) and immunofluorescence staining (f) showed that CSE-induced HBEC-derived EVs strikingly promoted the expression of fibrotic markers, that is, collagen type I and α-SMA, in LFs compared with non-treated HBEC-derived EVs. Furthermore, we found that CSE-induced HBEC-derived EVs had no remarkable effect on the expression of senescent markers, that is, p21Waf1/Cip1 and p16INK4A, in LFs compared with non-treated HBEC-derived EVs (e). These data suggest that HBEC-derived EVs have potential for modulating lung myofibroblast differentiation phenotypes in response to CSE in vitro.
Fig. 2. CSE-induced HBEC-derived EVs promote a lung myofibroblast differentiation phenotype. (a) Schematic representation of the conditioned medium collected (on days 4 and 6) after HBECs (passage ≤ 4) was incubated with or without a low concentration of CSE (1.0%) for 2 days. MC: medium change. (b) Cell proliferation assay of HBECs with or without 1.0% CSE (days 0–2). (c and d) Nanoparticle tracking analyses of the particle size (c) and counts (d) in non-treated HBEC-derived EVs or CSE-induced HBEC-derived EVs. (e) Western blot of senescent markers and fibrotic markers by CSE-induced HBEC-derived EVs or non-treated HBEC-derived EVs in primary lung fibroblasts (LFs). (f) Immunofluorescence staining for collagen type I (red) and α-SMA (green) in primary LFs with CSE-induced HBEC-derived EVs or non-treated HBEC-derived EVs was evaluated by confocal microscopy. DAPI (blue) was used for nuclear staining. Scale bar: 20 µm.
HBEC-derived EV miR-210 promotes myofibroblast differentiation in LFs
EVs can carry several different types of biomolecular information and they spread from cell to cell. In the present study, we focused on EV miRNAs because they have great potential as disease therapeutics and biomarkers (Citation23). We hypothesized that miRNAs shuttled by HBEC-derived EVs might be responsible for myofibroblast differentiation in LFs. To identify EV miRNAs that regulate cigarette smoke-induced lung myofibroblast differentiation, we conducted a miRNA microarray between CSE-induced HBEC-derived EVs and non-treated HBEC-derived EVs. After analysis of variance of miRNA microarray signal expression from the 2 EV groups, 22 miRNAs (17 downregulated miRNAs and 5 upregulated miRNAs in CSE-induced HBEC-derived EVs) exhibited significant expression changes (fold change >1.5) (a). Next, quantitative reverse transcription-PCR (qRT-PCR) validation of EV miRNAs from the 2 groups showed that only 8 miRNAs (let-7a, let-7b, let-7c, let-7f, let-7g, let-7i, miR-100 and miR-210) exhibited significant expression changes (b). Moreover, the expression levels of cellular miRNAs from CSE-induced HBECs and non-treated HBECs showed that the 8 miRNAs also exhibited significant changes (Supplementary Fig. 3).
Fig. 3. HBEC-derived EV miR-210 promotes lung myofibroblast differentiation. (a) A heat map of the EV miRNA microarray analysis revealed differentially expressed miRNAs (change >1.5-fold) in CSE-induced HBEC-derived EVs or non-treated HBEC-derived EVs. (b) qRT-PCR validation of EV miRNAs from the 2 EV groups. miR-16 was used as an internal control. (c) qRT-PCR analyses of miR-210 expression levels in non-smoker or smoker lungs (including non-COPD smokers and COPD patients; P=0.049). (d) miR-210-specific probe, scramble control probe and β-actin were hybridized in situ with normal lung tissue. Original magnification, 200×. (e) A transwell co-culture assay with transfected HBECs (top well) and primary lung fibroblasts (LFs) (bottom well). A 0.4-µm porous membrane is between the 2 wells, inhibiting cell–cell contact. (f) A co-culture assay to study the miRNA cargo from HBECs to primary LFs. HBECs were transfected with a Cy3-labelled miRNA (red) or a control precursor miRNA (non-labelled). Nuclei were counterstained with DAPI (blue). Scale bar: 50 µm. (g) miR-210 expression in LFs after 72 h co-culturing with HBECs transfected with miR-210 mimic (pre-miR-210) or miR-NC. RNU6B was used as an internal control. (h) Western blot of fibrotic markers in primary LFs (the bottom well) in the co-culture assay. HBECs were transfected with a precursor of miR-210 (pre-miR-210) or a control precursor miRNA (pre-miR-NC) and co-cultured with primary LFs for 72 h. *P<0.05. NC: negative control.
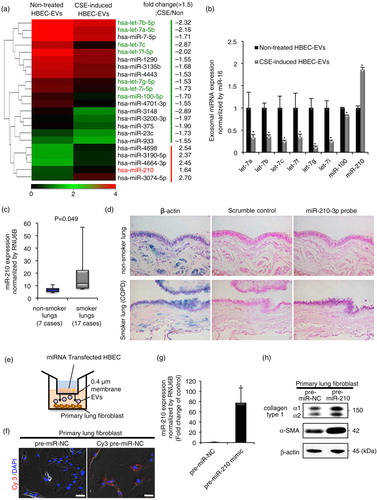
From the putative regulatory miRNAs, we focused on the upregulated miRNA-miR-210. To further examine whether miR-210 is a crucial mediator for cigarette smoke-induced myofibroblast differentiation in lung tissues, we compared the miR-210 expression level in the non-smoker group (n=7) with that in the smoker group (including non-COPD smokers and COPD patients; n=17) (Supplementary Table I). We also observed increased expression of miR-210 in the lung tissues of the smoker group compared with that in the non-smoker group (c; P=0.049). In agreement with the qRT-PCR analysis, in situ hybridization with a miR-210-locked nucleic acid probe revealed increased miR-210 expression in the bronchial epithelial cells of smoker lungs (COPD patients) compared with that of non-smoker lungs. In contrast, miR-210 expression was not detectable in the parenchymal and alveolar structures in lung tissues (d and Supplementary Fig. 4).
Based on these fundamental data, we hypothesized that EV miR-210 might functionally regulate myofibroblast differentiation through cell-to-cell communication. We conducted a co-culture assay to investigate the miRNA crosstalk between HBECs and LFs through EV cargo-mediated delivery (e). To visualize whether miRNAs released from HBECs are transported to LFs, we transfected HBECs with a Cy3-labelled pre-miR-NC and co-cultured the transfected HBECs with LFs for 36 h. In confocal microscopic findings, we detected Cy3-labelled miRNA in separated LFs (f). HBECs are unable to migrate through a 0.4-µm pore membrane, indicating that Cy3-labelled miRNA in HBECs has been transported and taken up into LFs via EVs. Next, after 72 h co-culture with HBECs transfected with miR-210 mimic or miR-NC, we measured miR-210 expression in LFs and evaluated the myofibroblast differentiation in the LFs. Indeed, HBEC-derived miR-210 was enriched in the LFs after co-culture (g). Notably, western blotting (h and Supplementary Fig. 5) and immunofluorescence staining (Supplementary Fig. 6) showed that HBEC-derived miR-210 increased the levels of collagen type I and α-SMA expression in LFs. These findings suggest that HBEC-derived EV miR-210 can promote myofibroblast differentiation through the horizontal transfer of EV cargo.
miR-210 directly targets ATG7 in LFs, leading to a modulating autophagy process
In general, miRNAs regulate various gene expressions by repressing the translation or promoting the degradation of their target genes (Citation24). To strategically identify direct miR-210 target genes, we first performed mRNA microarray analyses of normal foetal LF cells (MRC5 cell line) and primary LFs that were transiently transfected with the miR-210 mimic and the control together with in silico predictions (Supplementary Fig. 7). We detected 552 genes that were downregulated at least 1.5-fold in both MRC5 cells and primary LFs (Supplementary Table II). Subsequently, we selected highly predicted target genes using 2 computational algorithms, that is, TargetScanHuman 6.2 (www.targetscan.org) and miRanda (www.microrna.org). Overall, we identified 8 candidates: iron–sulphur cluster assembly enzyme (ISCU); nuclear receptor subfamily 1, group D, member 2 (NR1D2); NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 4, 9 kDa (NDUFA4); brain-derived neurotrophic factor (BDNF); glycerol-3-phosphate dehydrogenase 1-like (GPD1L); MID1 interacting protein 1(MID1IP1); SMG5 non-sense-mediated mRNA decay factor (SMG5) and autophagy-related 7 (ATG7).
Among the 8 predicted genes, we focused on the critical autophagy-related factor, ATG7, in this study. We chose this gene because there is growing evidence that alleviation of autophagy-mediated cellular stress plays a key role in the pathogenesis of a variety of pulmonary diseases (Citation11, Citation12). To elucidate the function of ATG7 in lung tissue, we first evaluated the expression levels of ATG7 in LF homogenates from non-COPD (non-smokers/light smokers; n=5) and COPD patients (n=4) (Supplementary Table III). Western blotting showed that relatively low accumulations of ATG7 were detected in LF homogenates from COPD patients as compared with those detected in LF homogenates from non-smokers/light smokers (a). To investigate the autophagy status in LF from COPD patients, western blotting for the conversion of LC3 from LC3-I to LC3-II was performed. Although only LC3-II expression was detected in LFs, LFs homogenates from COPD patients demonstrated a decrease in LC3-II expression (b). Remarkably, the ATG7 expression levels correlated significantly with the percentage of forced expiratory volume in 1 s/forced vital capacity (FEV1/FVC) (c) and inversely correlated with miR-210 expression (d). Based on the clinical relevance of these LF findings, we conducted small interfering RNA (siRNA)-based silencing of ATG7 and assessed cells for myofibroblast differentiation. Remarkably, we observed that siRNA-mediated knockdown of ATG7 resulted in increased levels of collagen type I and α-SMA expression in LFs (e).
Fig. 4. miR-210 directly targets ATG7 in lung fibroblasts (LFs), leading to a modulating autophagy process. (a) Western blot of ATG7 in LF homogenates from non-COPD (n=5) and COPD patients (n=4). The right panel shows the average taken from 3 independent experiments shown as the relative expression of ATG7 as compared with that of β-actin. (b) Western blot of LC3 in LF homogenates from non-COPD (n=5) and COPD patients (n=4) in the presence of protease inhibitors (E64d and pepstatin A). The right panel shows the average taken from 3 independent experiments shown as the relative expression of LC3-II as compared with that of β-actin. (c) The relationship between relative ATG7 expression normalized to β-actin and the percentages of FEV1/FVC (n=9). (d) The relationship between relative ATG7 expression normalized to β-actin and miR-210 expression normalized to RNU6B (n=9). (e) Western blotting of ATG7 and fibrotic markers in primary LFs transfected with ATG7-siRNA or siRNA-NC. (f) Western blot of ATG7 and fibrotic markers in primary LFs transfected with pre-miR-210 or pre-miR-NC. (g) Schematic miR-210 putative target sites in the 3′ UTRs of ATG7 and the sequence of mutant UTRs. (h) The effect of co-transfection of pre-miR-210 with wild-type (Wt) and mutant (Mut) psiCHECK2 vectors with each gene construct in MRC5 cells was measured using luciferase reporter assays. (i) LC3 western blotting of primary LFs transfected with pre-miR-210 or pre-miR-NC in the presence or absence of protease inhibitors (E64d and pepstatin A). (j) Fluorescence microscopic detection of pEGFP-LC3 dot formation in primary LFs. The right panel is the percentage of positive cells with more than 5 dot formations. (k) LC3 western blotting of primary LFs cultured with CSE-induced HBEC-derived EVs or non-treated HBEC-derived EVs in the presence or absence of protease inhibitors (E64d and pepstatin A). *P<0.05. **P<0.01. NC: negative control.
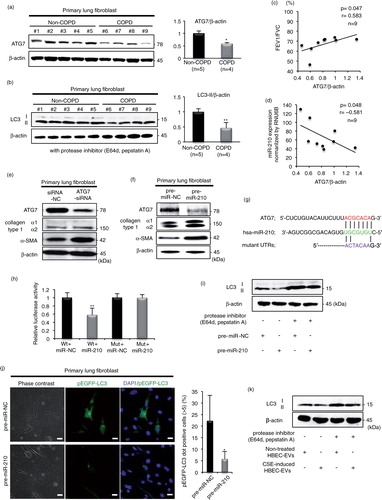
To determine whether miR-210 plays a role in lung myofibroblast differentiation through the direct regulation of ATG7, we performed experimental validation. The specific regulation of ATG7 expression by miR-210 was validated by qRT-PCR analysis, demonstrating a significant inhibitory effect of the miR-210 mimic on ATG7 expression in LFs (Supplementary Fig. 8). Western blotting detected the inhibition of ATG7 expression and increased levels of collagen type I and α-SMA expression treated by the miR-210 mimic as compared with that of the negative control cells (f). Next, the 3′ untranslated region (UTR) of the ATG7 gene was conjugated with luciferase for reporter assays. Particularly, the 3′ UTR of the ATG7 mRNA harboured sequences complementary to the miR-210 seed sequence (g). To verify whether ATG7 is a direct target of miR-210, we cloned the 3′ UTR into the vector downstream of the luciferase open-reading frame. In addition, to validate target specificity, we conducted site-directed mutagenesis of the 3′ UTR. When we co-transfected MRC5 cells with the cloned UTRs and the miR-210 mimic, we observed a consistent reduction in the luciferase activity of the 3′ UTR of ATG7. By contrast, co-transfection of the miR-210 mimic with the mutated forms of the 3′ UTRs resulted in no significant change in luciferase activity (h).
These data demonstrate that ATG7 is a potential direct target gene for miR-210, and this miRNA could negatively regulate ATG7 expression, resulting in modulation of the autophagy process in LFs. To investigate the regulatory role of autophagy by miR-210, western blotting for LC3 and the detection of EGFP-LC3 dot formation by fluorescence microscopy were performed. Remarkably, LFs transfected with pre-miR-210 demonstrated a decrease in LC3-II expression (i). To directly visualize this observation, we detected EGFP-LC3 dot formation in LFs using confocal microscopy. The miR-210 mimic significantly inhibited EGFP-LC3 dot formation in cells as compared with miR-NC (j). Our results show that miR-210 regulates autophagy through autophagosome formation, as shown by the decrease in LC3 and ATG7 protein expression and by the analysis of EGFP-LC3-positive autophagosome vesicles in LFs. Finally,to elucidate the regulatory role of autophagy by the EV cargo, LFs were cultured with CSE-induced HBEC-derived EVs or non-treated HBEC-derived EVs (5 µg/ml). Remarkably, LFs cultured with CSE-induced HBEC-derived EVs also demonstrated a decrease in LC3-II expression (k).
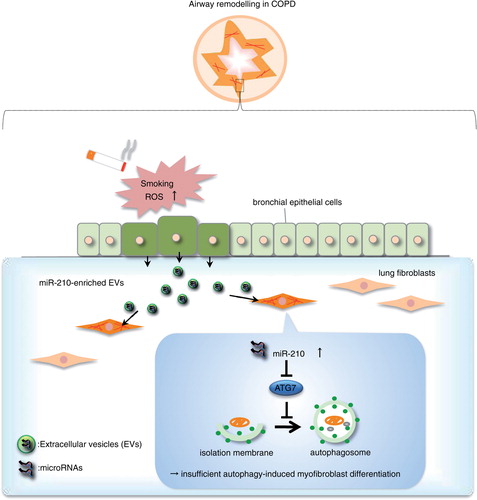
The above data show that transferred EV-mediated autophagy suppression promoted myofibroblast differentiation in response to smoke exposure. We concluded that this novel mechanism by the EV miRNA cargo might be involved in airway remodelling in COPD pathogenesis ().
Fig. 5. Proposed novel airway remodelling model for bronchial epithelial cells and fibroblast crosstalk in COPD.
Discussion
Our present study revealed a miRNA/EV-mediated cellular communication mechanism between bronchial epithelial cells and LFs that promotes the development of smoke-induced myofibroblast differentiation. Remarkably, we elucidated that the novel mechanism of myofibroblast differentiation in LFs is attributed to the CSE-induced HBEC-derived EV miR-210 regulating autophagy machinery.
Cigarette smoke contains a variety of harmful toxic components, such as carcinogenic and mutagenic chemicals, free radicals and reactive oxygen species (ROS), which drive the phenotypic alterations, including dysregulated expressions of miRNAs in bronchial epithelial cells, and may be involved in the regulation of various smoke-induced gene expression as a part of COPD pathogenesis (Citation25). To investigate this smoke-induced phenotypic alteration of miRNAs, we speculate that in vitro CSE exposure of HBECs appears to closely resemble that of human smokers (Citation26). To date, CSE-mediated miRNA alterations can be explained as an outcome of epigenetic disruption, such as DNA methylation and genetic alterations (Citation27). It has been reported that cigarette smoke exposure in the lungs of both mice and rats resulted in evident dysregulation of cellular miRNA expression profiles, mainly in the sense of downregulation such as with the let-7 family (Citation28). In this study, we found that 8 miRNAs (6 members of the let-7 family, miR-100 and miR-210) exhibited significant expression changes between CSE-induced HBEC-derived EVs and non-treated HBEC-derived EVs. Among these miRNAs, we focused on the function of EV miR-210 because the expression levels of miR-210 in CSE-induced HBECs and COPD lung samples (qRT-PCR and in situ hybridization) were higher than those of each control (c and d, Supplementary Figs. 3 and 4). In general, miR-210 has been known as a hypoxia-associated miRNA and appears to control a signalling network of multiple processes during hypoxic responses (Citation21). In addition to hypoxia, recent data have demonstrated that miR-210 induction is associated with various sources of ROS generation, such as mitochondrial ROS donors and platelet-derived growth factor-BB (Citation29). Indeed, we have already reported that CSE induces mitochondrial ROS production in HBECs (Citation30). Therefore, our data indicate that CSE-induced ROS production might influence cellular and EV miR-210 expression derived from HBECs, independent of hypoxia.
Trophic interactions between epithelium and the surrounding fibroblast are crucial in lung diseases. Changes in bronchial epithelial cells contribute to myofibroblast differentiation through paracrine interactions, resulting in airway wall thickening in COPD. In this study, we found that HBEC-derived EV miR-210 caused a marked increase in collagen type I and α-SMA expression in LFs, which are established features of myofibroblast differentiation via EVs. Additionally, we first found that miR-210 regulates autophagy via targeting a critical autophagy-related factor, ATG7. By analysing EGFP-LC3-positive autophagosome vesicles in LFs, we have demonstrated that miR-210 regulates autophagy through autophagosome formation, resulting in a decrease in ATG7 protein expression. Among more than 35 autophagy-related (Atg) genes that have been identified in yeast, there are important Atg proteins required for autophagosome formation that are highly conserved in mammals (Citation31). ATG7 is one of the key regulators of the autophagy process and is responsible for 2 major reactions involved in autophagosome formation and in vesicle progression (Citation32). However, we recognize that it is possible that miR-210 regulates other target genes that are responsible for myofibroblast differentiation or autophagy. Further investigations are needed to clarify the detailed pathogenesis of airway remodelling regulated by miRNA networks.
Autophagy is an orchestrated homeostatic process that involves the degradation and digestion of intracellular components by lysosomes (Citation33). Although the detailed cellular and molecular mechanisms underlying the development of COPD are complex and poorly understood, recent studies have showed that autophagy regulation is crucially involved in COPD pathogenesis (Citation34–Citation36). We have already reported that insufficient autophagy is involved in accelerated CSE-induced HBEC senescence (Citation30, Citation34) (Citation35). In LFs, it has been reported that autophagy inhibition by knockdown of LC3 and ATG5 induced increased α-SMA and type I collagen expression levels (Citation36). Additionally, another study has proposed that TGF-β-mediated autophagy inhibition in LFs is responsible for myofibroblast differentiation (Citation37). Differentiation of resident fibroblasts into myofibroblasts is a part of the sequence of events that follows insults to tissue repair. However, the sustained and extensive accumulation of myofibroblasts has been implicated in the mechanisms of the aberrant wound-healing process of fibrosis. Accordingly, the autophagic regulation of myofibroblast differentiation can be a critical determinant of airway fibrotic remodelling in COPD pathogenesis. Intriguingly, both the inhibition and activation of autophagy have been associated with myofibroblast differentiation during fibrosis development in various organs (Citation39–Citation40). Furthermore, a recent paper showed that long-term autophagy activation via starvation induces myofibroblast differentiation through mTOR2 activation and downstream upregulation of CTGF in foetal lung-derived fibroblasts (Citation41). In the present study, lower levels of ATG7 and LC3 were shown to have accumulated in LFs from COPD patients than in LFs from non-COPD patients. Furthermore, we demonstrated that knockdown of ATG7 resulted in increased levels of collagen type I and α-SMA expression in LFs, which in turn resulted in the promotion of myofibroblast differentiation and suggested that autophagy may have a tissue-specific and stress-dependent role in regulating myofibroblast differentiation. Accordingly, we speculate that epithelial cell-derived EV miR-210-mediated autophagy inhibition via ATG7 suppression may play a crucial role in the pathogenesis of small airway remodelling in COPD through myofibroblast accumulation.
Several recent studies have demonstrated that EVs can act as mediators of cell-to-cell communications affecting various physiological processes and pathological disorders, which often lead to alterations of gene expression in the recipient cells (Citation14). With respect to the autophagy regulation by EVs, there is only one report that breast cancer cell-derived EVs induce autophagy through ROS generation in mammary epithelial cells (Citation42). In this study, we found that CSE-induced HBEC-derived EVs regulated autophagy through miRNA transfer to LFs. In normal cellular physiology, EV secretion is a protective process due to its ability to remove harmful components during adverse conditions including smoking exposure (Citation17). Furthermore, exposure to various stressors can modify the proteomic and miRNA compositions of EVs to change the surrounding microenvironment for escaping from cell damage through EV cell-to-cell communication (Citation16). In human body, we speculate that routine smoking may secrete miR-210-enriched EVs for maintaining their own autophagy function in HBECs, thereby chronic transfer of the EVs results in airway fibrosis for repairing damaged lung microenvironment. Although here we focused on EV miRNA from among the various components packaged in EVs, it is important for analysing the details of other components such as harmful proteins in the CSE-induced HBEC-derived EVs, which may affect cellular phenotypes in LFs. In any event, our present study showed that EV miRNA released by bronchial epithelial cells are major components triggering myofibroblast differentiation.
In summary, we have proposed a novel mechanism of airway remodelling through autophagy regulation by the EV miRNA cargo in response to cigarette smoke exposure in COPD pathogenesis. Our findings suggest that stressor exposure modifies EV components as emerging factors, potentially controlling pathological lung disorders. In the present study, we understand the limitations of using large airway bronchial epithelial cells as an in vitro culture model to elucidate the pathogenesis of small airway disease; therefore, more relevant cell-culturing models using small airway epithelial cells or alveolar epithelial cells are needed in future studies to further confirm the physiological relevance of our results. In addition, we have to closely examine in vivo COPD models to test the mechanism found in vitro. Furthermore, EVs are released from several cells including various immune cells in human lungs (Citation43). Therefore, we have to carefully investigate their multiple EV crosstalking among a wide variety of cells during trigger exposure within the lung microenvironment. However, our data provide a compelling proof of concept for airway remodelling by EV miRNA cargo based on the clinical relevance of the full mechanism found in vitro. These findings demonstrate that smoke exposure modifies EV components and identify bronchial epithelial cell-derived miR-210 as a paracrine autophagy mediator of myofibroblast differentiation that has potential as a therapeutic target for COPD.
Authors’ contributions
YF, SI, KK, YY, TK and HH performed the experimental work, conducted data analysis and wrote the manuscript. JA, NK and KK assisted in the writing of the manuscript and provided helpful discussion. TO supervised this project.
Conflict of interest and funding
This study was supported in part by a grant-in-aid for the Third-Term Comprehensive 10-Year Strategy for Cancer Control of Japan, including awards of a Research Resident Fellowship from the Foundation for Promotion of Cancer Research (Japan), the Program for the Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NiBio) and Scientific Research from the Ministry of Education of Japan (to JA and KK) and Health and Labour Sciences Research grants from the Ministry of Health, Labour and Welfare of Japan (to JA and KK). The National Cancer Center Biobank is supported by the National Cancer Center Research and Development Fund, Japan.
Supplementary Material
Download PDF (13.9 MB)Acknowledgements
We thank Mizuyo Arashi for excellent technical assistance, Dr. Luc Gailhouste for carefully reading the manuscript, Dr. Odaka and Dr. Morikawa (Jikei University School of Medicine, Tokyo, Japan) for providing surgical participants, Dr. Harada (Jikei University School of Medicine, Tokyo, Japan) for supporting the pathological assessment, and Dr. Mizushima (Tokyo Medical and Dental University, Tokyo, Japan) and Dr. Yoshimori (Osaka University, Osaka, Japan) for providing LC3 cDNA.
Notes
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Related Research Data
References
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004; 350: 2645–53.
- Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008; 8: 183–92.
- Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, Satoh M et al. Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med. 2001; 163: 1476–83.
- Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med. 2012; 18: 684–92.
- Evans MJ, Van Winkle LS, Fanucchi MV, Plopper CG. The attenuated fibroblast sheath of the respiratory tract epithelial–mesenchymal trophic unit. Am J Respir Cell Mol Biol. 1999; 21: 655–7.
- Araya J, Cambier S, Morris A, Finkbeiner W, Nishimura SL. Integrin-mediated transforming growth factor-beta activation regulates homeostasis of the pulmonary epithelial–mesenchymal trophic unit. Am J Pathol. 2006; 169: 405–15.
- Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014; 24: 69–79.
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Ann Rev Cell Dev Biol. 2011; 27: 107–32.
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T et al. The role of autophagy during the early neonatal starvation period. Nature. 2004; 432: 1032–6.
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005; 169: 425–34.
- Araya J, Hara H, Kuwano K. Autophagy in the pathogenesis of pulmonary disease. Intern Med. 2013; 52: 2295–303.
- Mercado N, Ito K, Barnes PJ. Accelerated ageing of the lung in COPD: new concepts. Thorax. 2015; 70: 482–9.
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013; 200: 373–83.
- Kosaka N, Yoshioka Y, Hagiwara K, Tominaga N, Katsuda T, Ochiya T. Trash or treasure: extracellular microRNAs and cell-to-cell communication. Front Genet. 2013; 4: 173.
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007; 9: 654–9.
- Beninson LA, Fleshner M. Exosomes: an emerging factor in stress-induced immunomodulation. Semin Immunol. 2014; 26: 394–401.
- Baixauli F, Lopez-Otin C, Mittelbrunn M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol. 2014; 5: 403.
- Oglesby IK, McElvaney NG, Greene CM. MicroRNAs in inflammatory lung disease – master regulators or target practice?. Respir Res. 2010; 11: 148.
- Fujita Y, Takeshita F, Kuwano K, Ochiya T. RNAi therapeutic platforms for lung diseases. Pharmaceuticals. 2013; 6: 223–50.
- Fujita Y, Kuwano K, Ochiya T, Takeshita F. The impact of extracellular vesicle-encapsulated circulating microRNAs in lung cancer research. BioMed Res Int. 2014; 2014: 486413.
- Chan SY, Loscalzo J. MicroRNA-210: a unique and pleiotropic hypoxamir. Cell Cycle. 2010; 9: 1072–83.
- Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, Hill A et al. Squamous metaplasia amplifies pathologic epithelial–mesenchymal interactions in COPD patients. J Clin Invest. 2007; 117: 3551–62.
- Kosaka N, Takeshita F, Yoshioka Y, Hagiwara K, Katsuda T, Ono M et al. Exosomal tumor-suppressive microRNAs as novel cancer therapy: “exocure” is another choice for cancer treatment. Adv Drug Deliv Rev. 2013; 65: 376–82.
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010; 11: 597–610. [PubMed Abstract].
- Schembri F, Sridhar S, Perdomo C, Gustafson AM, Zhang X, Ergun A et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc Natl Acad Sci USA. 2009; 106: 2319–24.
- Mathis C, Poussin C, Weisensee D, Gebel S, Hengstermann A, Sewer A et al. Human bronchial epithelial cells exposed in vitro to cigarette smoke at the air–liquid interface resemble bronchial epithelium from human smokers. Am J Physiol Lung Cell Mol Physiol. 2013; 304: L489–503.
- Momi N, Kaur S, Rachagani S, Ganti AK, Batra SK. Smoking and microRNA dysregulation: a cancerous combination. Trends Mol Med. 2014; 20: 36–47.
- Izzotti A, Larghero P, Longobardi M, Cartiglia C, Camoirano A, Steele VE et al. Dose-responsiveness and persistence of microRNA expression alterations induced by cigarette smoke in mouse lung. Mutat Res. 2011; 717: 9–16.
- Kim JH, Park SG, Song SY, Kim JK, Sung JH. Reactive oxygen species-responsive miR-210 regulates proliferation and migration of adipose-derived stem cells via PTPN2. Cell Death Dis. 2013; 4: e588.
- Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. 2015; 11: 547–59.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011; 147: 728–41.
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Ann Rev Biochem. 2011; 80: 125–56.
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008; 451: 1069–75.
- Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology. 2012; 1: 630–41.
- Takasaka N, Araya J, Hara H, Ito S, Kobayashi K, Kurita Y et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J Immunol. 2014; 192: 958–68.
- Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2013; 304: L56–69.
- Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J et al. Autophagy in idiopathic pulmonary fibrosis. PLoS One. 2012; 7: e41394.
- Del Principe D, Vona R, Giordani L, Straface E, Giammarioli AM. Defective autophagy in fibroblasts may contribute to fibrogenesis in autoimmune processes. Curr Pharm Design. 2011; 17: 3878–87.
- Hernandez-Gea V, Friedman SL. Autophagy fuels tissue fibrogenesis. Autophagy. 2012; 8: 849–50.
- Del Principe D, Lista P, Malorni W, Giammarioli AM. Fibroblast autophagy in fibrotic disorders. J Pathol. 2013; 229: 208–20.
- Bernard M, Dieude M, Yang B, Hamelin K, Underwood K, Hebert MJ. Autophagy fosters myofibroblast differentiation through MTORC2 activation and downstream upregulation of CTGF. Autophagy. 2014; 10: 2193–207.
- Dutta S, Warshall C, Bandyopadhyay C, Dutta D, Chandran B. Interactions between exosomes from breast cancer cells and primary mammary epithelial cells leads to generation of reactive oxygen species which induce DNA damage response, stabilization of p53 and autophagy in epithelial cells. PLoS One. 2014; 9: e97580.
- Fujita Y, Yoshioka Y, Ito S, Araya J, Kuwano K, Ochiya T. Intercellular communication by extracellular vesicles and their microRNAs in asthma. Clin Ther. 2014; 36: 873–81.