Abstract
An estimated 80 million US adults have one or more types of cardiovascular diseases. Atherosclerosis is the single most important contributor to cardiovascular diseases; however, only 50% of atherosclerosis patients have currently identified risk factors. Chronic periodontitis, a common inflammatory disease, is linked to an increased cardiovascular risk. Dendritic cells (DCs) are potent antigen presenting cells that infiltrate arterial walls and may destabilize atherosclerotic plaques in cardiovascular disease. While the source of these DCs in atherosclerotic plaques is presently unclear, we propose that dermal DCs from peripheral inflamed sites such as CP tissues are a potential source. This review will examine the role of the opportunistic oral pathogen Porphyromonas gingivalis in invading DCs and stimulating their mobilization and misdirection through the bloodstream. Based on our published observations, combined with some new data, as well as a focused review of the literature we will propose a model for how P. gingivalis may exploit DCs to gain access to systemic circulation and contribute to coronary artery disease. Our published evidence supports a significant role for P. gingivalis in subverting normal DC function, promoting a semimature, highly migratory, and immunosuppressive DC phenotype that contributes to the inflammatory development of atherosclerosis and, eventually, plaque rupture.
Introduction
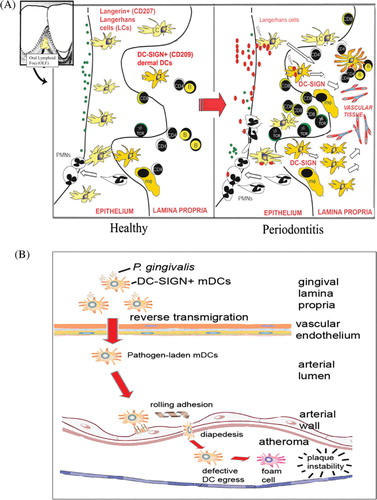
The pathological manifestations of chronic periodontitis (CP), namely, destruction of the soft and hard tissues that support the dentition culminating in tooth loss, have been previously described, as has the high prevalence of the CP in the general adult population Citation1 Citation2. Also well documented is the specific role of the anaerobic Gram-negative species, Porphyromonas gingivalis, in infection of the tissues around the dentition in CP and in initiation of CP Citation3. P. gingivalis, along with two other species, Tannerella forsythia and Treponema denticola, comprise the so-called red complex of pathogens Citation4 that function cooperatively within the subgingival plaque of CP Citation1 Citation2 Citation5 Citation6 Citation7 . P. gingivalis has been associated with several important systemic diseases such as cardiovascular disease, rheumatoid arthritis, preterm birth weight, and diabetes mellitus Citation8 Citation9. Our group is particularly interested in the influx and efflux of various dendritic cells (DCs) in response to P. gingivalis and their role in local and systemic inflammatory processes. DCs are very active antigen-capture cells when immature. When DCs mature, they become potent antigen-presenting cells and are very efficient at stimulating T-naïve cells to differentiate into T-cell effectors [reviewed in Steinman Citation10, Cutler Citation11]. DCs have been implicated in a number of allergic and inflammatory diseases in the periphery [reviewed in Velazquez and Teran Citation12] including atherosclerosis Citation13 Citation14. Being central to the development of immunologic memory and tolerance Citation15, the normal immunologic function of DCs is to patrol the periphery, capture infecting microbes and then migrate out to the secondary lymphoid organs where they can initiate and regulate the adaptive immune response. DC functions in this regard are tightly regulated and depend on the activation signals that DCs receive in the periphery [reviewed in Cutler and Jotwani Citation16]. These signals include inflammatory cytokines, chemokines, as well as pathogen-associated molecular patterns (PAMPS) of bacteria. The development of gingivitis and periodontitis involves the influx and efflux of different DC subsets at distinct stages of disease (). Dendritic Langerhans cells (LC) infiltrate the gingival epithelium in gingivitis and then efflux into the lamina propria in CP, where they begin to undergo maturation Citation17. Dermal dendritic cells (DC-SIGN + ) increase in the lamina propria in CP and become localized toward the lymphatics and vasculature Citation18 Citation19 Citation20 . LCs have been implicated in both the initiation and regulation of contact-hypersensitivity responses in mice Citation21 [reviewed in Igyarto and Kaplan Citation22], while dermal DCs have been implicated in many other inflammatory diseases including rheumatoid arthritis and inflammatory bowel disease Citation23. This review will focus on P. gingivalis and DCs and their respective contributions to the development (and instability) of atherosclerotic plaques.
Fig. 1. Schematic representation of how P. gingivalis-laden dendritic cells may promote atheroma formation and maturation. (A) Representation of the oral lymphoid foci, its organized inflammatory infiltrate. The left panel depicts the healthy oral biofilm in the gingival crevice, comprised predominantly of Gram-positive bacteria (green dots). Healthy gingival tissue is infiltrated with numerous Langerhans cells in the epithelium, with sparse dermal dendritic cells in the lamina propria. As disease progresses, the oral biofilm changes to a predominantly Gram-negative subgingival flora (red dots). In response, a dramatic loss (efflux) of Langerhans cells occurs from the epithelium toward the lamina propria. Also observed is an influx of myeloid-derived DC-SIGN+ dermal dendritic cells (DCs) into the lamina propria. Present are neutrophils, macrophages (mφ), B-cells, and CD4+ and CD8+ T-cells. The DCs form immune conjugates with CD4+ T-cells and also mobilize toward the vasculature. (B) Hypothetical model in periodontitis, showing mobilization of P. gingivalis-laden DC-SIGN+ myeloid DCs (mDCs) in the gingival lamina propria. These mDCs undergo reverse transmigration through the vascular endothelium. Once in the circulation, mDCs carrying P. gingivalis attach to endothelial integrins via DC-SIGN and, after rolling adhesion, undergo diapedesis between endothelial cells. As the atheroma continues to mature and DCs contribute to the foam cells and release MMP-9, the atheroma becomes highly unstable and, eventually, thrombus formation occurs.
Atherosclerosis and microbes
Atherosclerosis (ATH) is a progressive disease characterized by the accumulation of lipids, fibrous elements, and inflammatory cells in the large arteries. ATH constitutes the single most important contributor to the growing worldwide burden of cardiovascular disease. Only about 50% of patients with ATH have currently identified risk factors Citation24. This suggests how little we know about ATH risk. Inflammation in the arterial vessel wall is particularly important in the development of ATH. Four mechanisms have been proposed for how bacterial pathogens may induce or accelerate ATH [reviewed in Gibson et al. Citation25]. These include: (i) direct invasion of the vascular endothelium by pathogens in the blood, (ii) immunological sounding, (iii) molecular mimicry, and (iv) pathogen trafficking of microbes within leukocytes in peripheral blood.
Of particular relevance to atherogenesis are bacterial species that infect and survive within endothelial cells and within migrating leukocytes [reviewed in Rackley Citation26]. A prevailing hypothesis is that, regardless of their viability status, bacteria release PAMPs that serve as agonists for TLRs, thus activating inflammatory leukocytes and endothelial cells, and can contribute to the development of ATH Citation27. Large population studies support the role of bacterial species in ATH Citation28 Citation29. However, the results of clinical trials using antibiotics to treat cardiovascular disease have been disappointing Citation30 Citation31 Citation32 Citation33 . Many atherogenic bacteria, including P. gingivalis, are intracellular pathogens Citation34 Citation35. An apparent consequence of this is that these pathogens are less susceptible to antibiotics when sequestered inside host cells. P. gingivalis is 100-fold more resistant to moxofloxocin, 10-fold more resistant to clindamycin and metronidazole when inside host epithelial cells Citation36. Clindamycin- and azithromycin-resistant P. gingivalis isolates have been identified in human subjects with CP Citation37.
P. gingivalis has also been identified in human ATH plaques Citation38, as have other atherogenic bacteria such as Chlamydophila pneumoniae and Helicobacter pylori Citation27 Citation39 Citation40. Experimental infection with P. gingivalis accelerates ATH in animal models Citation35 Citation41. The FINRISK 1992 cohort study of 6,051 individuals implicates exposure to P. gingivalis or endotoxin in increased risk for cardiovascular diseases Citation42. A recent meta-analysis indicated that the level of systemic bacterial exposure in CP mediates ATH risk Citation43 Citation44. Another meta-analysis studied human cohort studies, case-control studies, and cross-sectional studies and concluded that CP is a significant risk factor for developing coronary artery disease (CAD) Citation44. The degree of increased risk of CAD conferred by CP appears comparable to smoking Citation45 and elevated serum triglycerides Citation46. Viable and invasive P. gingivalis, though in a dormant state, have been cultured from human ATH plaques Citation38. This has not been shown with other atherogenic bacteria such as C. pneumoniae and H. pylori, which have only been identified by DNA-based methods Citation27 Citation39 Citation40. Overall, several infectious agents have been shown to be disseminated by pathogen trafficking leukocytes, they include Streptococcus pyogenes Citation47, C. pneumoniae Citation48, and Listeria monocytogenes Citation49.
P. gingivalis virulence and targeting of dendritic cells (DCs)
P. gingivalis is a an amino acid fermentor with an absolute requirement for hemin Citation2. The bacteria utilizes its many virulence factors to fulfill its complex nutritional requirements, while still enabling it to evade and even modulate the host immune system Citation1 Citation2 Citation5. Several virulence factors including the polysaccharide capsule, fimbriae, proteases for opsonins C3, proteases for IgG, gingipains, bacterial lipopolysaccharides (LPS), toxins, and hemagglutinins enable P. gingivalis to persist in the oral mucosa and help facilitate some of the physiopathology of CP Citation19 Citation50 Citation51 Citation52 Citation53 Citation54 Citation55 Citation56 .
There are two fimbriae that are essential adhesins for the invasion and colonization of the oral mucosa by P. gingivalis. These adhesins, termed the major and minor fimbriae, are distinct antigenically by amino acid composition and by size Citation57 Citation58. The major fimbriae form long projections from the bacteria and have been shown in most reports to facilitate their adhesion to and invasion of the host cells. The major fimbriae is a 41 kDa protein, encoded by the fimA gene Citation59, and many of its cellular receptors have been identified as either the β-1 integrins (CD29) Citation60 Citation61 or the β-2 integrins (CD18) Citation62 Citation63 Citation64 . The minor fimbriae (though much shorter) are comprised of a 67 kDa protein encoded by the mfa1 gene Citation57. We have recently shown that the minor fimbriae targets DC specific ICAM-3 grabbing non-integrin (DC-SIGN or CD209) on monocyte-derived DCs for entry Citation5. DC-SIGN is a type II membrane protein in which the extracellular domain consists of a stalk that promotes tetramerization Citation65. It contains a C-terminal carbohydrate recognizing domain (CRD) that belongs to the C-type lectin superfamily Citation65. Early studies by Feinberg et al. showed that the DC-SIGN CRD preferentially binds to the high-mannose N-linked oligosaccharides GlcNAc (N-acetylglucosamine) and Manα1-3[Manα1-6] Man (mannose) Citation65. Furthermore, Appelmelk et al. showed that DC-SIGN also binds to fucose-containing Lewis blood antigens Citation66. Guo et al. utilized an extensive glycan array and showed that DC-SIGN will bind high mannose-containing glycans or glycans that contain terminal fucose residues Citation67. Previous studies showed that DC-SIGN is used by microorganisms such as Neisseria gonorrhoeae Citation68, Mycobacterium tuberculosis Citation66 Citation69 Citation70, Mycobacterium leprae, HIV Citation71, H. pylori Citation66, and P. gingivalis Citation5 to target DCs for entry and immune suppression.
Pathological consequences of DC-SIGN targeting
The ability of minor fimbriae to specifically target DC-SIGN on DCs has significant pathological and immunological repercussions. Periodontitis lesions contain an intense infiltrate of DC-SIGN+ DCs Citation17 Citation72. DCs in the lesions become activated and appear to mobilize toward the capillary-rich lamina propria Citation17. We propose that infection of DCs by P. gingivalis triggers reverse transmigration of infected gingival DCs into circulation and these may contribute to the pathogenesis of ATH (). Evidence exists for the presence of activated DC-SIGN+ myeloid DCs in rupture prone unstable plaques Citation13 Citation73. The sources of these DC-SIGN+ plaque DCs are not clear, but presumably may include DCs from inflamed peripheral tissues. Other sources of DCs may include CD14+ CD16− monocytes, CD14low CD16+ monocytes Citation74 that differentiate into DCs in situ Citation5. DCs that infiltrate rupture-prone atherosclerotic plaques express atherogenic markers including C1q (a classical complement pathway component involved in apoptotic cell clearance); HSP60 and HSP70 (chaperone proteins involved in autoimmune responses); chemokine receptors CCR2, CCR5, CX3CR1; and chemokines CXCL16, CCL19, and CCL21 (involved in DC transmigration and leukocyte homing) Citation74 Citation75 Citation76 . Also expressed are DC maturation markers CD40, CD80, and CD86 Citation73 Citation77. Matrix-metalloproteinase-9 (MMP-9), which are highly expressed in vulnerable regions of the atherosclerotic plaques and suggested to be causally involved in plaque rupture Citation78, is produced by leukocytes including DCs. The P. gingivalis LPS is a particularly potent inducer of MMP-9 but not TIMP-1 by DCs, suggesting that P. gingivalis induces an MMP-9/TIMP-1 imbalance in DCs Citation79. Our published data indicated that fimbriated strains of P. gingivalis infect DCs and induce atherogenic biomarkers in vitro, but the mechanisms are not presently clear Citation5.
Immunological consequence of targeting DC-SIGN on dendritic cells (DCs)
DC-SIGN targeting by minor fimbriae results in a dampening of the maturation status and the inflammatory cytokine profile of DCs. Conversely, removal of the minor fimbriae results in robust DC maturation coupled with a strong proinflammatory cytokine response Citation5. This regulation of DC immunogenic functions based on minor fimbriae expression extends to the T-cell effector response elicited by DCs. In DC-CD4+ T-cell coculture experiments, P. gingivalis strains expressing solely the minor fimbriae induced DCs to prime T-cells into a Th2 effector phenotype, whereas, strains expressing solely the major fimbriae induced DCs to prime T-cells into a Th1 effector phenotype Citation5. Intriguingly, the wild-type strain was able to stimulate a mixed or anergic T-cell effector phenotype Citation5. Furthermore, in vitro studies in the presence of DC-SIGN targeting agonists (e.g. mannan from S. cerevisiae or glycosylated HIV gp120) resulted in a diminished association of minor fimbriated strains with cells Citation5. When the wild-type strain of P. gingivalis was cocultured with glycosylated HIV gp120, we observed a dramatic uncoupling of DC maturation from DC inflammatory cytokine secretion Citation5. The presence of HIV or even Candida albicans (etiological agent of oral thrush) might act synergistically with P. gingivalis to further exasperate this uncoupling and act to greatly diminish DC maturation.
Mechanisms of DC mobilization, access to peripheral blood
Experimental studies demonstrate the important role that chemokines and chemokine receptors play in trafficking of leukocytes to and invasion of the arterial wall in ATH Citation80 Citation81 Citation82 Citation83 Citation84 . Immature DCs express inflammatory chemokine receptors () that direct their migration into infected tissues. In response to capture of antigens or to TLR-mediated recognition of microbes, DCs undergo a process called functional maturation in which they downregulate inflammatory chemokine receptors and upregulate homeostatic chemokine receptors (). This directs DC migration out of the tissues toward lymph nodes. When the DC maturation process is disrupted, as occurs in DC-SIGN ligation Citation5, activated DCs ostensibly undergo reverse transmigration into the blood (), which can lead to systemic inflammation. This process contributes to the initiation, progression, and instability of arterial plaque in patients with CAD. Recent studies have investigated the presence of blood DCs in patients with CAD, but the results are controversial Citation85 Citation86. Shi et al. found that the total peripheral blood CD11c + DCs were significantly higher in patients with CAD compared to healthy controls Citation85. Conversely, Yilmaz et al. found a decrease in circulating DCs in patients with CAD Citation86. However, the patient population and markers used for DC isolation were different. None of the studies reported the presence of CP on these patients, which might have affected these conflicting findings. An attempt has been made to correlate the presence of blood DCs as a risk factor for CAD Citation87. Further, much speculation has been made about the source of these DCs that infiltrate the atheromas.
Table 1. Summary of results of immunohistochemistry (IHC), cDNA microarray, and qRT-PCR analyses of MoDCs and gingival tissues
Fully matured DCs lose their ability to uptake and process antigens, stop migrating, and express CD83 and other molecules involved in antigen presentation to T-cells Citation88. Full maturation of DCs also results in loss of expression of many endocytic receptors, including DC-SIGN; thus, expression of DC-SIGN is one indicator that DC are not fully mature Citation89. There is evidence that a particular subset of blood DCs expressed DC-SIGN and that this receptor may be involved in the uptake and dissemination of HIV Citation90. Engering et al. showed that DC-SIGN+ blood DCs are able to stimulate proliferation of allogeneic T-cells, as well as infect these T-cells in trans Citation90. Potentially, DC-SIGN+ blood DCs can disseminate pathogens, increase systemic inflammation, and contribute to plaque instability.
Environmental regulation of fimbriae: possible role in systemic immunosuppression, dissemination of P. gingivalis
Wu et al. discovered that the major and minor fimbriae are regulated by a two component regulatory system termed FimS/FimR Citation91. It was also determined that while FimR binds directly to mfa1, it will only bind to the first gene of the fimA gene cluster, pg2130 Citation91 Citation92. Moreover, this two component regulatory system responds to environmental cues like heme and temperature Citation91. However, it is still not clear how these adhesive fimbriae are regulated in vivo. The possibility of major and minor fimbriae being differentially regulated in response to different environmental cues or stimuli may allow this organism to modulate the immune system to expand its ecological niche. Moreover, our observations of intact bacteria inside DC-SIGN rich vesicles might explain the dissemination of this organism to atherosclerotic plaques Citation5. We propose here that P. gingivalis interacts with DC-SIGN on dermal DCs from the gingiva mucosa (unpublished observation). This interaction facilitates uptake of P. gingivalis and immunomodulation of normal DC functions. Directed migration is disrupted and DCs then migrate through the endothelium instead of the lymphatic system (). Once in the endothelium, the DC undergoes partial maturation resulting in adherence to the endothelium and recruitment of other leukocytes. Soilleux et al. previously described the presence of immature (lacking CD83 but expressing HLA-DR and LAMP) DC-SIGN+ DCs on atherosclerotic plaques Citation73. Strikingly, this maturation profile is very similar to our results with minor fimbriated strains (Pg381 and DPG-3) and DCs Citation5. Thus, P. gingivalis infected DCs may exit into the bloodstream and migrate to the site of developing atheroma, where they adhere to and invade the arterial endothelium. Recent reports suggest that P. gingivalis is able to spread from infected epithelial, endothelial, and smooth muscle cells to new host cells where it multiplies Citation93. These mechanisms might explain how an oral opportunistic pathogen is able to disseminate throughout its host and potentially facilitate the formation of atherosclerotic plaques.
Conflict of interest and funding
This study was supported by US Public Health Service grants from the NIH/NIDCR R01 DE014328, R21 DE020916 to Christopher W. Cutler, F31 DE020014 to Amir E. Zeituni, and K23DE018187-01 to Julio Carrion.
References
- Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010; 8: 481–90.
- Pathirana RD, O'Brien-Simpson NM, Reynolds EC. Host immune responses to Porphyromonas gingivalis antigens. Periodontol 2000. 2010; 52: 218–37.
- Rylev M, Kilian M. Prevalence and distribution of principal periodontal pathogens worldwide. J Clin Periodontol. 2008; 35: 346–61.
- Ximenez-Fyvie LA, Haffajee AD, Socransky SS. Comparison of the microbiota of supra- and subgingival plaque in health and periodontitis. J Clin Periodontol. 2000; 27: 648–57.
- Zeituni AE, Jotwani R, Carrion J, Cutler CW. Targeting of DC-SIGN on human dendritic cells by minor fimbriated Porphyromonas gingivalis strains elicits a distinct effector T cell response. J Immunol. 2009; 183: 5694–704.
- Zeituni AE, McCaig W, Scisci E, Thanassi DG, Cutler CW. Native 67 kDa minor fimbriae of Porphyromonas gingivalis: a novel glycoprotein with DC-SIGN-targeting motifs. J Bacteriol. 2010; 192(16): 4103–10.
- Burt B. Position paper: epidemiology of periodontal diseases. J Periodontol. 2005; 76: 1406–19.
- Gibson FC III, Hong C, Chou HH, Yumoto H, Chen J, Lien E, et al.. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004; 109: 2801–6.
- Reddy MS. Reaching a better understanding of non-oral disease and the implication of periodontal infections. Periodontol 2000. 2007; 44: 9–14.
- Steinman RM. Dendritic cells and the control of immunity: enhancing the efficiency of antigen presentation. Mt Sinai J Med. 2001; 68: 160–6.
- Cutler CW Jr. Dendritic cells at the oral mucosal interface. J Dent Res. 2006; 85: 678–89.
- Velazquez JR, Teran LM. Chemokines and their receptors in the allergic airway inflammatory process. Clin Rev Allergy Immunol. 2010. Mar 30 [Epub ahead on print].
- Bobryshev YV. Dendritic cells in atherosclerosis: current status of the problem and clinical relevance. Eur Heart J. 2005; 26: 1700–1704.
- Niessner A, Weyand CM. Dendritic cells in atherosclerotic disease. Clin Immunol. 2010; 134: 25–32.
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010; 327: 656–61.
- Cutler CW, Jotwani R. Dendritic cells at the oral mucosal interface. J Dent Res. 2006; 85: 678–89.
- Jotwani R, Cutler CW. Multiple dendritic cell (DC) subpopulations in human gingiva and association of mature DCs with CD4+ T-cells in situ. J Dent Res. 2003; 82: 736–41.
- Bodineau A, Coulomb B, Tedesco AC, Seguier S. Increase of gingival matured dendritic cells number in elderly patients with chronic periodontitis. Arch Oral Biol. 2009; 54: 12–6.
- Jotwani R, Pulendran B, Agrawal S, Cutler CW. Human dendritic cells respond to Porphyromonas gingivalis LPS by promoting a Th2 effector response in vitro. Eur J Immunol. 2003; 33: 2980–6.
- Jotwani R, Cutler CW. Fimbriated Porphyromonas gingivalis is more efficient than fimbria-deficient P. gingivalis in entering human dendritic cells in vitro and induces an inflammatory Th1 effector response. Infect Immun. 2004; 72: 1725–32.
- Igyarto BZ, Jenison MC, Dudda JC, Roers A, Muller W, Koni PA, et al.. Langerhans cells suppress contact hypersensitivity responses via cognate CD4 interaction and langerhans cell-derived IL-10. J Immunol. 2009; 183: 5085–93.
- Igyarto BZ, Kaplan DH. The evolving function of Langerhans cells in adaptive skin immunity. Immunol Cell Biol. 2010; 88: 361–5.
- Kwon DS, Gregorio G, Bitton N, Hendrickson WA, Littman DR. DC-SIGN-mediated internalization of HIV is required for trans-enhancement of T cell infection. Immunity. 2002; 16: 135–44.
- Vita JA, Loscalzo J. Shouldering the risk factor burden: infection, atherosclerosis, and the vascular endothelium. Circulation. 2002; 106: 164–6.
- Gibson FC III, Yumoto H, Takahashi Y, Chou HH, Genco CA. Innate immune signaling and Porphyromonas gingivalis-accelerated atherosclerosis. J Dent Res. 2006; 85: 106–21.
- Rackley CE. Vascular abnormalities in the metabolic syndrome: mechanisms and therapy. Curr Atheroscler Rep. 2005; 7: 403–4.
- Erridge C. The roles of pathogen-associated molecular patterns in atherosclerosis. Trends Cardiovasc Med. 2008; 18: 52–6.
- Kiechl S, Egger G, Mayr M, Wiedermann CJ, Bonora E, Oberhollenzer F, et al.. Chronic infections and the risk of carotid atherosclerosis: prospective results from a large population study. Circulation. 2001; 103: 1064–70.
- Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Victor A, et al.. Impact of infectious burden on progression of carotid atherosclerosis. Stroke. 2002; 33: 2581–6.
- Celik T, Iyisoy A, Celik M, Isik E. The ongoing debate over the association between Chlamydia pneumoniae infection and coronary artery disease: antibiotic dilemma. Int J Cardiol. 2008; 130: 260–2.
- Aghajanian A, Wittchen ES, Allingham MJ, Garrett TA, Burridge K. Endothelial cell junctions and the regulation of vascular permeability and leukocyte transmigration. J Thromb Haemost. 2008; 6(9): 1453–60.
- O'Connor CM, Dunne MW, Pfeffer MA, Muhlestein JB, Yao L, Gupta S, et al.. Azithromycin for the secondary prevention of coronary heart disease events: the WIZARD study: a randomized controlled trial. JAMA. 2003; 290: 1459–66.
- Cannon CP, Braunwald E, McCabe CH, Grayston JT, Muhlestein B, Giugliano RP, et al.. Antibiotic treatment of Chlamydia pneumoniae after acute coronary syndrome. N Engl J Med. 2005; 352: 1646–54.
- Capestany CA, Tribble GD, Maeda K, Demuth DR, Lamont RJ. Role of the Clp system in stress tolerance, biofilm formation, and intracellular invasion in Porphyromonas gingivalis. J Bacteriol. 2008; 190: 1436–46.
- Gibson FC III, Genco CA. Porphyromonas gingivalis mediated periodontal disease and atherosclerosis: disparate diseases with commonalities in pathogenesis through TLRs. Curr Pharm Des. 2007; 13: 3665–75.
- Eick S, Pfister W. Efficacy of antibiotics against periodontopathogenic bacteria within epithelial cells: an in vitro study. J Periodontol. 2004; 75: 1327–34.
- RAMS TM FD, editor. Occurrence of Clindamycin-resistant Porphyromonas gingivalis in Human Periodontitis. San AntonioTX: International Association for Dental Research. 2003.
- Kozarov EV, Dorn BR, Shelburne CE, Dunn WA Jr., Progulske-Fox A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol. 2005; 25: e17–e18.
- Kaplan M, Yavuz SS, Cinar B, Koksal V, Kut MS, Yapici F, et al.. Detection of Chlamydia pneumoniae and Helicobacter pylori in atherosclerotic plaques of carotid artery by polymerase chain reaction. Int J Infect Dis. 2006; 10: 116–23.
- Watson C, Alp NJ. Role of Chlamydia pneumoniae in atherosclerosis. Clin Sci (Lond). 2008; 114: 509–31.
- Lalla E, Lamster IB, Hofmann MA, Bucciarelli L, Jerud AP, Tucker S, et al.. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2003; 23: 1405–11.
- Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler Thromb Vasc Biol. 2007; 27: 1433–9.
- Mustapha IZ, Debrey S, Oladubu M, Ugarte R. Markers of systemic bacterial exposure in periodontal disease and cardiovascular disease risk: a systematic review and meta-analysis. J Periodontol. 2007; 78: 2289–302.
- Bahekar AA, Singh S, Saha S, Molnar J, Arora R. The prevalence and incidence of coronary heart disease is significantly increased in periodontitis: a meta-analysis. Am Heart J. 2007; 154: 830–7.
- Trichopoulou A, Yiannakouris N, Bamia C, Benetou V, Trichopoulos D, Ordovas JM. Genetic predisposition, nongenetic risk factors, and coronary infarct. Arch Intern Med. 2008; 168: 891–6.
- Austin MA, Rodriguez BL, McKnight B, McNeely MJ, Edwards KL, Curb JD, et al.. Low-density lipoprotein particle size, triglycerides, and high-density lipoprotein cholesterol as risk factors for coronary heart disease in older Japanese–American men. Am J Cardiol. 2000; 86: 412–6.
- Loof TG, Rohde M, Chhatwal GS, Jung S, Medina E. The contribution of dendritic cells to host defenses against Streptococcus pyogenes. J Infect Dis. 2007; 196: 1794–803.
- MacIntyre A, Abramov R, Hammond CJ, Hudson AP, Arking EJ, Little CS, et al.. Chlamydia pneumoniae infection promotes the transmigration of monocytes through human brain endothelial cells. J Neurosci Res. 2003; 71: 740–50.
- Greiffenberg L, Goebel W, Kim KS, Weiglein I, Bubert A, Engelbrecht F, et al.. Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: InlB-dependent invasion, long-term intracellular growth, and spread from macrophages to endothelial cells. Infect Immun. 1998; 66: 5260–7.
- Cutler CW, Kalmar JR, Genco CA. Pathogenic strategies of the oral anaerobe, Porphyromonas gingivalis. Trends Microbiol. 1995; 3: 45–51.
- Lamont RJ, Jenkinson HF. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998; 62: 1244–63.
- Imamura T, Travis J, Potempa J. The biphasic virulence activities of gingipains: activation and inactivation of host proteins. Curr Protein Pept Sci. 2003; 4: 443–50.
- Mezyk-Kopec R, Bzowska M, Potempa J, Jura N, Sroka A, Black RA, et al.. Inactivation of membrane tumor necrosis factor alpha by gingipains from Porphyromonas gingivalis. Infect Immun. 2005; 73: 1506–14.
- Potempa J, Mikolajczyk-Pawlinska J, Brassell D, Nelson D, Thogersen IB, Enghild JJ, et al.. Comparative properties of two cysteine proteinases (gingipains R), the products of two related but individual genes of Porphyromonas gingivalis. J Biol Chem. 1998; 273: 21648–57.
- Sroka A, Sztukowska M, Potempa J, Travis J, Genco CA. Degradation of host heme proteins by lysine- and arginine-specific cysteine proteinases (gingipains) of Porphyromonas gingivalis. J Bacteriol. 2001; 183: 5609–16.
- Pulendran B, Kumar P, Cutler CW, Mohamadzadeh M, Van Dyke T, Banchereau J. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J Immunol. 2001; 167: 5067–76.
- Hamada N, Sojar HT, Cho MI, Genco RJ. Isolation and characterization of a minor fimbria from Porphyromonas gingivalis. Infect Immun. 1996; 64: 4788–94.
- Arai M, Hamada N, Umemoto T. Purification and characterization of a novel secondary fimbrial protein from Porphyromonas gingivalis strain 381. FEMS Microbiol Lett. 2000; 193: 75–81.
- Xie H, Lamont RJ. Promoter architecture of the Porphyromonas gingivalis fimbrillin gene. Infect Immun. 1999; 67: 3227–35.
- Yilmaz O, Watanabe K, Lamont RJ. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol. 2002; 4: 305–14.
- Nakagawa I, Amano A, Kuboniwa M, Nakamura T, Kawabata S, Hamada S. Functional differences among FimA variants of Porphyromonas gingivalis and their effects on adhesion to and invasion of human epithelial cells. Infect Immun. 2002; 70: 277–85.
- Hajishengallis G, Harokopakis E. Porphyromonas gingivalis interactions with complement receptor 3 (CR3): innate immunity or immune evasion?. Front Biosci. 2007; 12: 4547–57.
- Harokopakis E, Albzreh MH, Haase EM, Scannapieco FA, Hajishengallis G. Inhibition of proinflammatory activities of major periodontal pathogens by aqueous extracts from elder flower (Sambucus nigra). J Periodontol. 2006; 77: 271–9.
- Takeshita A, Murakami Y, Yamashita Y, Ishida M, Fujisawa S, Kitano S, et al.. Porphyromonas gingivalis fimbriae use beta2 integrin (CD11/CD18) on mouse peritoneal macrophages as a cellular receptor, and the CD18 beta chain plays a functional role in fimbrial signaling. Infect Immun. 1998; 66: 4056–60.
- Feinberg H, Mitchell DA, Drickamer K, Weis WI. Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science. 2001; 294: 2163–6.
- Appelmelk BJ, van Die I, van Vliet SJ, Vandenbroucke-Grauls CM, Geijtenbeek TB, van Kooyk Y. Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J Immunol. 2003; 170: 1635–9.
- Guo Y, Feinberg H, Conroy E, Mitchell DA, Alvarez R, Blixt O, et al.. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat Struct Mol Biol. 2004; 11: 591–8.
- Zhang P, Schwartz O, Pantelic M, Li G, Knazze Q, Nobile C, et al.. DC-SIGN (CD209) recognition of Neisseria gonorrhoeae is circumvented by lipooligosaccharide variation. J Leukoc Biol. 2006; 79: 731–8.
- Maeda N, Nigou J, Herrmann JL, Jackson M, Amara A, Lagrange PH, et al.. The cell surface receptor DC-SIGN discriminates between Mycobacterium species through selective recognition of the mannose caps on lipoarabinomannan. J Biol Chem. 2003; 278: 5513–6.
- Soilleux EJ, Sarno EN, Hernandez MO, Moseley E, Horsley J, Lopes UG, et al.. DC-SIGN association with the Th2 environment of lepromatous lesions: cause or effect?. J Pathol. 2006; 209: 182–9.
- Pohlmann S, Baribaud F, Lee B, Leslie GJ, Sanchez MD, Hiebenthal-Millow K, et al.. DC-SIGN interactions with human immunodeficiency virus type 1 and 2 and simian immunodeficiency virus. J Virol. 2001; 75: 4664–72.
- Jotwani R, Palucka AK, Al-Quotub M, Nouri-Shirazi M, Kim J, Bell D, et al.. Mature dendritic cells infiltrate the T cell-rich region of oral mucosa in chronic periodontitis: in situ, in vivo, and in vitro studies. J Immunol. 2001; 167: 4693–700.
- Soilleux EJ, Morris LS, Trowsdale J, Coleman N, Boyle JJ. Human atherosclerotic plaques express DC-SIGN, a novel protein found on dendritic cells and macrophages. J Pathol. 2002; 198: 511–6.
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al.. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007; 117: 185–94.
- Bobryshev YV, Lord RS. Expression of heat shock protein-70 by dendritic cells in the arterial intima and its potential significance in atherogenesis. J Vasc Surg. 2002; 35: 368–75.
- Cao W, Bobryshev YV, Lord RS, Oakley RE, Lee SH, Lu J. Dendritic cells in the arterial wall express C1q: potential significance in atherogenesis. Cardiovasc Res. 2003; 60: 175–86.
- Erbel C, Sato K, Meyer FB, Kopecky SL, Frye RL, Goronzy JJ, et al.. Functional profile of activated dendritic cells in unstable atherosclerotic plaque. Basic Res Cardiol. 2007; 102: 123–32.
- Muzzio ML, Miksztowicz V, Brites F, Aguilar D, Repetto EM, Wikinski R, et al.. Metalloproteases 2 and 9, Lp-PLA(2) and lipoprotein profile in coronary patients. Arch Med Res. 2009; 40: 48–53.
- Jotwani R, Eswaran SV, Moonga S, Cutler CW. MMP-9/TIMP-1 imbalance induced in human dendritic cells by Porphyromonas gingivalis. FEMS Immunol Med Microbiol. 2010; 58: 314–21.
- Price DT, Loscalzo J. Cellular adhesion molecules and atherogenesis. Am J Med. 1999; 107: 85–97.
- Reape TJ, Groot PH. Chemokines and atherosclerosis. Atherosclerosis. 1999; 147: 213–25.
- Reape TJ, Rayner K, Manning CD, Gee AN, Barnette MS, Burnand KG, et al.. Expression and cellular localization of the CC chemokines PARC and ELC in human atherosclerotic plaques. Am J Pathol. 1999; 154: 365–74.
- Weber C, Schober A, Zernecke A. Chemokines: key regulators of mononuclear cell recruitment in atherosclerotic vascular disease. Arterioscler Thromb Vasc Biol. 2004; 24: 1997–2008.
- Pan S, Kleppe LS, Witt TA, Mueske CS, Simari RD. The effect of vascular smooth muscle cell-targeted expression of tissue factor pathway inhibitor in a murine model of arterial thrombosis. Thromb Haemost. 2004; 92: 495–502.
- Shi H, Ge J, Fang W, Yao K, Sun A, Huang R, et al.. Peripheral-blood dendritic cells in men with coronary heart disease. Am J Cardiol. 2007; 100: 593–97.
- Yilmaz A, Weber J, Cicha I, Stumpf C, Klein M, Raithel D, et al.. Decrease in circulating myeloid dendritic cell precursors in coronary artery disease. J Am Coll Cardiol. 2006; 48: 70–80.
- Yilmaz A, Schaller T, Cicha I, Altendorf R, Stumpf C, Klinghammer L, et al.. Predictive value of the decrease in circulating dendritic cell precursors in stable coronary artery disease. Clin Sci (Lond). 2009; 116: 353–63.
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998; 392: 245–52.
- Figdor CG, van Kooyk Y, Adema GJ. C-type lectin receptors on dendritic cells and Langerhans cells. Nat Rev Immunol. 2002; 2: 77–84.
- Engering A, Van Vliet SJ, Geijtenbeek TB, Van Kooyk Y. Subset of DC-SIGN(+) dendritic cells in human blood transmits HIV-1 to T lymphocytes. Blood. 2002; 100: 1780–6.
- Wu J, Lin X, Xie H. Porphyromonas gingivalis short fimbriae are regulated by a FimS/FimR two-component system. FEMS Microbiol Lett. 2007; 271: 214–21.
- Wu J, Lin X, Xie H. OxyR is involved in coordinate regulation of expression of fimA and sod genes in Porphyromonas gingivalis. FEMS Microbiol Lett. 2008; 282: 188–95.
- Li L, Michel R, Cohen J, Decarlo A, Kozarov E. Intracellular survival and vascular cell-to-cell transmission of Porphyromonas gingivalis. BMC Microbiol. 2008; 8: 26–37.