
Abstract
After ischaemic injury and in patients with atherosclerosis, the pool of inflammatory macrophages is enlarged in the heart and in atherosclerotic plaques. Monocyte/macrophage-derived microparticles (MPs) are part of the pathological process of unstable atherosclerotic plaques. The present study focused on effects of MPs, produced by apoptotic murine RAW 264.7 macrophage cell line, in adult murine cardiomyocytes. Flow cytometry and western blot analysis showed that these MPs contained the soluble form of tumour necrosis factor alpha (TNF-α). Cardiomyocyte sarcomere shortening amplitudes and kinetics were reduced within 5 min of exposure to these MPs. Conversely, Ca2+ transient amplitude and kinetics were not modified. The contractile effects of MPs were completely prevented after pretreatment with nitric oxide synthase, guanylate cyclase or TNF-α inhibitors as well as blocking TNF-α receptor 1 with neutralizing antibody. Microscopy showed that, after 1 h, MPs were clearly surrounding rod-shaped cardiomyocytes, and after 2 h they were internalized into cardiomyocytes undergoing apoptosis. After 4 h of treatment with MPs, cardiomyocytes expressed increased caspase-3, caspase-8, Bax and cytochrome C. Thus, MPs from apoptotic macrophages induced a negative inotropic effect and slowing of both contraction and relaxation, similar to that observed in the presence of TNF-α. The use of specific inhibitors strongly suggests that TNF-α receptors and the guanylate cyclase/cGMP/PKG pathway were involved in the functional responses to these MPs and that the mitochondrial intrinsic pathway was implicated in their proapoptotic effects. These data suggest that MPs issued from activated macrophages carrying TNF-α could contribute to propagation of inflammatory signals leading to myocardial infarction.
The healthy myocardium hosts a large number of macrophages that are among the largest cardiac resident cell populations, with fibroblasts, myocytes and endothelial cells (for recent review, see Ref. (Citation1). Heart macrophages deserve attention because they perform numerous tasks in wound healing, regeneration and tissue remodelling. They are recognized as central actors in cancer, infection, rheumatoid arthritis, diabetes, obesity, atherosclerosis and stroke. Furthermore, after ischaemic injury and in patients with atherosclerosis, an increased production and supply of inflammatory monocytes likely enlarge the pool of inflammatory macrophages in the heart and in atherosclerotic plaques. These inflammatory macrophages may directly harm cardiomyocytes due to their ability to produce the tumour necrosis factor alpha (TNF-α), a pro-inflammatory cytokine that induces myocyte apoptosis (Citation2) and a negative inotropic effect (Citation3, Citation4), cardiac β-adrenergic dysfunction (Citation5), sphingosine production (Citation6), phospholipase A2 activation/increased arachidonic acid (Citation7) and inducible nitric oxide (NO)-synthase (NOS) activity (Citation3). Elevated expression levels of TNF-α in cardiomyocytes and increased plasma concentrations of TNF-α have been reported in patients with end-stage heart failure (Citation8). Experimental animal studies also showed that cardiac-specific overexpression of TNF-α results in the development of a dilated cardiomyopathy with ventricular hypertrophy, ventricular dilation and other cardiomyopathy-like phenotype (Citation9). Furthermore, high plasma levels of TNF-α are found in cardiac diseases in which left ventricular dysfunction develops, such as septic shock, reperfusion injury, myocardial infarction, myocarditis, cardiac allograft rejection and congestive heart failure (Citation10). TNF-α induces its biological effects by binding to 2 distinct sarcolemmal receptors, TNF receptors 1 and 2 (TNFR 1 and 2) (Citation11). Upon binding to TNF-α, the TNFRs recruit a number of molecules to form the TNFR complex, which in turn initiates the activation of downstream signalling cascade (Citation12). Both TNFR1 and TNFR2 are expressed in murine heart, but deleterious effects of TNF-α seem mainly initiated by activation of TNFR1 (reviewed in Ref. (Citation13). Thus, TNF-α/TNFR1 signalling induces mitochondrial dysfunction, reactive oxygen species (ROS) production, contractile abnormalities and cell death (Citation4, Citation11).
Microparticles (MPs) are extracellular vesicles of 0.1–1 µm in diameter, with pro-coagulant and pro-inflammatory properties (Citation14). Briefly, according to source cell type and triggering stimuli specificity (i.e. chemical/physical activation resulting in sustained high [Ca2+]i or apoptosis), MP formation occurs due to cytoskeleton disruption and subsequent plasma membrane blebbing. MPs contain nucleic acids, proteins and antigens from their cells of origin. Circulating MPs are present in the blood of healthy individuals, but MP formation increases with some diseased states, such as metabolic syndrome, sepsis, pre-eclampsia, rheumatoid arthritis, diabetes and in various clinical situations associated with thrombosis. Thus, several types of MPs including leukocyte-derived MPs are present in atherosclerotic plaques (Citation15, Citation16). Leukocyte-derived MPs are part of the pathological process of unstable atherosclerotic plaques and are thought to contribute to plaque rupture and subsequent myocardial infarction or stroke (Citation17). As shown by Leroyer et al. (Citation18), the majority of plaque MPs originates actually from leukocytes (more than 50%) of which almost 30% were from macrophages. Subsequently, it was shown that a large proportion (>90%) of these MPs are CD14+ indicating their monocyte/macrophage origin (Citation15).
In the present study, we have focused on MP released by apoptotic macrophages, testing the hypothesis that they could carry TNF-α and thus induce deleterious effects in adult myocardial cells. The murine RAW 264.7 cell line was used as a model for macrophages (Citation19) and we investigated the effects of MPs released via apoptosis induced by actinomycin D upon contractility in cardiomyocytes isolated from hearts of adult C57BL/6 mice.
Material and methods
Ethical approval
All experimental procedures were conducted in compliance with local guidelines for the care and use of laboratory animals (authorizations n°D49007002 on 17/07/2012). Furthermore, the study complied with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th edition, Washington (DC): National Academies Press (US); 2011. 2, Animal Care and Use Program).
Preparation of MPs
The murine RAW 264.7 macrophage-like cell line was obtained from BALB/c mice (American Type Culture Collection, Manasses, VA). Cells were maintained at 37°C in 5% CO2/95% air in DMEM (Lonza, Basel, Switzerland) supplemented with 10% decomplemented foetal calf serum (FCS, Invitrogen, Cergy Pontoise, France), 1% penicillin/streptomycin (GE Healthcare, Little Chalfont, Buckinghamshire, UK) and 1% l-glutamine (Lonza). Cells were seeded in culture plates (2×106/mL) and then treated with actinomycin D (1 µg/mL) for 24 h. Following treatment, supernatants were harvested and centrifuged twice at 1,500 g for 15 min and 5 min in order to eliminate cells and large debris, respectively. Remaining supernatant was subjected to 3 series of centrifugation (13,000 g for 1 h, then twice at 13,000 g for 45 min), to pellet MPs and the supernatant was replaced by 200 µL of 0.9% saline salt solution and stored at 4°C until subsequent use. Preliminary studies analyzing the concentration-dependent MP effects showed that 1 µg/mL corresponds to the maximally effective concentration in inducing functional effects and thus we used this concentration to analyze the impact of MPs upon contractility. With regard to apoptosis, we used 10 µg/mL, a concentration in accordance with that used to study apoptosis triggered by MPs from lymphocytes in bronchial epithelial cells (Citation20). Determination of the corresponding amount of MPs was carried out by measuring MP-associated proteins determined against bovine serum albumin standards (Lowry method).
Characterization of MPs
To determine MP concentration and the localization of TNF-α either on plasma membrane or inside, the MP labelling with TNF-α was carried out. Two microlitres of MPs were incubated 45 min with either 0.5 µL of anti-TNF-α alexa fluor® 488 (0.5 mg/mL) or anti-rat IgG1 kappa alexa fluor® 488 (0.5 mg/mL) (eBioscience, San Diego, CA) as an isotype-matched negative control. The presence of intraMP TNF-α was assessed after MP fixation (2% paraformaldehyde, 15 min, Electron Microscopy Sciences, Hatfield, PA) and permeabilization (0.1% saponin, 5 min). After incubation with antibodies, samples were diluted in 200 µL of 0.9% saline salt solution. Then, Flow-count fluorospheres (2 µL, equal to volume of MPs) (Beckman Coulter, Villepinte, France) were added in order to calculate MP count. The samples were analyzed in a flow cytometer 500 MPL System (Beckman Coulter) using the MXP software (Beckman Coulter). Sample analysis was stopped after the count of 10,000 events.
Isolated cardiomyocyte preparation
Ventricular cardiomyocytes were isolated from 8- to 10-week-old male adult C57BL/6 mice. Mice were pretreated with heparin (i.p. 100 U, 5 min, Heparine Choay, Sanofi Aventis, Paris). Then, mice were euthanized by CO2. The heart was rapidly excised, rinsed in ice-cold Hanks-HEPES buffer [in mM: NaCl 113, KCl 4.7, NaHCO3 12, KHCO3 10, Na2HPO4 0.6, KH2PO4 0.6, MgCl2 1.2, HEPES 10, glucose 5.5, taurine 29, supplemented with 2,3-butanedione monoxime (BDM) 10, pH 7.4], and then mounted on a Langendorff perfusion system and perfused (3 mL/min at 37°C) first with Hanks-HEPES buffer for 5 min to be cleared from blood and then with buffer supplemented with 10 mg/mL liberase TH (Roche Diagnostics GmbH, Manheim, Germany) for 6–9 min. The heart was placed into Hanks-HEPES buffer supplemented with BDM (10 mM) and FCS (10%). The atria were removed and the ventricles were gently dissociated with scissors and pipettes in the same medium. The suspension was filtered through a nylon mesh (200 µm) and incubated for decantation of the cells for 10 min. CaCl2 was progressively added in the Hanks-HEPES buffer in 4 steps of 5 min (0.125, 0.25, 0.50, 1 mM).
Ca2+ transient and cell shortening
Isolated cardiomyocytes were placed on laminin (Sigma-Aldrich)-precoated glass coverslips (Corning, NY) and loaded with Fura-2 AM (1 µM, Molecular Probes, Eugene, OR) at room temperature for 20 min and then washed with HEPES-buffered solution (in mM: NaCl 116, KCl 4.7, NaHCO3 4.4, Na2HPO4 0.6, KH2PO4 1.5, MgCl2 1.2, HEPES 20, glucose 10, taurine 20, pH 7.3) containing 1.25 mM CaCl2. Sarcomere shortenings and Ca2+ transients were studied using field stimulation (0.5 Hz, 0.1 ms, 7–15 V, 22°C) in the absence or in the presence of MPs from 5 min to 1 h. Sarcomere length and fluorescence (excitation at 340 and 380 nm, measured at 510 nm) were simultaneously recorded (IonOptix Myocyte Calcium and Contractility Recording System, Dublin, Ireland).
Inhibitors and chemicals
In some experiments, cardiomyocytes were preincubated for 30 min with different pharmacological inhibitors: L-NAME (100 µM, NOS inhibitor), 1H-[1.2.4]oxadiazolo[4.3-a]quinoxalin-1-one [ODQ, 25 µM, soluble guanylate cyclase (sGC) inhibitor], 6,7-dimethyl-3-[(methyl{2-[methyl({1-[3-(trifluoromethyl)phenyl]-1H-indol-3-yl}methyl)amino]ethyl}amino)methyl]-4H-chromen-4-one,-diHCl (SPD 304, 10 µM, TNF-inhibitor, Calbiochem, Darmstadt, Germany). A specific antibody to TNFR1 (purified anti-mouse TNFR type 1/p55, 20 µg/mL, BioLegend, San Diego, CA), a leaf purified Armenian hamster IgG (20 µg/mL, BioLegend) or TNF-α (50 ng/mL, Miltenyi Biotec, Bisley, UK) were also used. All drugs were prepared as a stock solution in appropriate solvents. All the drugs were used at already reported maximally active concentrations. On the day of the experiment, stock solutions were diluted to the desired concentration and the final concentration of DMSO was <0.1%. For the treatment with MPs (4 h) of cardiomyocytes further used in western blot, M199 culture medium was supplemented with creatine (5 mM), carnitine (5 mM), taurine (5 mM), HEPES (25 mM) and penicillin/streptomycin (1%). Unless stated, all chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Western blot
After treatment with MPs for 4 h, cardiomyocytes were homogenized and lysed. Proteins (20 µg) were separated on 4–12% SDS-polyacrylamide gel (Invitrogen) electrophoresis (150 V, 90 min). Blots were probed with polyclonal rabbit anti-cleaved caspase-3 (Cell Signaling Technology, Danvers, MA), mouse anti-caspase-8 (Cell Signaling), anti-Bax (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-cytochrome C (Santa Cruz). In another set of experiments, total proteins from MPs (10 and 20 µg) were also probed with a polyclonal goat anti-TNF-α (Santa Cruz). Mouse TNF-α recombinant protein was also used as a positive control (1, 2, 4, 8 and 10 ng) (Miltenyi Biotec). A polyclonal goat anti-α-tubulin (Santa Cruz) was used as a loading control. Membranes were washed 3 times in Tris-buffer solution (20 mM Tris, 61.5 mM NaCl) containing 0.1% Tween and incubated for 90 min at room temperature with the appropriate horseradish peroxidase-conjugated secondary antibody (Santa Cruz). Protein bands were detected by enhanced chemiluminescence plus (Amersham Biosciences, Piscataway, NJ). Immunoblots were quantified by densitometric analysis (Bio1D, Scientific Software Group, Provo, UT).
Interaction between MPs and cardiomyocytes
MPs were loaded with 2 µM PKH67 dye stock (Sigma-Aldrich) in NaCl (0.9%) solution, incubated for 2 min at room temperature and then added to an equal volume of FCS to stop the labelling. MPs were centrifugated (13,000 g, 45 min) and the pellet resuspended in NaCl (0.9%) solution. Fluorescence was checked by flow cytometry (Beckman Coulter). Isolated cardiomyocytes were then stimulated with MPs (10 µg/mL) labelled with PKH67 during 1, 2 and 4 h at room temperature. At the end of stimulations, cardiomyocytes were fixed with paraformaldehyde (2%, 20 min), washed and diluted with phalloidin-tetramethylrhodamine isothiocyanate (50 µg/mL, Sigma-Aldrich) during 90 min at room temperature, in order to label F-actin filaments. After washing with phosphate buffer solution, cells were observed by confocal microscopy (CLMS 700, Zeiss, ZEN fluorescence, Jena, Germany).
Statistics
Results were expressed as mean ± SEM and n represents the number of cells or mice for functional and western blot experiments, respectively. Statistical analysis was performed using ANOVA and subsequent Bonferroni post-hoc test for functional experiments, or ANOVA and Mann–Whitney U-test for western blot data (Prism® V5.0 software).
Results
Expression of TNF-α in MPs from apoptotic macrophages
Flow cytometry analysis of MPs (a) showed that 1 µg of MP proteins, the concentration used for the present functional study, corresponds to 52.14±5.4×103 MPs. As shown in b, no fluorescence was observed in MPs without TNF-α labelling. Upon labelling, only 1.8% of MPs present TNF-α on surface (c). Interestingly, we found that TNF-α is inside 11% of MPs as evidenced after permeabilization labelling (d and e).
Fig. 1. Tumour necrosis factor-α (TNF-α) expression in macrophage MPs. (a) Representative images of cytometer analysis showing MPs and counting beads represented on a forward light scatter (FS)/side light scatter (SS) dot-plot; dot plots of MP gated region are representations of FS vs. alexa fluor® 488 fluorescence signals from (b) unlabelled MPs, (c) intact MPs incubated in the presence of anti-TNF-α alexa fluor® 488, permeabilized MPs incubated (d) with isotype IgG1 alexa fluor® 488 or (e) with anti-TNF-α alexa fluor® 488. (f) Western blot showing TNF-α expression in MPs (10 and 20 µg) compared to a mouse recombinant TNF-α (1, 2, 4, 8 and 10 ng).
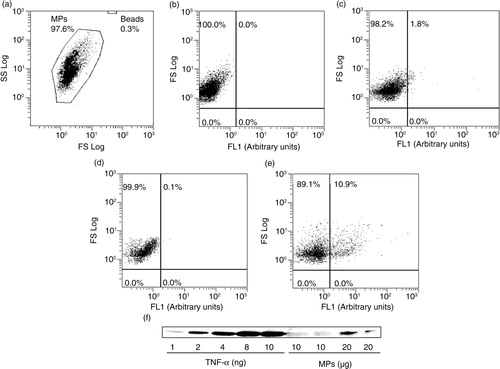
This expression was also confirmed by western blot analysis (f). Using the recombinant protein TNF-α at different growing concentrations as a positive control, these data indicated that MPs from apoptotic macrophages contain the soluble form of TNF-α. However, under our experimental conditions, we cannot exclude the presence of MPs bearing TNF-α at their membrane. Furthermore, the concentration of MPs used has been found in human atherosclerotic plaques (see Ref. 21).
Ca2+ transient and cell shortening
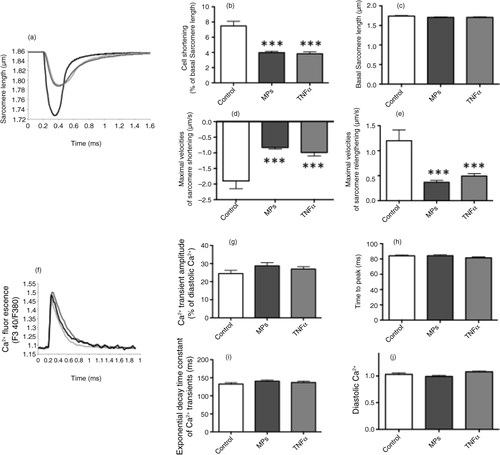
Isolated cardiomyocytes were loaded with Fura-2 (1 µM for 20 min) and paced at 0.5 Hz. Cell shortenings and changes in cytosolic Ca2+ were simultaneously recorded. The sarcomere shortening triggered by the electrical stimulation was reduced by MPs (1 µg/mL) and TNF-α (50 ng/mL) (a and b), without affecting the resting sarcomere length (c). The effect was apparent, maximal within 5 min of exposure to the MPs and TNF-α. The maximal velocities of sarcomere shortening and relengthening were also significantly reduced in the presence of MPs and TNF-α (d and e). Conversely, Ca2+ transient amplitude (f and g) and kinetics (h and i) were not significantly modified by MPs and TNF-α, and the diastolic Ca2+ level remained unchanged (j).
Fig. 2. Effects of macrophage MPs (1 µg/mL) and TNF-α (50 ng/mL) upon sarcomeric shortenings and Ca2+ transients of adult murine cardiomyocytes. (a) Representative traces of sarcomere shortenings of control and MP-treated cardiomyocytes (digital detection and fast Fourier transformation). (b) Sarcomere shortening (% of resting sarcomere length), (c) resting sarcomere length, maximal velocities of (d) sarcomere shortening and (e) relengthening, (f) representative traces of Ca2+ transients, (g) Ca2+ transient amplitude (F340/380), (h) time to peak and (i) exponential decay time constant of Ca2+ transients and (j) diastolic Ca2+ levels in control, MP- and TNF-α-treated cardiomyocytes. Data are expressed as mean±SEM (n=28–50). ***P<0.001 vs. Control (statistical analysis performed using ANOVA and subsequent Bonferroni post-hoc test).
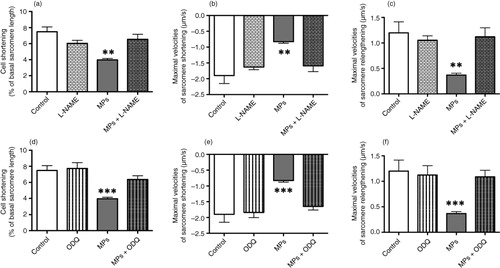
NO pathway is well documented as a negative modulator of cardiac inotropy (Citation22, Citation23). Thus, experiments were carried out to evaluate the impact of inhibitors of this pathway. The reduction of cell shortening amplitude and slowing of contraction kinetics observed when cells were perfused with 1 µg/mL MPs was completely prevented after pretreatment (30 min) of the NOS inhibitor, L-NAME (a, b and c) or of the sGC inhibitor, ODQ (d, e and f).
Fig. 3. Effects of macrophage MPs (1 µg/mL) upon sarcomeric shortenings of adult murine cardiomyocytes in the absence or in the presence of different inhibitors. (a) Sarcomere shortening (% of resting sarcomere length) and maximal velocities of (b) sarcomere shortening and (c) relengthening, in control, L-NAME (100 µM) alone-treated and MP-treated cardiomyocytes in the absence or in the presence of L-NAME. (d) Sarcomere shortening (% of resting sarcomere length) and maximal velocities of (e) sarcomere shortening and (f) relengthening, in control, ODQ (25 µM) alone-treated, and MP-treated cardiomyocytes in the absence or in the presence of ODQ. Data are expressed as mean±SEM (n=17–50). **P<0.01 vs. L-NAME and MPs+L-NAME. ***P<0.001 vs. ODQ and MPs + ODQ. Statistical analyses were performed using ANOVA and subsequent Bonferroni post-hoc test.
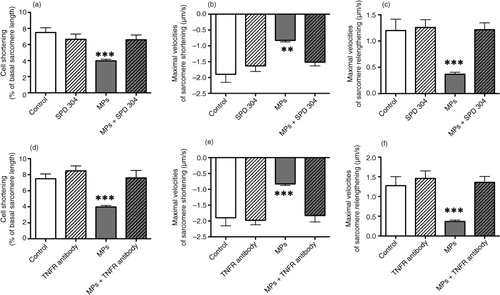
Because of the presence of TNF-α revealed by western blotting of MPs (see above), we also tested the effect of (a) SPD304, known to prevent TNF-α binding to its receptor (Citation24) and of (b) a specific antibody to TNFR1. In some control experiments, we also used a leaf purified Armenian hamster IgG (20 µg/mL) which has no effect on cardiomyocyte contractility (data not shown). Interestingly, the effects of MPs (1 µg/mL) upon cell shortening were completely prevented after pretreatment with SPD304 (10 µM, 30 min) (a, b and c) or the antibody to TNFR1 (20 µg/mL, 1 h) (d, e and f).
Fig. 4. Effects of macrophage MPs (1 µg/mL) upon sarcomeric shortenings of adult murine cardiomyocytes in the absence or the presence of different TNF-α blockers. (a) Sarcomere shortening (% of resting sarcomere length), maximal velocities of (b) sarcomere shortening and (c) relengthening in control, SPD 304 (10 µM) alone-treated, and MP-treated cardiomyocytes in the absence or in the presence of SPD 304. (d) Sarcomere shortening (% of resting sarcomere length), maximal velocities of (e) sarcomere shortening and (f) relengthening in control, TNFR1 antibody (20 µg/mL) alone-treated, and MP-treated cardiomyocytes in the absence or in the presence of TNFR1 antibody. Data are expressed as mean±SEM (n=12–50). **P<0.01, **P<0.001 vs. SPD 304 or TNFR1 antibody and MPs+SPD 304 or TNFR1 antibody. Statistical analyses were performed using ANOVA and subsequent Bonferroni post-hoc test.
The 3 inhibitors (L-NAME, ODQ, SPD 304) and TNFR1 antibody alone have no significant effects upon Ca2+ transients (data not shown) and cell shortening ( and 4) concomitantly measured.
Interaction between MPs and cardiomyocytes
Firstly, using flow cytometer, we checked whether MPs from apoptotic murine RAW 264.7 macrophage cell line, were effectively labelled with the probe PKH67. A right shift of fluorescence peaks clearly confirmed that MPs were efficiently labelled (data not shown).
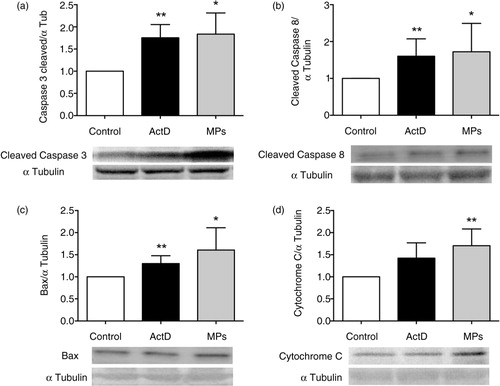
Then, cardiomyocytes from adult mice were treated with labelled MPs (10 µg/mL) during different durations of time (1, 2 and 4 h) and observed in confocal microscopy in a z-stack of serial imaging of 2 µm. After 1 h, MPs were clearly surrounding rod-shaped (i.e. alive) cardiomyocytes (). After 2 and 4 h of treatment, as illustrated on orthogonal sections in XZ and YZ planes, MPs were internalized into cardiomyocytes undergoing to the early phase of apoptosis (). Thus, cardiomyocyte apoptosis was also studied after 4 h of pretreatment with MPs (10 µg/mL) in terms of caspase-3, caspase-8, Bax and cytochrome C expressions. The caspase family of aspartate-specific cysteine proteases is the central executor of apoptosis. The expressions of 17 kDa-cleaved caspase-3 and of 18 kDa-cleaved caspase-8 fragments were similarly increased in MP-exposed murine adult cardiomyocytes (caspases vs. α-tubulin densitometric ratios increased, a and b). The expression of the proapoptotic protein Bax, which determines the ability of cells to undergo apoptosis, was also significantly increased in the presence of MPs and the proapoptotic effect of MPs was further confirmed by an increase of cytochome C on the cytosolic extract probably by an increase of cytochrome C release from mitochondria (c and d).
Fig. 5. Interactions between macrophage MPs and adult murine cardiomyocytes. Confocal images of (a) PKH67-labelled MPs (green), (b) phalloidin-labelled adult ventricular cardiomyocytes and (c) merge images after 1, 2 and 4 h of incubation. For each merge image, orthogonal sections are represented in the XY plane (central panel), in the XZ plane (upper panel) and in the YZ plane (right panel). White bars: 10 µm.

Fig. 6. Apoptosis of adult murine cardiomyocytes in the presence of macrophage MPs. Western blot showing increased expression of (a) 17 kDa-cleaved caspase-3, (b) 18 kDa-cleaved caspase-8, (c) Bax and (d) cytochrome C in MPs (10 µg/mL) or actinomycin D (ActD) (1 µg/mL)-exposed adult murine cardiomyocytes. Data are mean±SEM (n=5–12). *P<0.05 and **P<0.01 vs. control (statistical analyses were performed using ANOVA and Mann–Whitney U-test).
Discussion
In the present work, we used the murine RAW 264.7 cell line induced in apoptosis using actinomycin D to promote MP release. The present study showed that these MPs carry functional TNF-α and activate TNFR1 in isolated adult murine ventricular cardiomyocytes, leading to cell shortening without affecting calcium signalling via a mechanism sensitive to NO synthase inhibitor.
Thus, in the present study, MPs from apoptotic macrophages induced a negative inotropic effect and slowing of both contraction and relaxation, similar to the effects observed in the presence of TNF-α. It should already be noted that such effects were not observed in the presence of MPs obtained from apoptotic human lymphoid CEM T cell line where apoptosis was also induced by treatment with actinomycin D (Citation25). Furthermore, the negative inotropic effect of MPs from apoptotic macrophages was absent after pretreatment with a TNF-α inhibitor, suggesting that receptors to the cytokine were involved in the responses to MPs. Reduced contractility by cytokines has been described in whole heart, multicellular preparations and isolated ventricular cardiomyocytes, and has been correlated with NO induction by constitutive NOS in numerous studies (Citation3, Citation4, Citation26). However, the findings are contradictory in the time frame of effect of TNF-α. Most report effects occurring progressively, over 10 min (Citation4, Citation7) though relatively acute effects (1 min), have been reported (Citation27, Citation28). Furthermore, some studies have shown a biphasic effect of TNF-α, with an initial increase of systolic [Ca2+]i preceding an ultimate decrease (Citation7, Citation29). Furthermore, although most studies have reported a reduction in systolic [Ca2+]i and shortening (Citation4, Citation7, Citation28, Citation29), others have found no effect on systolic [Ca2+]i (Citation4) and even an increase (Citation7). As pointed out by Greensmith and Nirmalan (Citation28), the lack of homogeneity regarding the effects of TNF-α on contractility in cardiomyocytes may be as a result of the diverse models, temperatures and techniques used. In our study, the maximal effects of MPs from apoptotic murine RAW 264.7 macrophage cell line carrying TNF-α occurred within 5 min and we also showed that within 1 h of treatment, these MPs were still surrounding cardiomyocytes which strongly supports the view that the effects were most likely through receptor binding, that is, TNFRs. This point was reinforced by the complete blockade of the effect in the presence of the TNF-α inhibitor or TNFR1 antibody. Furthermore, the rapid effect of TNF-α by MPs containing the soluble form of the cytokine is not surprising. Indeed, MacKenzie et al. (Citation30) showed that shed microvesicles from human THP-1 monocytes were capable of releasing bioactive interleukin-1β (IL-1β) from their contents to activate IL-1 receptors at target cells (HeLa cells) within 5 min.
Reduction in contractile amplitude can result either from depressed Ca2+ transients or from altered Ca2+ myofilament sensitivity. In the present study, we never observed any change in the amplitude of Ca2+ transients. Our study also showed that the negative inotropic effect of MPs from apoptotic macrophages could be completely blocked by the NOS inhibitor L-NAME and the sGC inhibitor ODQ, suggesting that NO pathway is implicated in the observed effect. It also strongly suggests that the sGC/cGMP/PKG pathway is the sole mechanism for this MP-induced negative inotropy.
TNF-α induces cardiomyocyte apoptosis and this effect is mediated by an increase in oxidative stress (Citation31, Citation32). The pro-apoptotic role of TNF-α has been extensively investigated in various in vitro and in vivo models (Citation31–Citation33). TNF-α causes apoptotic cell death by the two different but interrelated pathways, so-called extrinsic (TNFR binding and caspase-3 activation) and intrinsic (overproduction of ROS and mitochondrial injury) pathways (Citation32). In the present study, chronic exposure to MPs from apoptotic macrophages induced apoptosis and increased the expression of proapoptotic proteins, such as cleaved caspase-3 and -8, Bax and cytochrome C, suggesting that the intrinsic pathway, involving the mitochondria, was particularly involved in the proapoptotic effects of these MPs. Human atherosclerotic plaques harbour MPs released from apoptotic cell death within the lesion (Citation18). Canault et al. (Citation21) also showed that these MPs carry catalytically active metalloprotease TNF-α converting enzyme (TACE/ADAM17), which significantly enhances the cell surface processing of the TACE/ADAM17 substrates TNF-α, and TNFR1. Present study suggests that MPs from apoptotic macrophages carry also per se TNF-α that would potentiate their proapoptotic property. Altogether, these data support the interaction between MPs bearing TNF-α and TNFR1 on the cardiomyocytes for the rapid cell shortening and most likely the internalization for the induction apoptosis.
During the acute phase of myocardial infarction, resident macrophages in the myocardial tissue are activated following neutrophil infiltration into the infarcted region. In addition, monocytes, induced by the expression of chemotactic factors, infiltrate the infarcted myocardium, differentiate to macrophages and secrete a variety of inflammatory cytokines participating to the evolution towards cardiac failure. Even if further works need to be carried out in Langendorff perfused hearts in order to add substantially to the physiological relevance of the present study, our report suggests that MPs issued from activated macrophages could carry TNF-α and then contribute to propagation of inflammatory signals leading to myocardial infarction. Thus, the present study allows supporting the hypothesis that MPs from human carotid atherosclerotic plaques may contain active TNF-α, which could contribute to MP-induced inflammatory signals in human atherosclerotic lesions.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
Acknowledgements
The authors are grateful to the ITMO “Physiopathologie, Métabolisme, Nutrition” Inserm, and the “Collectivités Territoriales Angevines” for financial support. The authors thank the “Plateforme de Métabolomique” of University of Angers for technical assistance and SCAHU staff (University of Angers) for animal handling. EM is recipient of a doctoral fellowship from Nanofar Erasmus Mundus Program.
References
- Frantz S, Nahrendorf M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc Res. 2014; 102: 240–8.
- Haudek SB, Taffet GE, Schneider MD, Mann DL. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest. 2007; 117: 2692–701.
- Balligand JL, Ungureanu D, Kelly RA, Kobzik L, Pimental D, Michel T et al. Abnormal contractile function due to induction of nitric oxide synthesis in rat cardiac myocytes follows exposure to activated macrophage-conditioned medium. J Clin Invest. 1993; 91: 2314–9.
- Goldhaber JI, Kim KH, Natterson PD, Lawrence T, Yang P, Weiss JN. Effects of TNF-α on [Ca2+]i and contractility in isolated adult rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 1996; 271: H1449–55.
- Vasudevan NT, Mohan ML, Gupta MK, Martelli EE, Hussain AK, Qin Y et al. Gβγ-independent recruitment of G-protein coupled receptor kinase 2 drives tumor necrosis factor α-induced cardiac β-adrenergic receptor dysfunction. Circulation. 2013; 128: 377–87.
- Oral H, Dorn GW II, Mann DL. Sphingosine mediates the immediate negative inotropic effects of tumor necrosis factor α in the adult mammalian cardiac myocyte. J Biol Chem. 1997; 272: 4836–42.
- Amadou A, Nawrocki A, Best-Belpomme M, Pavoine C, Pecker F. Arachidonic acid mediates dual effect of TNF-α on Ca2+ transients and contraction of adult rat cardiomyocytes. Am J Physiol Cell Physiol. 2002; 282: C1339–47.
- Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Eng J Med. 1990; 323: 236–41.
- Kubota T, McTierman CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP et al. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-α. Circ Res. 1997; 81: 627–35.
- Kleinbongard P, Schulz R, Heusch G. TNF-alpha in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail Rev. 2011; 16: 49–69.
- Torre-Amione G, Kapadia S, Lee J, Bies RD, Lebovitz R, Mann DL. Expression and functional significance of tumor necrosis factor receptors in human myocardium. Circulation. 1995; 92: 1487–93.
- Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002; 296: 1634–5.
- Prabhu SD. Cytokine-induced modulation of cardiac function. Circ Res. 2004; 95: 1140–53.
- Martinez MC, Tual-Chalot S, Leonetti D, Andriantsitohaina R. Microparticles: targets and tools in cardiovascular disease. Trends Pharmacol Sci. 2011; 32: 659–65.
- Mayr M, Grainger D, Mayr U, Leroyer AS, Leseche G, Sidibe A et al. Proteomics, metabolomics, and immunomics on microparticles derived from human atherosclerotic plaques. Circ Cardiovasc Genet. 2009; 2: 379–88.
- Rautou PE, Leroyer AS, Ramkhelawon B, Devue C, Duflaut D, Vion AC et al. Microparticles from human atherosclerotic plaques promote endothelial ICAM-1-dependent monocyte adhesion and transendothelial migration. Circ Res. 2011; 108: 335–43.
- Sarlon-Bartoli G, Bennis Y, Lacroix R, Marti P, Bartoli MD, Arnaud L et al. Plasmatic level of leukocyte-derived microparticles is associated with unstable plaque in asymptomatic patients with high-grade carotid stenosis. J Am Coll Cardiol. 2013; 62: 1436–41.
- Leroyer AS, Isobe H, Leseche G, Castier Y, Wassef M, Mallat Z et al. Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J Am Coll Cardiol. 2007; 49: 772–7.
- Gauley J, Pisetsky DS. The release of microparticles by RAW 264.7 macrophage cells stimulated with TLR ligands. J Leukoc Biol. 2010; 87: 1115–23.
- Qiu Q, Xiong W, Yang C, Gagnon C, Hardy P. Lymphocyte-derived microparticles induce bronchial epithelial cells’ proinflammatory cytokine production and apoptosis. Mol Immunol. 2013; 55: 220–30.
- Canault M, Leroyer AS, Peiretti F, Lesèche G, Tedgui A, Bonardo B et al. Microparticles of human atherosclerotic plaques enhance the shedding of the tumor necrosis factor-α converting enzyme/ADAM17 substrates, tumor necrosis factor and tumor necrosis factor receptor-1. Am J Pathol. 2007; 171: 1713–23.
- Hare JM. Nitric oxide and excitation–contraction coupling. J Mol Cell Cardiol. 2003; 35: 719–29.
- Hammond J, Balligand JL. Nitric oxide synthase and cyclic GMP signaling in cardiac myocytes: from contractility to remodeling. J Mol Cell Cardiol. 2012; 52: 330–40.
- He MM, Smith AS, Oslob JD, Flanagan WM, Braisted AC, Whitty A et al. Small-molecule inhibition of TNF-alpha. Science. 2005; 310: 1022–5.
- Paulis L, Fauconnier J, Cazorla O, Thireau J, Soleti R, Vidal B et al. Activation of Sonic hedgehog signaling in ventricular cardiomyocytes exerts cardioprotection against ischemia reperfusion injuries. Sci Rep. 2015; 5: 7983.
- Finkel MS, Oddis CV, Jacobs TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992; 257: 387–9.
- Sugishita K, Kinugawa K-I, Shimizu T, Harada K, Matsui H, Takahashi T et al. Cellular basis for the acute inhibitory effects of IL-6 and TNF-[alpha] on excitation-contraction coupling. J Mol Cell Cardiol. 1999; 31: 1457–67.
- Greensmith DJ, Nirmalan M. The effects of tumor necrosis factor-alpha on systolic and diastolic function in rat ventricular myocytes. Physiol Rep. 2013; 1: e00093.
- Cailleret M, Amadou A, Andrieu-Abadie N, Nawrocki A, Adamy C, Ait-Mamar B et al. N-acetylcysteine prevents the deleterious effect of tumor necrosis factor-(alpha) on calcium transients and contraction in adult rat cardiomyocytes. Circulation. 2004; 109: 406–11.
- MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North A, Surprenant A. Rapid secretion of interleukin-1β by microvesicle shedding. Immunity. 2001; 8: 825–35.
- Dhingra S, Sharma AK, Singla DK, Singal PK. p38 and ERK1/2 MAPKs mediate the interplay of TNF-α and IL-10 in regulating oxidative stress and cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007; 293: H3524–31.
- Dhingra S, Sharma AK, Arora RC, Slezak J, Singal PK. IL-10 attenuates TNF-α-induced NF κB pathway activation and cardiomyocyte apoptosis. Cardiovasc Res. 2009; 82: 59–66.
- Engel D, Peshock R, Armstrong RC, Sivasubramanian N, Mann DL. Cardiac myocyte apoptosis provokes adverse cardiac remodeling in transgenic mice with targeted overexpression. Am J Physiol Heart Circ Physiol. 2004; 287: H1303–11.