
Abstract
Background
Exosomes have been implicated in tumour progression and metastatic spread. Little is known of the effect of mechanical and innate immune interactions of malignant cell-derived exosomes on endothelial integrity, which may relate to increased extravasation of circulating tumour cells and, therefore, increased metastatic spread.
Methods
Exosomes isolated from non-malignant immortalized HCV-29 and isogenic malignant non-metastatic T24 and malignant metastatic FL3 bladder cells were characterized by nanoparticle tracking analysis and quantitative nanomechanical mapping atomic force microscopy (QNM AFM) to determine size and nanomechanical properties. Effect of HCV-29, T24 and FL3 exosomes on human umbilical vein endothelial cell (HUVEC) monolayer integrity was determined by transendothelial electrical resistance (TEER) measurements and transport was determined by flow cytometry. Complement activation studies in human serum of malignant and non-malignant cell-derived exosomes were performed.
Results
FL3, T24 and HCV-29 cells produced exosomes at similar concentration per cell (6.64, 6.61 and 6.46×104 exosomes per cell for FL3, T24 and HCV-29 cells, respectively) and of similar size (120.2 nm for FL3, 127.6 nm for T24 and 117.9 nm for HCV-29, respectively). T24 and FL3 cell-derived exosomes exhibited a markedly reduced stiffness, 95 MPa and 280 MPa, respectively, compared with 1,527 MPa with non-malignant HCV-29 cell-derived exosomes determined by QNM AFM. FL3 and T24 exosomes induced endothelial disruption as measured by a decrease in TEER in HUVEC monolayers, whereas no effect was observed for HCV-29 derived exosomes. FL3 and T24 exosomes traffic more readily (11.6 and 21.4% of applied exosomes, respectively) across HUVEC monolayers than HCV-29 derived exosomes (7.2% of applied exosomes). Malignant cell-derived exosomes activated complement through calcium-sensitive pathways in a concentration-dependent manner.
Conclusions
Malignant (metastatic and non-metastatic) cell line exosomes display a markedly reduced stiffness and adhesion but an increased complement activation compared to non-malignant cell line exosomes, which may explain the observed increased endothelial monolayer disruption and transendothelial transport of these vesicles.
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Exosomes are nanoscale (50–130 nm) extracellular vesicles of endosomal origin detectable in most biological fluids (Citation1, Citation2). The role of exosomes in intercellular communication is supported by a composition that includes functional miRNA, mRNA, bioactive lipids and proteins (Citation3), whose transfer results in altered phenotypes of the target cells (Citation4). This could have considerable implications in disease conditions, including cancer (Citation5).
Tumour-derived exosomes can increase tumour invasiveness and proliferation in an autocrine fashion (Citation6), in addition to interactions with host stromal cells, such as the conversion of fibroblasts to a myofibroblast phenotype resulting in extracellular matrix (ECM) remodelling that is conducive to tumour growth (Citation7). The presence of matrix metalloproteinases also allows for direct modulation of the ECM by tumour-derived exosomes in the primary tumour microenvironment and metastatic spread (Citation8). We and others have also demonstrated that exosome secretion may facilitate advantageous exocytosis and cellular removal of tumour suppressors, showing the complex role of exosomes within tumour progression (Citation9, Citation10).
Metastasis is the primary cause of mortality in cancer patients, and it is driven by circulatory tumour cell (CTC) extravasation across endothelium and consequent formation of tissue micrometastasis (Citation11). The release of exosomes into the interstitial space and subsequent dissemination throughout the body highlights a potential role for tumour-derived exosomes in formation of a pre-metastatic niche (Citation12, Citation13) beyond that of an auto/paracrine action at the primary tumour site. This potential is further supported by increased vascular leakiness and concomitant increased metastatic lesion formation in the lungs of mice after systemic administration of malignant cell-derived exosomes (Citation14).
Factors that contribute to the cellular extravasation across endothelia are poorly understood; mechanical properties and immune interactions, however, may play a role. An increase in paracellular permeability and increased eosinophil migration has been shown to occur as a result of activation of endothelial C5aR, mediated by C5a (Citation15), resulting in cell retraction that suggests a contribution from complement activation. Recent work links chronic intratumoural complement activation to tumour progression. For instance, the measurement of C4d levels in astrocytomas correlated with cancer severity grade (Citation16). Another study in immunocompetent mice bearing a syngeneic tumour has strongly indicated that intratumoural accumulation of complement activating nanoparticles can accelerate tumour growth through C5a generation (Citation17). The role of C5a liberation and C5a receptor in tumour growth was confirmed in C5 and C5a receptor knock-out animals (Citation17). Collectively these observations may indicate a potential role in intratumoural complement activation by tumour-derived exosomes in tumour growth.
This work investigates the mechanical properties using atomic force microscopy (AFM), complement activation and the effect on endothelial integrity and transport across a primary endothelial cell barrier model of non-malignant, isogenic malignant metastatic and malignant non-metastatic cell-derived exosomes.
The findings in this work support a role of malignant cell-derived exosomes in loss of endothelial integrity and complement activation that may have a role in metastatic lesion formation as well as tumour growth.
Materials and methods
Cell lines and culture conditions
Human urothelial HCV-29 cells originate from a histologically normal bladder mucosa obtained from a patient with a previous history of bladder papillomata treated with irradiation (Citation18). Metastatic human urothelial FL3 cell line is an isogenic derivative of poorly metastatic T24T and its non-metastatic parental cell line T24 generated through reiterative tail vein injections in mice (Citation19). It forms pulmonary metastases in mice, whereas T24 does not (Citation10, Citation20). The cell lines were maintained in DMEM +10% (v/v) fetal bovine serum (FBS) +100 U/mL penicillin+100 µg/mL streptomycin (P/S) (all Life Technologies, Carlsbad, CA, USA). Cell line authentication was performed using short tandem repeat (STR) profiling (Cell ID System, Promega, Madison, WI, USA) to confirm identity and 100% isogenic relationship between T24 and FL3 cells.
Human umbilical vein endothelial cells (HUVECs) (Sigma Aldrich, Saint Louis, MO, USA) were maintained in EGM-2 +5% (v/v) FBS (Lonza, Basel, Switzerland) and harvested for use at no more than 75% confluence and passage number 6. Cells were incubated at 37°C at 5% CO2.
Generation of exosome-depleted media
DMEM +10% (v/v) FBS +P/S was subjected to 18 h ultracentrifugation at 200,000 g to remove bovine vesicles present in FBS. Supernatant was decanted and filtered at 0.2 µm, prior to use.
Exosome generation and isolation
Cells were cultured to 70% confluence prior to washing with Dulbecco's phosphate-buffered saline (PBS), pH 7.4, osmolality 275–304 mOs/kg (Sigma Aldrich) followed by the addition of exosome-depleted media (EDM) for 48 h. At that point EDM was harvested and subjected to sequential centrifugation at 300 g for 10 min, 2,000 g for 15 min, 15,000 g for 30 min, a final pelleting step of 100,000 g for 90 min followed by a washing step in PBS prior to re-pelleting at 100,000 g for 90 min (Citation21) using an Optima™ L-80 XP with a Ti-60 fixed angle rotor (Beckman Coulter, Brea, CA, USA). All exosomes were isolated on the day investigations were to be performed to reduce the possibility of degradation. The method of isolation has previously been optimized and assessed for exosomal markers and markers of contamination (Citation21, Citation22).
Nanoparticle tracking analysis
Exosome size and concentration was analysed by nanoparticle tracking analysis (NTA) using an LM-10 (NanoSight, Malvern Instruments, Malvern, UK). Measurements were performed in triplicates of 30-s video captures with camera level 13 and detection threshold 5 for all analysis. The data were analysed using software version 3.0.
HUVEC monolayer transendothelial electrical resistance experiments
xCELLigence E-16 plates (Roche, Basel, Switzerland) were coated with sterile 0.1% (w/v) gelatin (Sigma Aldrich) for 1 h at 37°C, followed by washing in PBS prior to use. 2.5×104 HUVEC cells were seeded per well in 100 µL EGM-2 media. The E-plate was then placed in the incubator and connected to the xCELLigence system (Roche). Cells were then cultured for 18 to 21 h to allow monolayer formation, which was assessed by impedance measurements (reported as cell index, CI) using RTCA v. 1.2 software (Roche). After reaching a CI plateau, indicative of an established monolayer, the measurements were paused and the E-plate moved to a pre-warmed holder followed by aspiration of media. Subsequently exosomes were applied to pre-warmed (37°C) EGM-2 media at the concentrations indicated. E-plates were then returned to the incubator and transendothelial electrical resistance (TEER) measurements acquired at 10 min intervals. Exosome dosing was repeated every hour for a total of 3 doses. Changes in CI were analysed using RTCA software v. 1.2 with calculation of delta-CI at T=7 h from first exosome addition.
Resistance to anoikis
HCV-29, T24 and FL3 cells were seeded for 24 h in 24-well Corning Costar ultra-low attachment plates (Corning Life Sciences, Corning, NY, USA). They were subsequently harvested and viability assessed using NucleoCounter NC-3000 (Chemometec, Allerod, Denmark), with staining by acridine orange for cell detection and nucleic acid stain DAPI for detection of non-viable cells. Viability index was calculated as the number of viable cells after 24 h against the number of cells seeded.
Fluorescent labelling of exosomes
HCV-29, T24 and FL3 cells were cultured in DMEM+10% FBS+P/S to 70% confluence and incubated with 4 µM DPPE-Rhod (Avanti Polar Lipids, Alablaster, AL, USA) in cold Opti-Mem (Life Technologies) media for 1 h. Cells were then washed 3 times in PBS and incubated with EDM. Fluorescent exosomes were collected by ultracentrifugation using the standard protocol (Citation21). 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE)-rhodamine fluorescent exosome size was analysed by NTA using an LM-10 (NanoSight, Malvern Instruments, Malvern, UK) and the fluorescent content measured using a FLUOstar Optima plate reader (BMG Labtech, Ortenberg, Germany). Plasma membrane integrity and cell viability were evaluated by measuring lactate dehydrogenase (LDH) release and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reduction assay with appropriate kits according to the manufacturer's protocol.
LDH release assay on HUVEC cells
Media from HUVEC cells 7 h post-exosome application was removed and centrifuged at 300 g for 5 min to remove cells. Then 50 µL of supernatant was transferred to a new 96-well plate and 50 µL LDH reagent (Roche) was added. The remaining cells were lysed by 0.1% (v/v) Triton-X and 50 µL lysate transferred for LDH quantification. Plates were incubated for 30 min in the dark at room temperature prior to absorbance reading at 490 nm. Induction of plasma membrane damage was calculated as percent released LDH (media) per total LDH (media+cells). HUVEC cells treated with 0.1% (v/v) Triton-X for 15 min served as the positive control for cell death.
Transwell transport of fluorescently labelled exosomes
HUVEC cells were seeded at a density of 100,000 cells/cm2 in apical well of 6.5 mm Transwell inserts (Corning Life Sciences) with a 0.4-µm pore diameter, precoated for 1 h at 37°C with sterile 0.1% (w/v) gelatin prior to washing once in PBS. Cells were incubated for 18 h in EGM-2 media, at which point TEER measurements were taken to confirm establishment of a monolayer. Inserts in media alone were used as background control for resistance measurements. Fluorescent exosomes were harvested and samples to be added to HUVEC cells were normalized to fluorescent vesicle number derived from fluorescent NTA analysis. Exosomes were added to HUVEC cells in Hanks’ balanced salt solution and incubated for 7 h prior to harvest of apical and basal well media for fluorescent analysis on a fluorescent plate reader. The fluorescent signal detected was normalized to the fluorescent intensity of the input material. Cells were then washed twice in PBS then incubated in trypsin to detach cells. DMEM+10% (v/v) FBS was added to apical wells to neutralize trypsin; cells were removed and spun at 200 g for 7 min. The supernatant was removed and resuspended in PBS+2% (v/v) FBS, then immediately analysed on a 2-laser, 6-colour Gallios flow cytometer (Beckman Coulter) with gating for single cells and analysis of fluorescent signal in FL2 channel.
Quantitative nanomechanical mapping atomic force microscopy in liquid
Exosome samples were drop-cast on freshly cleaved muscovite mica in PBS. In order to investigate exosomes’ physicochemical properties, dynamic nanomechanical mapping (DNM) (Citation23) experiments were carried out using PeakForce tapping quantitative nanomechanical mapping atomic force microscopy (PeakForce QNM AFM) in liquid (Bruker, Santa Barbara, CA, USA). Due to the fragile nature of exosomes, soft cantilevers with sharp tips (ScanAsyst-Fluid+, nominal spring constant ≈0.7 N/m, nominal tip radius ≈2 nm) were used. Cantilevers were calibrated in liquid and recalibrated after each sample in order to reduce systematic errors. Scan parameters such as scan speed, feedback gains and tip load were optimized for the highest possible resolution without damaging the sample or the tip. Images were acquired from multiple channels simultaneously, including but not limited to height, adhesion, stiffness, deformation, and so on. These images were later processed and the data analysed using commercial software (SPIP, Aalborg, Denmark).
In PeakForce mode, each time the tip interacts with the surface, force–time and force–distance curves are generated, with the maximum force applied being controlled. The tip presses on the surface (nanoindentation) up to the set PeakForce value. The sample stiffness, a measure of its rigidity, was derived from the ratio of the applied force versus the displacement produced by the same force along the same degree of freedom. Snap-off is the point where the interaction force reaches its minimum as the tip breaks contact with the sample, from which adhesion was calculated. Adhesion generally implies the “stickiness” of the sample.
In vitro complement activation assays
Details for human serum preparation, characterization and functional assessment of complement pathways were in accordance with our previous studies (Citation24). To measure complement activation in vitro, we determined the exosome-induced rise of human serum complement products C5a and SC5b-9 using the respective ELISA kits (Quidel, San Diego, CA, USA) according to the manufacturer's protocols as described previously (Citation24, Citation25). Briefly, complement activation was initiated by adding the required quantity of exosomes to undiluted serum in Eppendorf tubes in a shaking water bath at 37°C for 30 min, unless stated otherwise. Reactions were terminated by quickly cooling samples on ice and adding 25 mM ethylenediaminetetraacetic acid (EDTA). After centrifugation, the supernatant was used for the determination and quantification of complement activation products C5a and SC5b-9. Control plasma incubation contained PBS (the same volume as the exosome samples) to assess background levels, and zymosan (1 mg/mL) was used as a positive control for monitoring complement activation throughout. In selected experiments, exosome-mediated complement activation was monitored in the presence of EGTA/Mg2+ (10.0 mM/2.5 mM). The efficacy of exosome treatments was established by comparison with baseline levels using paired t-test; correlations between 2 variables were analysed by linear regression, and differences between groups (when necessary) were examined using ANOVA followed by multiple regression with a Student-Newman-Keuls test.
Unless otherwise stated, all experiments within this work were performed in biological triplicates and were representative of at least 2 independent experiments.
Results
Exosome characterization and mechanical properties
To investigate possible differences in the mechanical properties of exosomes related to tumourigenicity, exosomes from human (a) immortalized non-malignant urothelial cells (HCV-29), (b) malignant but non-metastatic urothelial cells (T24) and (c) a malignant metastatic isogenic derivate of T24 (FL3) were isolated and characterized. Upon differential centrifugation, exosomes from all 3 cell lines displayed similar vesicle size (mean average of triplicate analysis) as determined by NTA (117.9 nm for HCV-29, 127.6 nm for T24 and 120.2 nm for FL3, respectively) (a–c). In addition, the cell lines produced comparable quantities of exosomes when normalized to cell number (6.46, 6.64 and 6.61×104 exosomes per cell for HCV-29, FL3 and T24 cells, respectively) (d). The isolation procedure has been validated previously to be suitable for the harvest of exosomes as assessed by NTA and Western blotting for known markers of exosomes (CD81, Syntenin and TSG101) and possible contaminating organelles (Calreticulin and VDAC1) (Citation21, Citation22).
Fig. 1. Nanoparticle tracking analysis (NTA) size distributions of vesicles harvested from (a) FL3, (b) HCV-29 and (c) T24 cells. (d) Vesicle number harvested assessed by NTA normalized to cell number; vesicles produced per cell were not significantly different among any cell line, determined by Student's paired t-tests. (e) Resistance to anoikis assay assessing malignant capability of HCV-29, FL3 and T24 cells. p-values between pairs of groups were determined by Student's paired t-tests, *p<0.05; N.S., not significant.
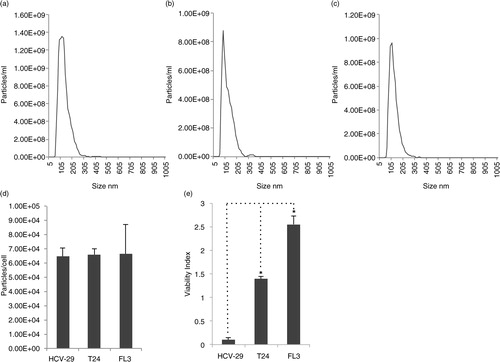
FL3 cells, in contrast to T24 cells, have previously been shown to induce solid tumours and pulmonary metastases in nude mice (Citation19, Citation20). We further examined the capability of the cell lines to avoid anoikis, a cell death programme induced upon cellular detachment. Anoikis is a prerequisite for cancer cell survival in the bloodstream during the metastatic cascade. We have previously observed increased resistance to anoikis of FL3 compared to T24 cells (Citation20). These results were confirmed in this study (e). In addition, non-malignant HCV-29 cells displayed little resistance to anoikis compared with T24 and FL3 cells (e).
AFM, especially with DNM, has been used widely for biomolecular imaging due to its high resolution and high speed but, most importantly, its capability for liquid operation, which enables label-/stain-free, non-destructive and non-invasive measurements (Citation23). Prior to DNM, AFM was used primarily as an imaging technique. A sample work of topographic imaging revealed the 3-dimensional structure of exosomes (Citation26). AFM was employed for both topography measurements and force spectroscopy of exosomes (Citation27). The suitability of AFM for the determination of mechanical properties relating to malignancy has been demonstrated by the study of T24 and HCV-29 cells elsewhere (Citation28). This work is the first to our knowledge that investigates, using AFM, the biomechanical properties of non-malignant and malignant isogenic metastatic and non-metastatic cell-derived exosomes.
Visualization by QNM AFM in liquid confirmed the presence of exosome-sized spherical vesicles as shown by AFM height images (). Exosomes derived from malignant and non-malignant cells were of a similar size under AFM analysis, confirming that shown by NTA. Nanomechanical analysis showed a marked decrease in the stiffness of malignant cell-derived exosomes – 95 MPa (non-metastatic-T24) and 280 MPa (metastatic-FL3), compared with non-malignant cell exosomes (1527 MPa for HCV-29) (Table ). Exosomes derived from non-metastatic cells displayed the least stiffness. Assessment of adhesion showed a decrease for malignant (477pN T24 and 219pN FL3) compared to non-malignant (803pN HCV-29) cell-derived exosomes (Table ).
Fig. 2. Atomic force microscopy height images and nanomechanical maps showing stiffness and adhesion profile of exosomes derived from non-malignant HCV-29 (a), malignant non-metastatic T24 (b), and malignant metastatic FL3 (c) cell-derived exosomes. Analysis performed on n=50 vesicles per cell line.
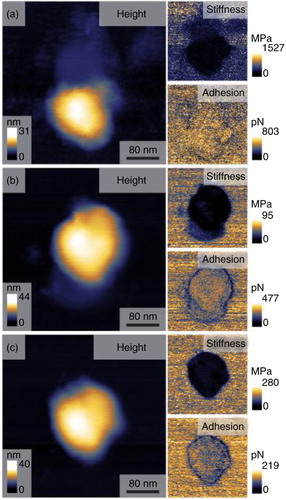
Table I. Mechanical properties of malignant and non-malignant cell exosomes
Exosome-mediated endothelial disruption
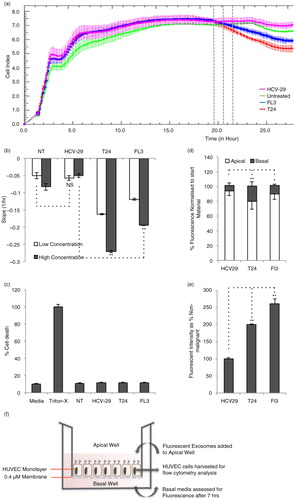
Investigation into the biological effect of malignant versus non-malignant cell-derived exosomes was performed through the measurement of disruption of the TEER of HUVEC monolayers after the application of exosomes. Exosome concentrations were normalized after quantification by NTA with 8×108 and 1.6×109 exosomes administered for the low and high dose, respectively. The formation of a stable HUVEC monolayer followed by application of exosomes and subsequent disruption, shown through a decrease in TEER measurements, can be seen for malignant cell-derived exosomes (a). The assessment of disruption was calculated as the decrease in TEER cell index against time in hours. The quantification of the slopes was taken at T=7 h post-administration of the first dose. Disruption of the endothelial barrier was dose-dependent when applying malignant cell-derived exosomes (from T24 and FL3 cells), but not by immortalized non-malignant cell-derived exosomes (HCV-29) (b). In addition, malignant cell-derived exosomes disrupted the monolayer significantly more than non-malignant exosomes (b). As a control, no release of cytosolic LDH from HUVEC cells could be detected following 7 h of exosome incubation (c). This suggests that the reduction in impedance measurements was not a consequence of reduced cell viability.
Fig. 3. xCELLigence-RTCA TEER measurements showing (a) human umbilical vein endothelial cell (HUVEC) monolayer formation followed by transendothelial electrical resistance measurements post-application of non-malignant (HCV-29), malignant non-metastatic (T24) and malignant metastatic (FL3) cell-derived exosomes (representation of high dose administration). (b) Quantification of slopes at T=7 h post-administration. (c) Viability of HUVEC cells post-exosome administration assessed by lactate dehydrogenase release assay; viability results were not significantly different for exosome treated or control cells by Student's paired t-test, p<0.05. (d) Transport of fluorescently labelled malignant and non-malignant cell-derived exosomes through HUVEC monolayers. (e) Flow cytometric analysis of HUVEC monolayer cells after application of fluorescently labelled exosomes. (f) Schematic of fluorescent exosome transwell assay. Statistical analyses were determined by Student's paired t-test, to calculate significance (*p<0.05, **p<0.01, N.S., not significant).
Transport of exosomes across an intact endothelial monolayer was examined by fluorescent labelling of exosomes. A pre-labelling system was employed through the introduction of lissamine rhodamine head group-labelled DPPE to cells prior to exosome secretion. Fluorescent exosomes were produced with incorporated fluorescent DPPE in the exosomal membrane and were subsequently harvested by standard ultracentrifugation protocol. The suitability of rhodamine-DPPE for the pre-labelling of exosomes was assessed and optimized using the HEK293 cell line (Supplementary Fig. 1). The use of a pre-labelling system reduces the contamination of exosome samples with dye micelles, which is a common problem with intercalating lipid dyes used in a post-labelling system, requiring multiple purification steps that are labour intensive (Citation29) and may result in a loss of material.
The addition of fluorescently labelled exosomes to the apical well of HUVEC monolayers and the subsequent harvest from the basal well (set-up depicted in f) 7 h later shows a significant increase in transport observed for malignant cell-derived exosomes (FL3, 11.6%; T24, 21.4%) compared to non-malignant (HCV-29, 7.2%), with non-metastatic cell-derived exosomes displaying the most transport across HUVEC monolayers (21.4%) (d). HUVEC cells used for fluorescent transport assays were harvested and analysed using flow cytometry to assess for exosome uptake and association. An increased uptake of greater than twofold was observed in HUVEC cells receiving malignant cell-derived exosomes (T24 and FL3) compared with non-malignant exosomes (HCV-29) (e).
Exosome complement activation
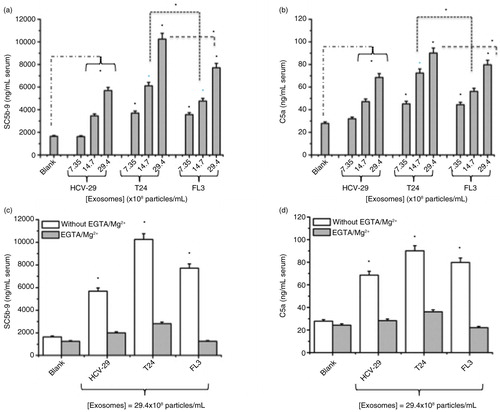
Exosome-mediated complement activation was monitored by measuring rises in serum level of 2 established complement markers. These were the anaphylatoxin and chemoattractive agent C5a and soluble SC5b-9, which is the S protein-bound form of the lytic C5b-9 complex and a measure of whole complement activation. The results in show that all exosomes induced complement activation in a concentration-dependent manner. However, exosomes of the malignant non-metastatic (T24) and malignant metastatic (FL3) cell lines were more potent in activating complement than those derived from non-malignant HCV-29 cells (a and b). Furthermore, exosomes of the malignant non-metastatic T24 cells were more effective in activating complement than those of the metastatic cells when compared at concentrations above 14.7×108 exosomes/mL of serum. Exosome-mediated complement activation was shown to be predominantly through the calcium-sensitive pathways (classical and lectin pathways), since generation of C5a and SC5b-9 was mostly abrogated in the presence of EGTA/Mg2+, which only allows operation of the alternative pathway turnover (c and d).
Fig. 4. Exosome-mediated complement activation in a typical human serum. (a,b) Concentration-dependent complement activation of different exosomes in human serum. Zymosan (1 mg/mL) was used as positive control for monitoring complement activation, which yielded 31.3 µg/mL SC5b-9 and 0.75 µg/mL C5a, respectively. (c,d) Contribution of Ca-sensitive pathways and the alternative pathway turnover in exosome-mediated complement activation in the same serum as panel (a). In (a) and (b) *p<0.05 comparing T24 and FL3 with the corresponding incubations of HCV-29 exosomes. In (a) and (b) p<0.05 for all samples compared with the background level (blank) with the exception of HCV-29 at the concentration of 7.35×108 exosomes/mL of serum. *p<0.05 for selected pairs. In (c) and (d), p<0.05 compared with Blank (control). Statistical analyses were determined by Student's paired t-test, to calculate significance (*p<0.05).
Discussion
CTCs have the ability to establish micrometastases at distant sites; however, this ability is dependent on their survival in the bloodstream, as well as their capability to extravasate the endothelial lining and invade the recipient tissue in which outgrowth takes place (Citation30). The predominant mechanism of extravasation by CTCs is by paracellular transport through the endothelium (Citation30). A role for malignant exosomes in increased vascular leakiness of murine lungs resulting in metastatic lesion formation has been previously demonstrated by Peinado et al. (Citation14), although the biological mechanism by which malignant exosomes facilitate this effect is unknown.
This work investigates the mechanical and biological properties of malignant cell-derived exosomes that may contribute to increased endothelial disruption compared to non-malignant cell-derived exosomes. The use of malignant isogenic metastatic and non-metastatic-derived bladder carcinoma allows for the observation of biological and mechanical properties of exosomes related uniquely to the metastatic capability of exosome-producing cells (Citation19).
The endothelium is a selectively permeable single cell barrier between the blood/lymph and the surrounding tissue. Endothelial layers are classified as continuous, fenestrated or discontinuous distinguished by the size of fenestrations. Most veins and arteries are comprised of continuous endothelial cells separated by tight and adherens junctions forming a barrier permeable only to small molecule diffusion (Citation31). The events and mechanisms preceding CTC crossing of continuous endothelium are not fully elucidated, but a requirement for disruption of the junctions of the endothelium has been shown (Citation30).
In this work, malignant metastatic, malignant non-metastatic and non-malignant urothelial cell line-derived exosomes were of similar size and number per cell, allowing subsequent biological and mechanical differences to be assessed. It is important to ensure that the correct isolation method is used in order to specifically isolate exosomes and provide homogeneity within the field. The Executive Committee of the International Society for Extracellular Vesicles has, thus, compiled a position paper with guidelines for experimental design to assure effects studied are mediated by extracellular vesicles and not cellular contaminants (Citation32). The paper recommends the use of flotation gradients or antibody capture to ensure purity; however, it highlights the lack of an optimized gold-standard method for isolation and purification of extracellular vesicles. In this work we used ultracentrifugation followed by a PBS washing step. We cannot completely dismiss the potential for co-isolation of non-vesicular contaminants and, thus, we cannot exclude a potential role of such contaminants in the functional assays performed here. Our previous findings, however, showing an enrichment of exosomal markers and absence of contamination markers (Citation21, Citation22), would suggest that vesicles are mediating the observed functions. The use of AFM for the investigation of mechanical properties of malignant and non-malignant cell-derived exosomes makes this the first study of its kind to date, to our knowledge, and it revealed a dramatic reduction in stiffness and adhesion of malignant cell-derived exosomes. Increased adhesion results from an increased interaction between tip and sample during tip retraction, possibly due to surface constituents of the sample. To elucidate the component of the non-malignant exosome membrane responsible for the increased adhesion, further investigation using modified AFM tips would be required. Reduced adhesion, however, may allow for improved transport of malignant exosomes due to reduced “stickiness” and reduced interaction with biological surfaces. The reduced stiffness of malignant cell-derived exosomes correlates with work in the literature performed on HCV-29 and T24 cells, revealing a stiffness of malignant cells that is reduced by an order of magnitude (Citation28). The increased transport of fluorescent malignant cell-derived exosomes through an intact HUVEC monolayer may suggest that the reduced stiffness allows for increased paracellular transport, such as reduced rigidity contributing to transport across biological membranes in the paracellular transport of leukocytes across the endothelia (Citation33) and the increased transdermal transport of ultraflexible liposomes (Citation34). Endothelial disruption by other mechanisms such as enzymatic or transfer of protein/nucleic acid cargo to HUVEC cells, however, cannot be dismissed. Assessment of uptake/association of fluorescently labelled exosomes by flow cytometry revealed an increased uptake of malignant cell-derived exosomes compared to non-malignant. The highest cell uptake of exosomes was observed for metastatic cell-derived exosomes, which correlates with the literature (Citation35).
The use of HUVEC cells in a monolayer system provides an in vitro model resembling the continuous endothelium present in blood vessels (Citation36). Combined with the xCELLigence-RTCA system, this presents a suitable method for the assessment of endothelial disruption by malignant and non-malignant cell-derived exosomes through the measurement of changes in TEER. The observed decreases in TEER of HUVEC monolayers allied with no decrease in cell viability, when treated with malignant cell-derived exosomes, suggests that the monolayer is disrupted at the junction level or through cellular retraction. Unexpectedly, the ability of non-metastatic cell-derived exosomes to disrupt the endothelial monolayer was greater than that of metastatic-derived exosomes, although the reduced resistance to anoikis shown by T24 cells compared to FL3 suggests that CTCs derived from non-metastatic tumour cells may be incapable of taking advantage of such disruption due to a reduced survival in circulation (Citation37). The focus of this study on the later stages of metastatic spread, namely the disruption of vascular endothelia allowing for CTC extravasation, does not detect events relevant to metastasis at the earlier stages of tumour invasion and intravasation. Here, differences between metastatic and non-metastatic cell-derived exosomes may be more pronounced, such as effects on the cellular proliferation rate in the primary tumour, local migration and invasion, stimulation of angiogenesis and so on. The increased transport across HUVEC monolayers and increased uptake of fluorescently labelled malignant cell-derived exosomes may present a mechanism for disruption and contribution to the formation of metastatic niche, although further investigation is required. We conclude that both malignant metastatic and malignant non-metastatic cell-derived exosomes have the ability to disrupt endothelial barriers, which may promote metastasis through increased extravasation of CTCs.
The complement system is a branch of the innate immune system activated in response to invasion by pathogenic microorganisms. Stimulation causes the sequential activation of the complement protein cascade, ultimately resulting in deposition of the membrane attack complex to lyse invading pathogens in addition to complementing the adaptive immune response through immune cell recruitment and microbial opsonization. Methods of activation occur in response to antigen bound antibody (classical pathway), microbial sugars such as mannose/lectin through mannose binding lectin or ficolin (lectin pathway) or direct binding of complement component C3b to pathogens (alternative pathway). Little is known of the interaction of malignant cell-derived exosomes with the innate immune system (Citation38). However, it has been shown that non-malignant cell-derived exosomes express complement regulatory proteins such as decay-accelerating factor (DAF) and MAC-inhibitory protein (CD59), suggesting that non-malignant exosomes are poor activators of the complement system (Citation39). Our results with exosomes derived from HCV-29 cells, which moderately activate complement, are in line with these suggestions. Furthermore, more potent activation of the complement system by both metastatic and non-metastatic malignant cell-derived exosomes is interesting, since intratumoural complement activation by such exosomes may promote tumour growth through recruitment of immunosuppressive cells as well as promoting angiogenesis (Citation40–Citation42). Finally, recent work has suggested a role for complement in endothelial cell retraction (Citation43). Collectively, these observations and suggestions seem to agree with results observed in TEER measured disruption, however, further work is required in non-bladder cancer cells to elucidate whether the properties investigated are ubiquitous among all tumour types.
In conclusion, this work demonstrates differences in mechanical and complement activation properties of malignant and non-malignant cell-derived exosomes that may contribute to increased endothelial disruption as well as tumour growth. The data suggests that malignant exosomes induce endothelial leakiness, which may be a prerequisite for subsequent tumour cell transendothelial migration during the metastatic cascade. This work has relevance for the study of metastatic spread and may present tumour-derived exosomes as a therapeutic target in cancer therapy.
Conflict of interest and funding
The authors declare no conflict of interest. SMM acknowledges financial support by the Danish Agency for Science, Technology and Innovation, references 09-065736 (Det Strategiske Forskningsråd) and 12-126894 (Technology and Production).
Supplementary figure 1
Download PDF (407.8 KB)Acknowledgements
We thank Dan Theodorescu (University of Colorado Cancer Center) for providing metastatic derivate urothelial FL3 cells. We thank Susanne Bruun for technical assistance.
Notes
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
References
- van Niel G, Porto-Carreiro I, Simoes S, Raposo G. Exosomes: a common pathway for a specialized function. J Biochem. 2006; 140: 13–21.
- Lässer C. Kosaka N. Identification and analysis of circulating exosomal microRNA in human body fluids. Circulating microRNAs. Methods in molecular biology. Vol. 1024. 2013; , Springer Protocols: Humana Press. 109–28.
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007; 9: 654–9.
- Bobrie A, Colombo M, Raposo G, Théry C. Exosome secretion: molecular mechanisms and roles in immune responses. Traffic. 2011; 12: 1659–68.
- Al-Nedawi K. The Yin–Yang of microvesicles (exosomes) in cancer biology. Front Oncol. 2014; 4: 172.
- Skog J, Wurdinger T, van Rijn S, Meijer D, Gainche L, Sena-Esteves M, etal. Glioblastoma microvesicles transport RNA and protein that promote tumor growth and provide diagnostic biomarkers. Nat Cell Biol. 2008; 10: 1470–6.
- Webber JP, Spary LK, Sanders AJ, Chowdhury R, Jiang WG, Steadman R, etal. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene. 2015; 34: 290–302.
- Mu W, Rana S, Zöller M. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia. 2013; 15: 875-IN4.
- Chairoungdua A, Smith DL, Pochard P, Hull M, Caplan MJ. Exosome release of β-catenin: a novel mechanism that antagonizes Wnt signaling. J Cell Biol. 2010; 190: 1079–91.
- Ostenfeld MS, Jeppesen DK, Laurberg JR, Boysen AT, Bramsen JB, Primdal-Bengtson B, etal. Cellular disposal of miR23b by RAB27-dependent exosome release is linked to acquisition of metastatic properties. Cancer Res. 2014; 74: 5758–71.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100: 57–70.
- Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, etal. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015; 17: 816–26.
- Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011; 71: 3792–801.
- Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, etal. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012; 18: 883–91.
- Schraufstatter IU, Trieu K, Sikora L, Sriramarao P, DiScipio R. Complement C3a and C5a induce different signal transduction cascades in endothelial cells. J Immunol. 2002; 169: 2102–10.
- Makela K, Helen P, Haapasalo H, Paavonen T. Complement activation in astrocytomas: deposition of C4d and patient outcome. BMC Cancer. 2012; 12: 565.
- Moghimi SM. Cancer nanomedicine and the complement system activation paradigm: anaphylaxis and tumour growth. J Contr Release. 2014; 190: 556–62.
- Ostrowski K, Dziedzic-Goclawska A, Strojny P, Grzesik W, Kieler J, Christensen B, etal. Fourier analysis of the cell shape of paired human urothelial cell lines of the same origin but of different grades of transformation. Histochemistry. 1986; 84: 323–8.
- Nicholson BE, Frierson HF, Conaway MR, Seraj JM, Harding MA, Hampton GM, etal. Profiling the evolution of human metastatic bladder cancer. Cancer Res. 2004; 64: 7813–21.
- Gildea JJ, Golden WL, Harding MA, Theodorescu D. Genetic and phenotypic changes associated with the acquisition of tumorigenicity in human bladder cancer. Genes Chromosomes Cancer. 2000; 27: 252–63.
- Jeppesen DK, Hvam ML, Primdahl-Bengtson B, Boysen AT, Whitehead B, Dyrskjøt L, etal. Comparative analysis of discrete exosome fractions obtained by differential centrifugation. J Extracell Vesicles. 2014; 3: 25011. doi: http://dx.doi.org/10.3402/jev.v3.25011.
- Jeppesen DK, Nawrocki A, Jensen SG, Thorsen K, Whitehead B, Howard KA, etal. Quantitative proteomics of fractionated membrane and lumen exosome proteins from isogenic metastatic and nonmetastatic bladder cancer cells reveal differential expression of EMT factors. Proteomics. 2014; 14: 699–712.
- Zhang S, Aslan H, Besenbacher F, Dong M. Quantitative biomolecular imaging by dynamic nanomechanical mapping. Chem Soc Rev. 2014; 43: 7412–29.
- Andersen AJ, Robinson JT, Dai H, Hunter AC, Andresen TL, Moghimi SM. Single-walled carbon nanotube surface control of complement recognition and activation. ACS Nano. 2013; 7: 1108–19.
- Banda N, Mehta G, Chao Y, Wang G, Inturi S, Fossati-Jimack L, etal. Mechanisms of complement activation by dextran-coated superparamagnetic iron oxide (SPIO) nanoworms in mouse versus human serum. Particle Fibre Toxicol. 2014; 11: 64.
- Palanisamy V, Sharma S, Deshpande A, Zhou H, Gimzewski J, Wong DT. Nanostructural and transcriptomic analyses of human saliva derived exosomes. PLoS One. 2010; 5: e8577.
- Sharma S, Rasool HI, Palanisamy V, Mathisen C, Schmidt M, Wong DT, etal. Structural-mechanical characterization of nanoparticle exosomes in human saliva, using correlative AFM, FESEM, and force spectroscopy. ACS Nano. 2010; 4: 1921–6.
- Lekka M, Laidler P, Gil D, Lekki J, Stachura Z, Hrynkiewicz AZ. Elasticity of normal and cancerous human bladder cells studied by scanning force microscopy. Eur Biophys J. 1999; 28: 312–6.
- van der Vlist EJ, Nolte-'t Hoen ENM, Stoorvogel W, Arkesteijn GJA, Wauben MHM. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat Protocols. 2012; 7: 1311–26.
- Reymond N, d'Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013; 13: 858–70.
- Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007; 100: 158–73.
- Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C, etal. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014; 3: 26913. doi: http://dx.doi.org/10.3402/jev.v3.26913.
- Muller WA. Mechanisms of transendothelial migration of leukocytes. Circ Res. 2009; 105: 223–30.
- Pandit J, Garg M, Jain NK. Miconazole nitrate bearing ultraflexible liposomes for the treatment of fungal infection. J Liposome Res. 2014; 24: 163–9.
- Bijnsdorp IV, Geldof AA, Lavaei M, Piersma SR, van Moorselaar RJA, Jimenez CR. Exosomal ITGA3 interferes with non-cancerous prostate cell functions and is increased in urine exosomes of metastatic prostate cancer patients. J Extracell Vesicles. 2013; 2: 22097. doi: http://dx.doi.org/10.3402/jev.v2.22097.
- Kazakoff P, McGuire T, Hoie E, Cano M, Iversen P. Anin vitro model for endothelial permeability: Assessment of monolayer integrity. In Vitro Cell Dev Biol Anim. 1995; 31: 846–52.
- Kim Y-N, Koo KH, Sung JY, Yun U-J, Kim H. Anoikis resistance: an essential prerequisite for tumor metastasis. Int J Cell Biol. 2012; 2012: 306879.
- Benito-Martin A, Di Giannatale A, Ceder S, Peinado H. The new deal: a potential role for secreted vesicles in innate immunity and tumor progression. Front Immunol. 2015; 6: 66.
- Clayton A, Harris CL, Court J, Mason MD, Morgan BP. Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of CD55 and CD59. Eur J Immunol. 2003; 33: 522–31.
- Moghimi SM, Farhangrazi ZS. Just so stories: the random acts of anti-cancer nanomedicine performance. Nanomedicine. 2014; 10(8): 1661–6.
- Kurihara R, Yamaoka K, Sawamukai N, Shimajiri S, Oshita K, Yukawa S, etal. C5a promotes migration, proliferation, and vessel formation in endothelial cells. Inflamm Res. 2010; 59: 659–66.
- Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Parsa AT. Cancer and the complement cascade. Mol Cancer Res. 2010; 8: 1453–65.
- Kerr H, Richards A. Complement-mediated injury and protection of endothelium: lessons from atypical haemolytic uraemic syndrome. Immunobiology. 2012; 217: 195–203.