Abstract
Both lesions of endodontic origin and periodontal diseases involve the host response to bacteria and the formation of osteolytic lesions. Important for both is the upregulation of inflammatory cytokines that initiate and sustain the inflammatory response. Also important are chemokines that induce recruitment of leukocyte subsets and bone-resorptive factors that are largely produced by recruited inflammatory cells. However, there are differences also. Lesions of endodontic origin pose a particular challenge since that bacteria persist in a protected reservoir that is not readily accessible to the immune defenses. Thus, experiments in which the host response is inhibited in endodontic lesions tend to aggravate the formation of osteolytic lesions. In contrast, bacteria that invade the periodontium appear to be less problematic so that blocking arms of the host response tend to reduce the disease process. Interestingly, both lesions of endodontic origin and periodontitis exhibit inflammation that appears to inhibit bone formation. In periodontitis, the spatial location of the inflammation is likely to be important so that a host response that is restricted to a subepithelial space is associated with gingivitis, while a host response closer to bone is linked to bone resorption and periodontitis. However, the persistence of inflammation is also thought to be important in periodontitis since inflammation present during coupled bone formation may limit the capacity to repair the resorbed bone.
Introduction
Periapical lesions of endodontic origin and periodontitis are two common conditions found in the oral cavity that share pathologic mechanisms involving interactions between immune cells and bone. Lesions of endodontic origin are associated with bacterial contamination and necrosis of the dental pulp, which typically progress through four stages: Citation1 exposure of the dental pulp to the oral cavity with subsequent bacterial colonization, Citation2 inflammation and necrosis of the dental pulp, Citation3 the development of inflammation in the periapical area, and Citation4 periapical resorption of bone and formation of granulomas or cysts. Osteolytic lesions in periodontitis are initiated by bacterial plaque in the gingival sulcus and on the tooth surface. Periodontitis similarly occurs in four stages: Citation1 bacterial accumulation of a biofilm and presence in the gingival sulcus (colonization), Citation2 bacterial penetration of epithelium and connective tissue in the gingiva adjacent to tooth surface (invasion), Citation3 stimulation of a host response that involves activation of the acquired and innate immune response (inflammation), and Citation4 destruction of connective tissue attachment to the tooth surface and bone that is irreversible (irreversible tissue loss). Both oral diseases demonstrate similar patterns of development, bone resorption associated with bacteria that adhere to and invade soft tissue stimulating an inflammatory response and subsequent osteoclastogenesis.
Lesions of endodontic origin
Endodontic lesions typically develop from exposure of the pulpal tissue to oral bacteria as a result of deficiencies in the integrity of a tooth. This may result from carious lesions that dissolve the mineralized dental tissue, fractures of the tooth structure, as well as iatrogenic and other circumstances that allow bacteria to penetrate into the pulpal tissues. In most cases, these events lead to infection within the dental pulp, which causes the development of inflammation that spreads from the exposed area. The inflammation is often followed by pulpal tissue necrosis, leading to chronic infection, the spread of inflammation to the tooth apex, and bone resorption. The inflammatory response involves the recruitment and activation of leukocytes of both the innate and adaptive immune responses, with resultant osteoclastogenesis and formation of an osteolytic lesion at the apex of the tooth.
Inflammation and resorption of bone at the tooth apex, in most cases, is a consequence of the interaction between microbial infection and the host response. The critical role of bacteria in the development of periapical lesions has been demonstrated by mechanical exposure of the dental pulp to the oral cavity in germ-free animals. In these germ-free animals, pulp exposure heals with an initial or transitory inflammatory response within the pulpal tissue, followed by a reparative response from pulpal cells, and leading to the formation of a new dentin-like matrix bridging the exposed site. In contrast, mechanical pulp exposure in animals with normal oral bacteria causes an infection of the dental pulp, with pulpal tissue necrosis and chronic infection that prevents the repair process Citation1. The infection persists as the necrotic tissue of the dental pulp is inaccessible to leukocytes and, hence, represents a protected reservoir of bacteria Citation2 Citation3. The chronic inflammation stimulated by bacteria and their products in the periapical area of the tooth leads to localized bone resorption that is ‘uncoupled’ so there is no reparative bone formation without treatment. The result is formation and expansion of granulomas or cysts in the apical tissues Citation4.
Understanding the pathogenic mechanisms underlying the development of endodontic lesions is confounded by the persistence of a ‘bacterial reservoir’ that exists in the pulp canal and necrotic tissue. The bacterial presence stimulates an inflammatory response to resist infection. During this response a number of cell types present release cytokines, chemokines, leukotrienes, and prostaglandins into the area. These inflammatory mediators reinforce the recruitment of polymorphonuclear leukocytes (PMNs) and other leukocytes, creating an interesting dichotomy of activity and consequences as to the essential protective or destructive roles mediated Citation3. As would be expected, the host response plays a critical and protective role in lesions of endodontic origin in limiting the spread of infection into the fascial planes. Consistent with this expectation, specific inhibitors of inflammatory cytokines tend to cause the formation of larger osteolytic lesions since they compromise the ability of the host to protect itself from the reservoir of bacteria in the necrotic pulp. This increase in lesion dimensions occurs even though the blocked inflammatory cytokines also play an important role in osteoclastogenesis. The use of inhibitors or mice with targeted genetic deletions may not necessarily reveal the role of a particular cytokine or cell type that plays an important role in activating osteoclastogenesis since its inhibition or knockout may also increase susceptibility to bacterial infection. If the impact on resistant infection is greater, the larger lesion will be produced even though direct effect on deletion or inhibition should reduce osteoclast formation. The reverse is also true. For example, an enhanced host response in an animal model for periapical endodontic lesions demonstrates increased numbers of PMNs and monocytes with a reduction in the extent of apical bone resorption, even though the host response may contribute to the bone resorption Citation5. In another example, the deletion of tumor necrosis factor (TNF) or IL-1 receptor signaling causes larger osteoclast lesion formation even though both cytokines stimulate bone resorption. This occurs because deletion of TNF or IL-1 signaling impairs the antibacterial activity of the host response that is critical in lesions of endodontic origin Citation6. In particular, IL-1 receptor signaling is needed to prevent the spread of infection from necrotic pulp into fascial planes and to protect the host from significant morbidity and mortality that would result Citation6. Thus, there is considerable complexity in examining the impact of cytokine signaling since cytokines have both destructive roles as well as an important protective function in antibacterial defense Citation6.
The control of the periapical infection seems to be a critical aspect of this process, since the absence of the pleiotropic enzyme inducible nitric oxide synthase (iNOS) also results in larger lesions with the recruitment of a greater number of inflammatory cells and frequently associated with periapical abscesses development Citation7. This contrasts with periodontal disease, in which a protected bacterial reservoir does not exist and the use of inhibitors or mice with targeted deletions of the host response typically do not compromise the antibacterial defenses sufficiently to complicate the analysis. Thus, lesions of endodontic origin appear to be at an increased susceptibility to bacterial infection with inhibition of the host response in contrast to periodontal disease.
Leukocytes and endodontic lesions
The initiation of an inflammatory cascade in lesions of endodontic origin includes the complex interplay of multiple cell types involving the activation of endothelial cells, PMNs, macrophages, lymphocytes, and osteoclasts leading to rapid bone destruction. The complex host response involves cells of both the innate and adaptive immune response. The rapid destruction of bone found with endodontic lesions is initiated by multiple bacteria or their products including lipopolysaccharides (LPSs) Citation3. Bacteria are thought to stimulate resorption through the induction of proinflammatory cytokines such as IL-1β, IL-1α,? receptor activator of nuclear factor kappa-B ligand (RANKL), or TNF-α Citation8 Citation9. The initial activation of the host response occurs through stimulation of toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD) receptors Citation10. Both TLRs and NODs are highly expressed on multiple cell types associated with the endodontic lesions including monocytes/macrophages, granulocytes, pulp fibroblasts, osteoclast precursors, and mesenchymal cells Citation10 Citation11. Activation of these receptors leads to the stimulation of multiple proinflammatory cytokines, including IL-1, TNF-α, and IL-6, and has been associated with enhanced RANKL production, osteoclastogenesis, and bone resorption Citation10 Citation11.
Multiple studies have reinforced the concept that the development of bone resorption in lesions of endodontic origin involves the adaptive immune response. The predominant cell types in endodontic lesions in a rat model were shown to be T-cells followed by B-cells and monocytes/macrophages Citation12. Multiple T-cell responses have been associated with endodontic lesions, including Th1 (IL-2 and IFN-γ), Th2 (IL-4 and IL-5), T-regulatory cells (Tregs; IL-10 and TGF-β), and Th17 (IL-17A) lymphocytes Citation13 Citation14 Citation15 . In fact, the key transcription factors essential for Th1, Th2, and Tregs differentiation, T-bet, GATA-3, and FOXp3, respectively, have been found in periapical lesions Citation14 as well as IL-17A, the prototypical cytokine produced by Th17 cells Citation13.
The importance of the adaptive immune response in protecting the host during formation of endodontic lesions has been demonstrated in numerous studies. The exposure of the dental pulp in severe combined immunodeficient (SCID) mice showed periapical lesions of similar size to that found in normal control mice Citation16. However, approximately one-third of the immunodeficient mice with endodontic lesions developed orofacial abscesses. Interestingly, two studies identified contrasting results utilizing nu/nu rats with a deficient T-cell response. While one study showed greater bone resorption following endodontic infections, suggesting a critical protective role, the other study failed to identify a difference in the amount of bone resorption Citation17 Citation18. Evidence of a protective role for IFN-γ, the prototypical Th1-cytokine, was demonstrated as the absence of IFN-γ resulted in increased bone resorption compared to wild-type mice Citation19. Emerging evidence suggests that the majority of Th17 cells also express IFN-γ, supporting a role for both Th1 and Th17 proinflammatory responses in the pathogenesis of periapical periodontitis Citation13. An examination of the Th2 response with genetic deletion of IL-4 failed to identify an effect, suggesting more complex redundancies or that Th2 responses are not critical in protection or bone resorption Citation19. However, the anti-inflammatory cytokine IL-10 has been demonstrated to be a protective factor against periapical bone resorption. Periapical lesions in mice with genetic ablation of IL-10 were increased in size compared with wild-type mice, consistent with a protective role for IL-10 Citation19. Furthermore, IL-10 mRNA levels in human periapical granulomas have been positively correlated with the expression of proteins, SOCS1 and SOCS3, which act as negative regulators of the inflammatory signaling Citation20. Interestingly, Tregs, as a potential source of IL-10, were found in the periapical lesions following endodontic infection consistent with a regulatory role in lesion development Citation15 Citation21.
Cytokines
The initial rapid destruction of bone in the apical area of the root has been associated with the production of prostaglandins, in particular PGE2, through the cyclooxygenase pathway Citation22. These findings provide clarification to an earlier report that indomethacin reduces the extent of bone resorption in endodontic lesions Citation23. Endodontic lesions have been associated with multiple proinflammatory cytokines and chemokines. Cytokines that participate in the formation of osteolytic lesions are shown in and . Interleukins (IL), particularly IL-1α and IL-β are produced in periapical lesions by several types of cells including macrophages, osteoclasts, PMNs, and fibroblasts Citation24 Citation25. The role for IL-1 in stimulating periapical bone destruction was demonstrated using interleukin-1 receptor antagonists to show a 60% reduction in lesion development Citation26. It appears that much of the induced osteoclastogenic activity in periapical lesions is specifically related to the formation of interleukin-1α Citation27. However, when IL-1 receptor signaling is completely deleted there is increased lesion size and systemic morbidity Citation5. In addition, TNF-α expression has been identified in lesions of endodontic origin by cells such as PMNs, monocytes/macrophages, and fibroblasts and may contribute to lesion formation Citation3. The IL-6 has been observed in exudates from human periapical lesions, with osteoblasts, fibroblasts, macrophages, PMNs, and T lymphocytes identified as expressing IL-6 protein Citation28 Citation29. IL-6 has been shown to play a protective role since endodontic lesions in IL-6 deficient animals are increased in size compared with control mice Citation30. The role of cytokines in formation of endodontic and periodontal osteolytic lesions is shown in and .
Fig. 1. RANKL/OPG balance is an important factor in regulating bone resorption in periodontal and periapical environments. Osteoclast differentiation and activation are driven by the interaction of RANK (receptor activator of nuclear factor-kB) with its ligand, RANKL. Osteoprotegerin, OPG, is a decoy receptor of RANKL that inhibits RANK-RANKL engagement. In homeostatic conditions (left side), RANKL and OPG levels are thought to be in balance so that there is limited osteoclastogenesis and bone resorption. With an inflammatory stimulus, the RANKL/OPG ratio increases in periodontal and periapical tissues and leads to stimulation of osteoclast activity and pathologic bone resorption.
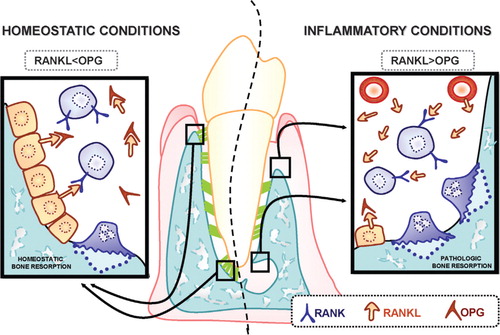
Fig. 2. Cytokine regulation of matrix degradation and bone resorption in periodontal and periapical environments. The presence of microbial pathogens in periodontal and periapical environments trigger an initial production of proinflammatory cytokines, such as TNF-α and IL1β, which stimulate expression and activation of matrix metalloproteinases (MMPs) that degrade extracellular connective tissue matrix. Cytokines such as TNF-α can stimulate osteoclastogenesis independently while other cytokines stimulate RANKL expression that leads to formation of osteoclasts and osteoclast activity. The combined innate and adaptive immune responses are likely to lead to the high levels of inflammation and bone resorption. These proinflammatory cytokines are thought to generate an amplification loop that contributes to periodontal and periapical lesion progression. Conversely, cytokines produced by Th2 cells and Tregs, such as IL-4 and IL-10 have the opposite effect, in part, through stimulating production of tissue inhibitors of matrix metalloproteinases (TIMPs) and OPG as well as restrain inflammatory cytokine production.
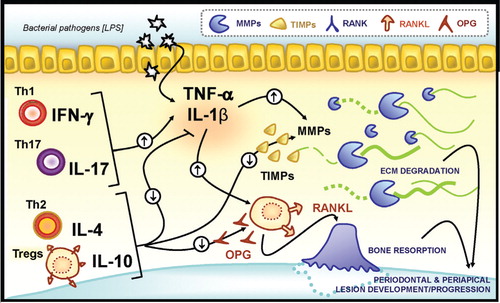
Table 1. Cytokine effects on bone lesions associated with endodontic or periapical lesions
Table 2. Comparison of inflammatory component roles in bone resorption
Neutrophils are active in the development of bone loss associated with endodontic lesions. This has been demonstrated in animals with some neutropenia having a considerable decrease in periapical lesion formation Citation31. The recruitment of PMNs with chemokines has also been implicated in the pathogenesis of periapical lesions. IL-8/CXCL8 chemokine expression is prominent in periapical lesions, consistent with heavy infiltration by PMNs Citation32.
The recruitment of monocytes is critical in the antimicrobial defense in lesions of endodontic origin. Chemokines and chemokine receptors stimulate innate and adaptive immunity in the periapical environment and in the development of granulomas associated with these lesions Citation33. Genetic deletion of MCP-1/CCL2, identified in monocytes/macrophages and bone lining cells in endodontic lesions, significantly reduces monocytic infiltrate while increasing the amount of bone resorption Citation34 Citation35. Similarly, the absence of the MCP-1 receptor (CCR2) or genetic ablation of CC chemokine receptor five (CCR5) results in an increased amount of apical bone resorption and is associated with higher levels of the osteolytic factors, such as RANKL and cathepsin K Citation19 Citation36. Interestingly, activation of the MCP-1/CCL2-CCR2 axis appears to play an active role in mediating the migration of monocytes/macrophages while limiting the infiltration of PMNs Citation36.
The RANKL and osteoprotegerin (OPG) expression demonstrate a heterogeneous pattern in periapical granulomas, ranging from a high ratio of RANKL to OPG consistent with bone resorption to a low ratio seen in sites with minimal bone resorption Citation37. These disparate findings in levels of RANKL/OPG ratio may be indicative of an expanding lesion with active bone resorption or a stable lesion with minimal bone resorption Citation37 Citation38. A description of the role of the RANK-OPG axis in stimulating osteoclastogenesis and bone resorption is shown in .
Periodontal diseases
The periodontium is a complex set of tissues that are in close proximity with a complex biofilm harboring diverse and numerous bacterial species Citation39 Citation40. Periodontal diseases include gingivitis and periodontitis. While it is a consensus that periodontal diseases are stimulated by bacterial adherence to the tooth surface, there is controversy about which bacteria stimulate the irreversible breakdown of periodontal tissues in periodontitis Citation40 Citation41. Recent evidences from studies that do not rely upon bacterial culture techniques suggest that there are approximately 700 bacterial species in the oral cavity Citation39. As a general rule, the bacteria that might cause periodontitis have classically been identified as gram-negative anaerobic bacteria that survive in the gingival sulcus, the space between the tooth surface and the adjacent gingival epithelium Citation42. Much attention has been spent on Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis, which have been linked to localized aggressive ‘juvenile periodontitis’ and ‘adult periodontitis’, respectively Citation42 Citation43. However, recent approaches to bacterial identification have suggested that a reevaluation of pathogenic species is warranted.
The presence of periodontal pathogens is required but not sufficient for disease initiation. In fact, studies clearly demonstrate that cytokines induced by the host response play a critical role in periodontal tissue breakdown Citation44 Citation45 Citation46 Citation47 . Host-microbe interactions start in the gingival epithelium and stimulate an inflammatory response that confers efficient protection against bacteria (i.e. the systemic consequences of acute infection are rare). However, the host mediator release results in a clinical outcome represented by the onset of gingivitis. Because the gingival epithelium and underlying connective tissue are chronically exposed to bacteria or their products, both the innate and the acquired immune response are chronically activated in connective tissue adjacent to epithelium lining the gingiva. In most cases, tissue destruction caused by activation of the host response is reversible and associated with gingivitis. On the other hand, under certain conditions that are not fully understood, the disease can progress and cause destruction of the underlying connective tissue attachment of the gingiva toward the tooth surface and from tooth to bone. Indeed, periodontitis is distinguished from gingivitis by the irreversible nature of the attachment loss. One of the most important uncertainties regarding periodontitis is its chronic nature. Periodontitis may represent a series of brief insults, or ‘bursts’, which accumulate and appear to be chronic over time with extended periods of remission. However, the length of time of the ‘burst’ is unknown. Alternatively, there may be relative constant stimulation over time, but it is not known how long the chronic destructive period lasts in the chronic model. In spite of evidences for both models Citation48 Citation49 Citation50 , the nature of periodontal disease progression remains uncertain. This problem has plagued human studies since it is difficult to know whether an individual is undergoing active periodontal breakdown at any given point in time. Furthermore, the relative absence of longitudinal studies has made the interpretation of results with human patients difficult since relationships between a given variable and irreversible periodontal breakdown are difficult to establish in cross-section studies.
Animal models have established a clear causal relationship between bacteria and periodontitis. In an animal model, a ligature is tied around the teeth allowing plaque accumulation and bacterial penetration, which leads to subsequent inflammation and alveolar bone resorption Citation51. In fact, gnotobiotic rats treated identically do not exhibit periodontal bone loss Citation52, demonstrating the essential role of bacteria in this model. Additional evidence is provided by the treatment with antibiotics or topical application of antimicrobial agents, which reduce bone resorption in the ligature model, while increased colonization by gram negative bacteria enhances bone resorption Citation51. In other animal models, the inoculation of periodontal pathogens into the oral cavity of rodents induces bone loss. In several studies, the introduction of P. gingivalis by oral lavage stimulates alveolar bone resorption Citation51. Similarly, introduction of A. actinomycetemcomitans in rodents leads to colonization and loss of the alveolar bone Citation43 Citation51. Thus, experiments with animal models support human studies demonstrating the role of bacteria in inflammation onset and periodontitis.
Since the presence of bacteria is required, but not sufficient to trigger periodontitis development, the recognition of microbial components as ‘danger signals’ by host cells and the subsequent production of inflammatory mediators is an essential step in periodontitis pathogenesis. Indeed, one of the critical components of the host response to bacteria or their products is a family of receptors called the toll-like receptors (TLRs). The TLRs activate the innate immune response binding to various microbial components (i.e. diacyl lipopeptides, peptidoglycan, LPS, flagellin, bacterial DNA, etc.) Citation53. After TLR activation, an intracellular signaling cascade leads to the activation of transcription factors, such as nuclear factor-κ B (NF-κB), activator protein-1 (AP-1), and subsequent production of various cytokines and chemokines Citation53. Recent studies describe a role for both TLR-2 and TLR-4 in the recognition of A. actinomycetemcomitans, whose impact range from stimulating inflammatory cytokine expression and inflammatory cell migration to inducing osteoclastogenesis and alveolar bone loss Citation54 Citation55. Besides TLRs, the nucleotide-binding oligomerization domain (NOD) receptors and the inflammasome system have been pointed out as potential accessory molecules that trigger the host response against periodontal pathogens Citation56.
Animal models also provided the initial clear evidences for the role of some host immune inflammatory factors in the progression of periodontal diseases. When the host response is altered by treatment with specific inflammatory inhibitors or genetic manipulation, the severity of periodontal connective tissue and bone loss stimulated by periodontal bacteria is clearly reduced. The first concrete evidence that inhibition of an inflammatory response reduces periodontal diseases was carried out in a dog model Citation57 in which the inhibition of prostaglandins significantly reduced alveolar bone loss. Subsequent studies have used a number of techniques to demonstrate that cytokines play an important role in periodontitis. Non-human primates treated with inhibitors to two major proinflammatory cytokines, IL-1 and TNF, exhibit reduced periodontal bone loss and loss of attachment compared to control animals Citation44 Citation45 Citation58. Similarly, RANKL inhibition decreases alveolar bone loss in several models of periodontal disease Citation46 Citation59 Citation60. It is hypothesized that periodontal disease progression is due to a combination of several factors, including the presence of periodontopathic bacteria, high levels of proinflammatory cytokines and prostaglandins, the production and activation of MMPs and RANKL, and relatively low levels of interleukin-10 (IL-10), transforming growth factor-β (TGF-β), tissue inhibitors of metalloproteinase (TIMPs), and osteoprotegerin (OPG) Citation61. A description of inflammatory cytokines and cell types that participate in bone resorption and destruction of connective tissue matrix is shown in .
Inflammatory mediators
The cyclooxygenase enzymes, COX-1 and COX-2, catalyze the conversion of arachidonic acid to prostaglandins. COX-1 is constitutively expressed and leads to the generation of prostaglandins that are particularly important in homeostasis. COX-2 is inducible and leads to the formation of prostaglandins involved in inflammatory processes. Prostaglandins (PGs), potent stimulators of bone formation and resorption, are produced by bone cells, fibroblasts, gingival epithelial cells, endothelial cells, and inflammatory cells Citation62. Prostaglandin E2 production is elevated in individuals with periodontitis compared with healthy subjects Citation63. When applied topically to the gingival sulcus, prostaglandin E2 induces a marked increase in osteoclasts. Moreover, it is synergistic with lipopolysaccharide stimulating osteoclastogenesis Citation64.
Both PGE-2 and leukotriene B-4 were found in gingival crevicular fluid of individuals with localized aggressive periodontitis. Furthermore, P. gingivalis stimulates increased PGE-2 levels and increased COX-2 expression by infiltrated leukocytes in vivo Citation65. In an animal ligature induced periodontitis model, both a non-selective COX inhibitor and a selective COX-2 inhibitor reduced osteoclast numbers and alveolar bone loss when compared to non-treatment Citation66. Many clinical trials have explored the use of a COX-2 inhibitor as an adjunct to periodontal therapy. These inhibitors improved the clinical outcome after periodontal therapy compared to periodontal therapy alone Citation67. However, they are not clinically used in the treatment of human patients due to side effects. Lipoxins and resolvins, products of omega-3 fatty acids, induce resolution of inflammation, and to protect against periodontal bone loss stimulated by bacteria in animal models Citation68.
The induction of experimental periodontitis is associated with the expression of innate immune cytokines Citation69. IL-1 stimulates the expression of proresorptive cytokines, such as RANKL and TNF-α and proteinases that participate in periodontal connective tissue destruction and bone resorption. In the periodontium, IL-1 is produced by several types of cells including PMNs, monocytes, and macrophages Citation70 Citation71. In patients with periodontitis, IL-1β expression is elevated in gingival crevicular fluid at sites of recent bone and attachment loss Citation71 Citation72. The IL-1β is also found to be higher in gingiva from individuals with a history of periodontitis than in samples from healthy individuals Citation72.
Using a non-human primate model, Delima et al. showed that IL-1 inhibition significantly reduced inflammation, connective tissue attachment loss, and bone resorption induced by periodontal pathogens when compared to controls Citation45. In other studies, IL-1 receptor deficient mice had less P. gingivalis LPS-induced osteoclastogenesis compared to similarly treated wild-type mice Citation73. In a different approach, the exogenous application of recombinant human IL-1β in a rat model of experimental periodontitis accelerated alveolar bone destruction and inflammation over a 2-week period Citation74. In addition, transgenic mice overexpressing IL-1α in gingival epithelium developed a periodontitis-like syndrome, leading to the loss of attachment and destruction of periodontal bone Citation75. Taken together, these studies strongly support the role of IL-1 in promoting alveolar bone destruction in periodontitis.
The TNF refers to two associated proteins, TNF-α and TNF-β. TNF-α levels are upregulated in gingival crevicular fluid at sites where bone and attachment loss have recently occurred Citation71 Citation76 Citation77 Citation78 . TNF-α was also found to be higher in diseased periodontal tissue samples than in tissue samples from healthy individuals Citation72. A cause and effect relationship between TNF-α and periodontal bone loss has been demonstrated. Administration of recombinant TNF-α accelerates periodontal destruction in a rat periodontitis model Citation79. On the other hand, P. gingivalis induced osteoclastogenesis is reduced in TNF receptor-deficient mice compared to wild-type controls, indicating that osteoclast formation is dependent on TNF-α regulated pathway as part of the host response to bacterial challenge Citation80. Garlet et al. recently showed that TNFR-1 knockout mice developed significantly less inflammation, indicated by chemokine and chemokine receptors downregulation, and less alveolar bone loss in association with RANKL downregulation in response to A. Actinomycetemcomitans oral inoculation Citation81. Furthermore, mRNA levels of IL-1β, IFN-γ, and RANKL in gingival tissues were significantly lower in TNFR-1 knockout mice than in wild-type infected mice. In contrast, A. actinomycetemcomitans levels quantified by real-time PCR were significantly greater in TNF receptor ablated mice than in wild-type controls and were associated with lower levels of the PMN-related antimicrobial mediator myeloperoxidase in experimental mice Citation81. Thus, the absence of TNFR-1 resulted in a lower production of cytokines in response to A. actinomycetemcomitans infection even in the presence of higher levels of periodontal pathogens. Based on similar studies, it can be implied that the local production of TNF-α plays a role in upregulating the host response to bacteria and stimulating bone resorption during periodontitis. It is also possible that oral bacteria in addition to causing local pathology may contribute to systemic conditions by enhancing cytokine production subsequent to bacteremias. Interestingly, P gingivalis LPS stimulates a strong local inflammatory response but a weak systemic inflammatory response Citation82. Inhibition of IL-1 and TNF-α together significantly reduces the progression of inflammation toward bone, osteoclastogenesis, and periodontal tissue destruction Citation44. In the gingiva, a higher expression of IL-6 is found in gingival crevicular fluid and in the gingiva in mononuclear cells and T-cells from periodontitis patients than in healthy controls Citation83 Citation84. The LPS from the periodontal pathogen A. actinomycetemcomitans induces IL-6 expression, osteoclastogenesis, and bone loss Citation85. Oral inoculation of P. gingivalis in mice with genetically deleted IL-6 have decreased bone loss compared to wild-type mice, suggesting that IL-6 contributes to the progression of bacteria-induced bone loss Citation47.
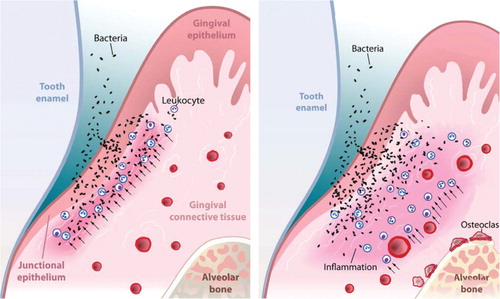
After a host response triggered by microbial recognition, the spatial orientation of the subsequent leukocyte infiltration into periodontal tissues is likely to contribute to periodontal disease. Histologically, the inflammatory infiltrate is observed even in the presence of minimal clinical signs of inflammation, but when inflammation is restricted to the connective tissue closest to the gingival epithelium, gingivitis is present Citation86. However, in animal models, when the inflammatory infiltrate moves closer to bone, osteoclastogenesis is induced and periodontal bone loss takes place Citation44. This suggests that the periodontal disease development might be determined by the progression of the inflammatory infiltrate toward bone. The impact of spatial location of the inflammatory infiltrate on bone resorption is shown in .
Fig. 3. Spatial relationship between an inflammatory infiltrate and periodontal bone loss. In periodontitis, bacteria attach to the tooth surface and invade the adjacent epithelium and connective tissue. This causes formation of an inflammatory infiltrate indicated by the black arrows. If the inflammatory infiltrate is at a distance from bone (left panel), osteoclastogenesis is not stimulated. However, if the infiltrate moves closer to bone (right panel), osteoclasts are induced and bone resorption occurs.
Among the mediators potentially involved in leukocyte diapedesis and subsequent spatial localization in periodontal environment, chemokines have been investigated with special interest in the last decade. Chemokines are small chemotactic cytokines that stimulate the recruitment of inflammatory cells Citation33 Citation87. They are divided into two major families based on their structure, CC and CXC chemokines, which basically bind to the two major classes of receptors, CC chemokine receptors (CCR) and CXC chemokine receptors (CXCR). Chemokines are produced by several resident and inflammatory cell types in the periodontium Citation33. Some chemokines can stimulate one or more steps of bone resorption, including the recruitment, differentiation, or fusion of precursor cells to form osteoclasts or enhance osteoclast survival Citation33 Citation87. They could also affect periodontal bone loss by recruiting cells, such as neutrophils, which protect against bacterial invasion.
Chemokines are found in gingival tissue and crevicular fluid. The IL-8/CXCL8, a chemoattractant of PMNs, is found at higher levels in gingival crevicular fluid prior to clinical signs of inflammation following cessation of tooth brushing. Moreover, in subjects with a history of periodontitis, IL-8/CXCL8 in gingiva and gingival crevicular fluid are increased and correlated with disease severity Citation88.
One of the most abundant expressed chemokines is macrophage inflammatory protein-1α (MIP-1α/CCL3), which is localized in the connective tissue subjacent to gingival epithelium Citation89. MIP-1α/CCL3-positive cells increase with increasing severity of periodontal disease. It is a ligand for the chemokine receptors CCR1 and CCR5 and is associated with the recruitment of monocytes/macrophages and dendritic cells via CCR1 and lymphocytes polarized into Th1 phenotype by CCR5 Citation90. Thus, MIP-1α/CCL3 has a potential role in stimulating bone resorption through effects on macrophages and Th1 cells. Moreover, CCR1+ and CCR5+ cell populations may affect resorption since they include osteoclast precursors Citation91 Citation92. This is consistent with findings that MIP-1α/CCL3 directly stimulates osteoclasts differentiation Citation93 and stromal-cell derived factor-1 (SDF1/CXCL12) regulates osteoclast function and is found in the periodontium Citation94.
A number of chemokines have been detected in gingiva or in gingival crevicular fluid including regulated upon activation normal T-cell expressed and secreted (RANTES/CCL5). RANTES/CCL5 is found in greater levels in active periodontal lesions compared to inactive sites Citation89 Citation95. That RANTES/CCL5 may be involved in periodontal bone resorption is supported by findings that it binds to CCR1 and CCR5 Citation91 Citation96. Monocyte chemoattractant protein-1 (MCP-1/CCL2) may also contribute to periodontitis. MCP-1/CCL2 levels are directly correlated with gingival inflammation Citation97 Citation98. It stimulates monocyte/macrophage recruitment and activity and has been implicated as a chemoattractant for osteoclast precursors Citation91 Citation96.
RANKL stimulates osteoclastogenesis and bone resorption. A number of studies have established that RANKL inhibition decreases periodontal bone resorption Citation46 Citation59 Citation60 and establishes a role for RANKL in periodontitis. Osteoprotegerin (OPG) is a molecule that is also upregulated by inflammatory conditions and blocks RANKL by binding to it. A high ratio of RANKL/OPG creates proresorptive conditions while a low RANKL to OPG ratio is antiresorptive. During bacteria stimulated periodontal bone loss there is an initial increase in the ratio of RANKL/OPG Citation69. After the initial bone loss, antiresorptive factors are produced including OPG, as well as IL-4 and IL-10, reducing the RANKL/OPG ratio Citation69. This relationship is shown in . The RANKL/OPG ratio has been examined in gingival tissues or gingival crevicular fluid. It has been shown that periodontitis is associated with an increase in RANKL. The RANKL/OPG ratio greater than 1 predominates in chronic periodontitis lesions while a ratio of 0.5 or less is found in chronic gingivitis lesions Citation37.
RANKL is upregulated in both pathologic and physiologic bone resorption. In pathologic inflammatory bone disease, RANKL expression has been shown to have the highest level in B-cells, followed by T-cells, and then monocytes Citation99. This indicates that activated T- and B-cells can be the cellular source of RANKL for bone resorption in diseased gingival tissue. In physiologic bone, remodeling bone-lining cells such as osteoblasts or their precursors appear to be an important source of RANKL.
The IFN-γ is a lymphokine, produced by lymphocytes and natural killer cells that has been implicated in periodontal bone loss. Its expression is associated with Th1 lymphocytes. Mice with a genetic ablation of IFN-γ have less P. gingivalis induced bone loss compared to wild-type controls Citation47. T-cells are an important source of IFN-γ in periodontitis Citation84 and have been linked to increased RANKL expression Citation100.
Lymphocytes also produce cytokines that are anti-inflammatory, such as IL-4 and IL-10 Citation101. These cytokines are associated with a Th2 response and reduce the severity of experimental periodontitis Citation102. However, a direct link between Th1 lymphocytes enhancing periodontal disease and Th2 lymphocytes reducing it is not necessarily straightforward since there are components of a Th2 response that are also prodestructive Citation103.
Innate immune cells have been shown to play an important role in periodontal bone resorption Citation61 Citation104. Monocytes and macrophages produce several cytokines and lytic enzymes that stimulate the breakdown of connective tissue and bone resorption Citation61. PMNs have been shown to have both protective and destructive effects Citation105. A protective role is inferred from findings that neutrophil disorders including cyclic neutropenia, Chédiak-Higashi syndrome, and leukocyte adhesion deficiency syndrome promote periodontal diseases Citation105. The production of reactive oxygen species and cytokines implicates them in the destructive phase. Monocytes and PMNs produce a respiratory burst that generates superoxides, hydrogen peroxide, hydroxyl radicals, hypochlorous acid, and chloramines. These products contribute to bacterial killing based on evidence that impaired production of iNOS and MPO are associated with increased levels of periodontal pathogens Citation81 Citation106. In addition, PMNs and monocytes/macrophages release elastases and collagenases that break down connective tissue Citation107 and are linked to the development of periodontal lesions Citation107.
Dendritic cells of monocytic lineage are another group of innate immune cells that function to present antigen to lymphocytes and also promote inflammation by the production of chemokines and cytokines Citation108. They have been implicated in periodontal disease Citation108 Citation109 Citation110 Citation111 . Oral bacteria induce dendritic cells to produce cytokines such as IL-1β, IL-12, IFN-γ, TNF-α, and TNF-β Citation109 Citation110. Dendritic cells can form to osteoclasts Citation112. For example, A. actinomycetemcomitans stimulates dendritic cells in vitro to form osteoclasts in a RANKL dependent manner Citation113.
Oral bacteria stimulate cells of the adaptive immune response as shown by the presence of activated T- and B-lymphocytes in periodontal disease tissues Citation61. As discussed above, lymphocytes produce cytokines that promote bone resorption directly through RANKL or indirectly through IFN-γ. There is evidence to suggest that lymphocytes are involved in mediating bacteria stimulated periodontal bone resorption. When severe combined immunodeficient (SCID) mice that lack B- and T-lymphocytes are challenged with P. gingivalis, there is considerably less bone resorption than in wild-type normal mice Citation114. Moreover, (SCID) mice engrafted with human CD4(+) T-cells from individuals with aggressive early onset periodontal disease and subsequently challenged with A. actinomycetemcomitans exhibit enhanced periodontal bone loss Citation46. This bone loss is mediated by RANKL. Similarly, the adoptive transfer of B-cells from A. actinomycetemcomitans immunized rats followed by an injection of A. actinomycetemcomitans into the gingiva, stimulates greater alveolar bone resorption than control mice that have received B-cells from non-immunized mice Citation59. The increased resorption was shown to be RANKL mediated Citation59. The results of these experiments indicate that cells of the adaptive immune response significantly contribute to periodontitis.
In addition to Th1 and Th2 CD4+ lymphocytes, there are two other T-cell subsets that have been identified, Th17 and Tregs (regulatory T-cells). The Th17 lymphocytes produce IL-17, which in turn stimulate RANKL-mediated osteoclastogenesis Citation115. They have been implicated in rheumatoid arthritis, periodontal disease, and loosening of joint prostheses Citation115. Under experimental conditions, IL-17 appears to have an important protective function since genetic deletion of IL-17 receptors enhance periodontal bone loss stimulated by P. gingivalis in vivo Citation116. This may be due to the role of IL-17 in stimulating chemokines that induce recruitment of neutrophils. However, humans with periodontitis have increased levels of Th17 cells and IL-17 mRNA, compared to healthy tissues suggesting but not proving that IL-17 contributes to the destructive process Citation117. The Tregs modulate activation, proliferation, and effector function of conventional T-cells Citation118 and have been identified in periodontal tissues Citation119 Citation120 Citation121 . Because they are associated with the production of IL-10, TGF-β, and the inhibitory molecule CTLA-4, Tregs may reduce periodontal disease progression Citation119.
Uncoupled bone formation and periodontitis
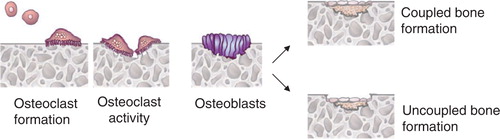
Bone remodeling involves a process of bone resorption followed by bone formation, a process referred to as coupling Citation122. Bacteria induced bone resorption in a healthy adult should be followed by an equivalent amount of bone formation. In periodontitis, there is a failure to form an adequate amount of new bone following resorption resulting in net bone loss. Thus, a critical aspect of periodontitis is uncoupling so that bacteria induced bone loss is not followed by an equivalent amount of new bone formation resulting in net bone loss. The impact of uncoupling on creating net bone loss is shown in .
Fig. 4. The role of coupling in periodontal lesion development. Bone formation occurs after bone resorption so that the two processes are coupled. Thus, the resorption pit is occupied by osteoblasts that form new bone. In a normal healthy individual, the amount of bone formed equals the amount resorbed. In pathologic bone resorption, the amount of bone that forms is less than that resorbed so that there is net bone loss. This may be due to the impact of inflammation on bone formation. Inflammation could potentially interfere with coupling by reducing proliferation of osteoblast precursors, inhibiting differentiation of osteoblasts, decreasing osteoblast numbers by stimulating apoptosis, or by interfering with the production of bone matrix.
The same process that stimulates bone resorption, inflammation, may be responsible for uncoupling. It is possible that under conditions where inflammation is in close proximity to and along the bone, it will affect osteoblast numbers or function and interfere with the coupling process. In an experimental model, the injection of P. gingivalis into connective tissue induces bone resorption followed by bone formation Citation123 Citation124. If the inflammation is prolonged by induction of the adaptive immune response, the capacity to form new bone is diminished and uncoupling occurs Citation123. Similarly, prolonged inflammation in diabetic animals interferes with bone formation in the periodontium following bacteria stimulated bone resorption Citation124. This interpretation is additionally supported by evidence that the application of cytokines in vivo stimulates bone resorption but also limits bone formation. Therefore, several lines of animal experimentation support the concept that inflammation uncouples bone formation from bone resorption. Thus, inflammation may not only stimulate the formation of osteoclasts and bone resorption, but also affect bone by altering the function of osteoblasts and limiting reparative bone formation.
Conclusions
Polymicrobial infection in lesions of endodontic origin stimulates bone resorption by interacting with the leukocytes of the innate and adaptive immune responses. In endodontic lesions, the presence of inflammation suppresses bone formation so that lesion resolution does not occur until the causal bacteria are entombed by treatment and the inflammation subsides. Periodontitis is caused by a host response to the presence of bacteria or their products that invade connective tissue. The host defense, including innate and adaptive immunity, is responsible for combating bacteria invading the periodontal tissue. In humans, plaque accumulation occurs even in health so that there is a continuous state of inflammation in gingival tissue adjacent to teeth. By using animal models and specific inhibitors, both the innate and adaptive immune response have been conclusively shown to participate in the formation of periodontal lesions. Cytokines generated that induce osteolytic lesions are shown in and a comparison of the effect of cytokine deletion or inhibition in the formation of lesions of endodontic origin and in periodontitis is shown in . It is also possible that the inflammation associated with periodontal bone resorption affects coupled bone formation contributing to net bone loss (see ).
Conflict of interest and funding
Funding for this work was provided in part by the National Institute of Dental and Craniofacial Research.
Related Research Data
References
- Kakehashi S, Stanley HR, Fitzgerald RJ. The effects of surgical exposures of dental pulps in germ-free and conventional laboratory rats. Oral Surg Oral Med Oral Pathol. 1965; 20: 340–9.
- Ricucci D, Siqueira JF Jr, Bate AL, Pitt Ford TR. Histologic investigation of root canal-treated teeth with apical periodontitis: a retrospective study from twenty-four patients. J Endod. 2009; 35: 493–502.
- Nair PN. Pathogenesis of apical periodontitis and the causes of endodontic failures. Crit Rev Oral Biol Med. 2004; 15: 348–81.
- Liapatas S, Nakou M, Rontogianni D. Inflammatory infiltrate of chronic periradicular lesions: an immunohistochemical study. Int Endod J. 2003; 36: 464–71.
- Stashenko P, Wang C, Riley E, Wu Y, Ostroff G, Niederman R. Reduction of infection-stimulated peripical bone resorption by the biological response modifier PGG glucan. J Dent Res. 1995; 74: 323–30.
- Graves D, Chen C, Douville C, Jiang Y. Interleukin-1 receptor signaling rather than that of tumor necrosis factor is critical in protecting the host from the severe consequences of a polymicrobe anaerobic infection. Infect Immun. 2000; 8: 4746–51.
- Fukada SY, Silva TA, Saconato IF, Garlet GP, Avila-Campos MJ, Silva JS, et al.. iNOS-derived nitric oxide modulates infection-stimulated bone loss. J Dent Res. 2008; 87: 1155–9.
- Jiang Y, Mehta C, Hsu T, Alsulaimani F. Bacteria induce osteoclastogenesis via an osteoblast-independent pathway. Infect Immun. 2002; 70: 3143–8.
- Gilles J, Carnes D, Dallas M, Holt S, Bonewald L. Oral bone loss is increased in ovariectomized rats. J Endodon. 1997; 23: 419–22.
- Hirao K, Yumoto H, Takahashi K, Mukai K, Nakanishi T, Matsuo T. Roles of TLR2, TLR4, NOD2, and NOD1 in pulp fibroblasts. J Dent Res. 2009; 88: 762–7.
- Bar-Shavit Z. Taking a toll on the bones: regulation of bone metabolism by innate immune regulators. Autoimmunity. 2008; 41: 195–203.
- Stashenko P, Yu SM, Wang CY. Kinetics of immune cell and bone resorptive responses to endodontic infections. J Endod. 1992; 18: 422–6.
- Colic M, Gazivoda D, Vucevic D, Vasilijic S, Rudolf R, Lukic A. Proinflammatory and immunoregulatory mechanisms in periapical lesions. Mol Immunol. 2009; 47: 101–13.
- Fukada SY, Silva TA, Garlet GP, Rosa AL, da Silva JS, Cunha FQ. Factors involved in the T helper type 1 and type 2 cell commitment and osteoclast regulation in inflammatory apical diseases. Oral Microbiol Immunol. 2009; 24: 25–31.
- Colic M, Gazivoda D, Vucevic D, Majstorovic I, Vasilijic S, Rudolf R, et al.. Regulatory T-cells in periapical lesions. J Dent Res. 2009; 88: 997–1002.
- Teles R, Wang CY, Stashenko P. Increased susceptibility of RAG-2 SCID mice to dissemination of endodontic infections. Infect Immun. 1997; 65: 3781–7.
- Tani N, Kuchiba K, Osada T, Watanabe Y, Umemoto T. Effect of T-cell deficiency on the formation of periapical lesions in mice: histological comparison between periapical lesion formation in BALB/c and BALB/c nu/nu mice. J Endod. 1995; 21: 195–9.
- Wallstrom JB, Torabinejad M, Kettering J, McMillan P. Role of T cells in the pathogenesis of periapical lesions. A preliminary report. Oral Surg Oral Med Oral Pathol. 1993; 76: 213–8.
- De Rossi A, Rocha LB, Rossi MA. Interferon-gamma, interleukin-10, intercellular adhesion molecule-1, and chemokine receptor 5, but not interleukin-4, attenuate the development of periapical lesions. J Endod. 2008; 34: 31–8.
- Menezes R, Garlet TP, Trombone AP, Repeke CE, Letra A, Granjeiro JM, et al.. The potential role of suppressors of cytokine signaling in the attenuation of inflammatory reaction and alveolar bone loss associated with apical periodontitis. J Endod. 2008; 34: 1480–4.
- Alshwaimi E, Purcell P, Kawai T, Sasaki H, Oukka M, Campos-Neto A, et al.. Regulatory T cells in mouse periapical lesions. J Endod. 2009; 35: 1229–33.
- McNicholas S, Torabinejad M, Blankenship J, Bakland L. The concentration of prostaglandin E2 in human periradicular lesions. J Endod. 1991; 17: 97–100.
- Torbinejad M, Clagett J, Engel D. A cat model for the evaluation of mechanisms of bone resorption: induction of bone loss by simulated immune complexes and inhibition by indomethacin. Calcif Tissue Int. 1979; 29: 207–14.
- Fouad AF. IL-1 alpha and TNF-alpha expression in early periapical lesions of normal and immunodeficient mice. J Dent Res. 1997; 76: 1548–54.
- Tani-Ishii N, Wang CY, Stashenko P. Immunolocalization of bone-resorptive cytokines in rat pulp and periapical lesions following surgical pulp exposure. Oral Microbiol Immunol. 1995; 10: 213–9.
- Stashenko P, Wang CY, Tani-Ishii N, Yu SM. Pathogenesis of induced rat periapical lesions. Oral Surg Oral Med Oral Pathol. 1994; 78: 494–502.
- Wang C, Stashenko P. The role of interleukin-1 alpha in the pathogenesis of periapical bone destruction in a rat model system. Oral Microbiol Immunol. 1993; 8: 50–6.
- Takeichi O, Saito I, Tsurumachi T, Moro I, Saito T. Expression of inflammatory cytokine genes in vivo by human alveolar bone-derived polymorphonuclear leukocytes isolated from chronically inflamed sites of bone resorption. Calcif Tissue Int. 1996; 58: 244–8.
- Walker KF, Lappin DF, Takahashi K, Hope J, Macdonald DG, Kinane DF. Cytokine expression in periapical granulation tissue as assessed by immunohistochemistry. Eur J Oral Sci. 2000; 108: 195–201.
- Huang GT, Do M, Wingard M, Park JS, Chugal N. Effect of interleukin-6 deficiency on the formation of periapical lesions after pulp exposure in mice. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001; 92: 83–8.
- Yamasaki M, Kumazawa M, Kohsaka T, Nakamura H. Effect of methodtrexate-induced neutropenia on rat periapical lesion. Oral Surg Oral Med Oral Pathol. 1994; 77: 655–61.
- Huang GT, Potente AP, Kim JW, Chugal N, Zhang X. Increased interleukin-8 expression in inflamed human dental pulps. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999; 88: 214–20.
- Silva TA, Garlet GP, Fukada SY, Silva JS, Cunha FQ. Chemokines in oral inflammatory diseases: apical periodontitis and periodontal disease. J Dent Res. 2007; 86: 306–19.
- Rahimi P, Wang C, Stashenko P, Lee S, Lorenzo J, Graves D. Monocyte chemoattractant prtein-1 expression and monocyte recruitment in osseous inflammation in the mouse. Endocrinology. 1995; 136: 2752–9.
- Chae P, Im M, Gibson F, Jiang Y, Graves D. Mice lacking monocyte chemoattractant protein 1 have enhanced susceptibility to an interstitial polymicrobial infection due to impaired monocyte recruitment. Infect Immun. 2002; 70: 3164–9.
- Garlet TP, Fukada SY, Saconato IF, Avila-Campos MJ, Silva TAD, Garlet GP, et al.. CCR2 deficiency results in increased osteolysis in experimental periapical lesions in mice. J Endod. 2010; 36: 244–50.
- Menezes R, Garlet TP, Letra A, Bramante CM, Campanelli AP, Figueira Rde C, et al.. Differential patterns of receptor activator of nuclear factor kappa B ligand/osteoprotegerin expression in human periapical granulomas: possible association with progressive or stable nature of the lesions. J Endod. 2008; 34: 932–8.
- Kawashima N, Suzuki N, Yang G, Ohi C, Okuhara S, Nakano-Kawanishi H, et al.. Kinetics of RANKL, RANK and OPG expressions in experimentally induced rat periapical lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007; 103: 707–11.
- Paster BJ, Olsen I, Aas JA, Dewhirst FE. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000. 2006; 42: 80–7.
- Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005; 366: 1809–20.
- Sakamoto M, Umeda M, Benno Y. Molecular analysis of human oral microbiota. J Periodontal Res. 2005; 40: 277–85.
- Haffajee AD, Teles RP, Socransky SS. The effect of periodontal therapy on the composition of the subgingival microbiota. Periodontol 2000. 2006; 42: 219–58.
- Fine DH, Kaplan JB, Kachlany SC, Schreiner HC. How we got attached to Actinobacillus actinomycetemcomitans: a model for infectious diseases. Periodontol 2000. 2006; 42: 114–57.
- Assuma R, Oates T, Cochran D, Amar S, Graves D. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immunol. 1998; 160: 403–9.
- Delima A, Spyros K, Amar S, Graves DT. Inflammation and tissue loss caused by periodontal pathogens is reduced by IL-1 antagonists. J Infect Dis. 2002; 186: 511–6.
- Teng Y, Nguyen H, Gao X, Kong Y, Gorczynski R, Singh B, et al.. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J Clin Invest. 2000; 106: R59–67.
- Baker P, Dixon M, Evans R, Dufour L, Johnson E, Roopenian D. CD4(+) T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infect Immun. 1999; 67: 2804–9.
- Socransky SS, Haffajee AD, Goodson JM, Lindhe J. New concepts of destructive periodontal disease. J Clin Periodontol. 1984; 11: 21–32.
- Cohen ME, Ralls SA. Distributions of periodontal attachment levels. Mathematical models and implications. J Periodontol. 1988; 59: 254–8.
- Gilthorpe MS, Zamzuri AT, Griffiths GS, Maddick IH, Eaton KA, Johnson NW. Unification of the “burst” and “linear” theories of periodontal disease progression: a multilevel manifestation of the same phenomenon. J Dent Res. 2003; 82: 200–5.
- Graves DT, Fine D, Teng YT, Van Dyke TE, Hajishengallis G. The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol. 2008; 35: 89–105.
- Rovin S, Costich ER, Gordon HA. The influence of bacteria and irritation in the initiation of periodontal disease in germfree and conventional rats. J Periodontal Res. 1966; 1: 193–204.
- Mahanonda R, Pichyangkul S. Toll-like receptors and their role in periodontal health and disease. Periodontol 2000. 2007; 43: 41–55.
- Gelani V, Fernandes AP, Gasparoto TH, Garlet TP, Cestari TM, Lima HR, et al.. The role of toll-like receptor 2 in the recognition of Aggregatibacter actinomycetemcomitans. J Periodontol. 2009; 80: 2010–9.
- Lima HR, Gelani V, Fernandes AP, Gasparoto TH, Torres SA, Santos CF, et al.. The essential role of TLR4 in the control of Aggregatibacter actinomycetemcomitans infection in mice. J Clin Periodontol. 2010; 37: 248–54.
- Uehara A, Takada H. Functional TLRs and NODs in human gingival fibroblasts. J Dent Res. 2007; 86: 249–54.
- Williams R, Jeffcoat M, Kaplan M, Goldhaber P, Johnson H, Wechter W. Flurbiprofen: a potent inhibitor of alveolar bone resorption in beagles. Science. 1985; 227: 640–2.
- Delima A, Oates T, Assuma R, Schwartz Z, Cochran D, Amar S, et al.. Soluble antagonists to interleukin-1 (IL-1) and tumor necrosis factor (TNF) inhibits loss of tissue attachment in experimental periodontitis. J Clin Periodontol. 2001; 28: 233–40.
- Han X, Kawai T, Eastcott JW, Taubman MA. Bacterial-responsive B lymphocytes induce periodontal bone resorption. J Immunol. 2006; 176: 625–31.
- Jin Q, Cirelli JA, Park CH, Sugai JV, Taba M Jr, Kostenuik PJ, et al.. RANKL inhibition through osteoprotegerin blocks bone loss in experimental periodontitis. J Periodontol. 2007; 78: 1300–8.
- Graves D. Cytokines that promote periodontal tissue destruction. J Periodontol. 2008; 79: 1585–91.
- Fracon RN, Teofilo JM, Satin RB, Lamano T. Prostaglandins and bone: potential risks and benefits related to the use of nonsteroidal anti-inflammatory drugs in clinical dentistry. J Oral Sci. 2008; 50: 247–52.
- Noguchi K, Ishikawa I. The roles of cyclooxygenase-2 and prostaglandin E2 in periodontal disease. Periodontol 2000. 2007; 43: 85–101.
- Miyauchi M, Ijuhin N, Nikai H, Takata T, Ito H, Ogawa I. Effect of exogenously applied prostaglandin E2 on alveolar bone loss—histometric analysis. J Periodontol. 1992; 63: 405–11.
- Pouliot M, Clish CB, Petasis NA, Van Dyke TE, Serhan CN. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: a role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry. 2000; 39: 4761–8.
- Bezerra MM, de Lima V, Alencar VB, Vieira IB, Brito GA, Ribeiro RA, et al.. Selective cyclooxygenase-2 inhibition prevents alveolar bone loss in experimental periodontitis in rats. J Periodontol. 2000; 71: 1009–14.
- Pinho Mde N, Pereira LB, de Souza SL, Palioto DB, Grisi MF, Novaes AB Jr, et al.. Short-term effect of COX-2 selective inhibitor as an adjunct for the treatment of periodontal disease: a clinical double-blind study in humans. Braz Dent J. 2008; 19: 323–8.
- Hasturk H, Kantarci A, Ohira T, Arita M, Ebrahimi N, Chiang N, et al.. RvE1 protects from local inflammation and osteoclast-mediated bone destruction in periodontitis. FASEB J. 2006; 20: 401–3.
- Garlet GP, Cardoso CR, Silva TA, Ferreira BR, Avila-Campos MJ, Cunha FQ, et al.. Cytokine pattern determines the progression of experimental periodontal disease induced by Actinobacillus actinomycetemcomitans through the modulation of MMPs, RANKL, and their physiological inhibitors. Oral Microbiol Immunol. 2006; 21: 12–20.
- Hou LT, Liu CM, Liu BY, Lin SJ, Liao CS, Rossomando EF. Interleukin-1beta, clinical parameters and matched cellular-histopathologic changes of biopsied gingival tissue from periodontitis patients. J Periodontal Res. 2003; 38: 247–54.
- Salvi GE, Brown CE, Fujihashi K, Kiyono H, Smith FW, Beck JD, et al.. Inflammatory mediators of the terminal dentition in adult and early onset periodontitis. J Periodontal Res. 1998; 33: 212–25.
- Stashenko P, Jandinski J, Fujiyoshi P, Rynar J, Socransky S. Tissue levels of bone resorptive cytokines in periodontal disease. J Perio. 1991; 62: 504–9.
- Chiang C, Kyritsis G, Graves D, Amar S. Interleukin-1 and tumor necrosis factor activities partially account for calvarial bone resorption induced by local injection of lipopolysaccharide. Infect Immun. 1999; 67: 4231–6.
- Koide M, Suda S, Saitoh S, Ofuji Y, Suzuki T, Yoshie H, et al.. In vivo administration of IL-1 beta accelerates silk ligature-induced alveolar bone resorption in rats. J Oral Pathol Med. 1995; 24: 420–34.
- Dayan S, Stashenko P, Niederman R, Kupper TS. Oral epithelial overexpression of IL-1alpha causes periodontal disease. J Dent Res. 2004; 83: 786–90.
- Kurtis B, Tuter G, Serdar M, Akdemir P, Uygur C, Firatli E, et al.. Gingival crevicular fluid levels of monocyte chemoattractant protein-1 and tumor necrosis factor-alpha in patients with chronic and aggressive periodontitis. J Periodontol. 2005; 76: 1849–55.
- Bostrom L, Linder L, Bergstrom J. Clinical expression of TNF-alpha in smoking-associated periodontal disease. J Clin Periodontol. 1998; 25: 767–73.
- Lee HJ, Kang IK, Chung CP, Choi SM. The subgingival microflora and gingival crevicular fluid cytokines in refractory periodontitis. J Clin Periodontol. 1995; 22: 885–90.
- Gaspersic R, Stiblar-Martincic D, Osredkar J, Skaleric U. Influence of subcutaneous administration of recombinant TNF-alpha on ligature-induced periodontitis in rats. J Periodontal Res. 2003; 38: 198–203.
- Graves D, Oskoui M, Volejnikova S, Naguib G, Cai S, Desta T, et al.. Tumor necrosis factor modulates fibroblast apoptosis, PMN recruitment, and osteoclast formation in response to P. gingivalis infection. J Dent Res. 2001; 80: 1875–9.
- Garlet GP, Cardoso CR, Campanelli AP, Ferreira BR, Avila-Campos MJ, Cunha FQ, et al.. The dual role of p55 tumour necrosis factor-alpha receptor in Actinobacillus actinomycetemcomitans-induced experimental periodontitis: host protection and tissue destruction. Clin Exp Immunol. 2007; 147: 128–38.
- Liu R, Desta T, Raptis M, Darveau RP, Graves DT. P. gingivalis and E. coli lipopolysaccharides exhibit different systemic but similar local induction of inflammatory markers. J Periodontol. 2008; 79: 1241–7.
- Graves D, Liu R, Alikhani M, Al-Mashat H, Trackman P. Diabetes-enhanced inflammation and apoptosis – impact on periodontal pathology. J Dent Res. 2006; 85f: 15–21.
- Takeichi O, Haber J, Kawai T, Smith DJ, Moro I, Taubman MA. Cytokine profiles of T-lymphocytes from gingival tissues with pathological pocketing. J Dent Res. 2000; 79: 1548–55.
- Rogers JE, Li F, Coatney DD, Rossa C, Bronson P, Krieder JM, et al.. Actinobacillus actinomycetemcomitans lipopolysaccharide-mediated experimental bone loss model for aggressive periodontitis. J Periodontol. 2007; 78: 550–8.
- Page R, Schroeder H. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab Invest. 1976; 34: 235–49.
- Aggarwal R, Ghobrial IM, Roodman GD. Chemokines in multiple myeloma. Exp Hematol. 2006; 34: 1289–95.
- Tsai CC, Ho YP, Chen CC. Levels of interleukin-1 beta and interleukin-8 in gingival crevicular fluids in adult periodontitis. J Periodontol. 1995; 66: 852–9.
- Gemmell E, Carter CL, Seymour GJ. Chemokines in human periodontal disease tissues. Clin Exp Immunol. 2001; 125: 134–41.
- Alnaeeli M, Park J, Mahamed D, Penninger JM, Teng YT. Dendritic cells at the osteo-immune interface: implications for inflammation-induced bone loss. J Bone Miner Res. 2007; 22: 775–80.
- Bonecchi R, Galliera E, Borroni EM, Corsi MM, Locati M, Mantovani A. Chemokines and chemokine receptors: an overview. Front Biosci. 2009; 14: 540–51.
- Zhang X, Teng YT. Interleukin-10 inhibits gram-negative-microbe-specific human receptor activator of NF-kappaB ligand-positive CD4 + -Th1-cell-associated alveolar bone loss in vivo. Infect Immun. 2006; 74: 4927–31.
- Watanabe T, Kukita T, Kukita A, Wada N, Toh K, Nagata K, et al.. Direct stimulation of osteoclastogenesis by MIP-1alpha: evidence obtained from studies using RAW264 cell clone highly responsive to RANKL. J Endocrinol. 2004; 180: 193–201.
- Hosokawa Y, Hosokawa I, Ozaki K, Nakae H, Murakami K, Miyake Y, et al.. CXCL12 and CXCR4 expression by human gingival fibroblasts in periodontal disease. Clin Exp Immunol. 2005; 141: 467–74.
- Gamonal J, Acevedo A, Bascones A, Jorge O, Silva A. Characterization of cellular infiltrate, detection of chemokine receptor CCR5 and interleukin-8 and RANTES chemokines in adult periodontitis. J Periodontal Res. 2001; 36: 194–203.
- Garlet G, Martins W Jr, Ferreira B, Milanezi C, Silva J. Patterns of chemokine and chemokine receptors expression in different forms of human periodontal disease. J Periodontal Res. 2003; 38: 210–7.
- Hanazawa S, Kawata Y, Takeshita A, Kumada H, Okithu M, Tanaka S, et al.. Expression of monocyte chemoattractant protein 1 (MCP-1) in adult periodontal disease: increased monocyte chemotactic activity in crevicular fluids and induction of MCP-1 expression in gingival tissues. Infect Immun. 1993; 61: 5219–24.
- Yu X, Graves D. Fibroblasts, mononuclear phagocytes and endothelial cells express MCP-1 in inflamed human gingiva. J Periodontol. 1995; 66: 80–8.
- Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, et al.. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol. 2006; 169: 987–98.
- Teng YT, Mahamed D, Singh B. Gamma interferon positively modulates Actinobacillus actinomycetemcomitans-specific RANKL+ CD4+ Th-cell-mediated alveolar bone destruction in vivo. Infect Immun. 2005; 73: 3453–61.
- Agnello D, Lankford CS, Bream J, Morinobu A, Gadina M, O'Shea JJ, et al.. Cytokines and transcription factors that regulate T helper cell differentiation: new players and new insights. J Clin Immunol. 2003; 23: 147–61.
- Eastcott JW, Yamashita K, Taubman MA, Harada Y, Smith DJ. Adoptive transfer of cloned T helper cells ameliorates periodontal disease in nude rats. Oral Microbiol Immunol. 1994; 9: 284–9.
- Gemmell E, Seymour GJ. Immunoregulatory control of Th1/Th2 cytokine profiles in periodontal disease. Periodontol 2000. 2004; 35: 21–41.
- Kantarci A, Oyaizu K, Van Dyke TE. Neutrophil-mediated tissue injury in periodontal disease pathogenesis: findings from localized aggressive periodontitis. J Periodontol. 2003; 74: 66–75.
- Dennison DK, Van Dyke TE. The acute inflammatory response and the role of phagocytic cells in periodontal health and disease. Periodontol 2000. 1997; 14: 54–78.
- Garlet GP, Cardoso CR, Campanelli AP, Garlet TP, Avila-Campos MJ, Cunha FQ, et al.. The essential role of IFN-gamma in the control of lethal Aggregatibacter actinomycetemcomitans infection in mice. Microbes Infect. 2008; 10: 489–96.
- Lee W, Aitken S, Sodek J, McCulloch CA. Evidence of a direct relationship between neutrophil collagenase activity and periodontal tissue destruction in vivo: role of active enzyme in human periodontitis. J Periodontal Res. 1995; 30: 23–33.
- Strober W. The multifaceted influence of the mucosal microflora on mucosal dendritic cell responses. Immunity. 2009; 31: 377–88.
- Vernal R, Leon R, Herrera D, Garcia-Sanz JA, Silva A, Sanz M. Variability in the response of human dendritic cells stimulated with Porphyromonas gingivalis or Aggregatibacter actinomycetemcomitans. J Periodontal Res. 2008; 43: 689–97.
- Jotwani R, Cutler CW. Fimbriated Porphyromonas gingivalis is more efficient than fimbria-deficient P. gingivalis in entering human dendritic cells in vitro and induces an inflammatory Th1 effector response. Infect Immun. 2004; 72: 1725–32.
- Madianos PN, Bobetsis YA, Kinane DF. Generation of inflammatory stimuli: how bacteria set up inflammatory responses in the gingiva. J Clin Periodontol. 2005; 32: 57–71.
- Leibbrandt A, Penninger JM. RANK/RANKL: regulators of immune responses and bone physiology. Ann NY Acad Sci. 2008; 1143: 123–50.
- Alnaeeli M, Penninger JM, Teng YT. Immune interactions with CD4+ T cells promote the development of functional osteoclasts from murine CD11c+ dendritic cells. J Immunol. 2006; 177: 3314–26.
- Baker P, Evans R, Roopenian D. Oral infection with Porphyromonas gingivalis and induced alveolar bone loss in immunocompetent and severe combined immunodeficient mice. Arch Oral Biol. 1994; 39: 1035–40.
- Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009; 361: 888–98.
- Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, et al.. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood. 2007; 109: 3794–802.
- Cardoso CR, Garlet GP, Crippa GE, Rosa AL, Junior WM, Rossi MA, et al.. Evidence of the presence of T helper type 17 cells in chronic lesions of human periodontal disease. Oral Microbiol Immunol. 2009; 24: 1–6.
- Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008; 8: 523–32.
- Cardoso CR, Garlet GP, Moreira AP, Junior WM, Rossi MA, Silva JS. Characterization of CD4+ CD25+ natural regulatory T cells in the inflammatory infiltrate of human chronic periodontitis. J Leukoc Biol. 2008; 84: 311–8.
- Ernst CW, Lee JE, Nakanishi T, Karimbux NY, Rezende TM, Stashenko P, et al.. Diminished forkhead box P3/CD25 double-positive T regulatory cells are associated with the increased nuclear factor-kappaB ligand (RANKL + ) T cells in bone resorption lesion of periodontal disease. Clin Exp Immunol. 2007; 148: 271–80.
- Nakajima T, Ueki-Maruyama K, Oda T, Ohsawa Y, Ito H, Seymour GJ, et al.. Regulatory T-cells infiltrate periodontal disease tissues. J Dent Res. 2005; 84: 639–43.
- Parfitt A. The coupling of bone formation to bone resorption: a critical analysis of the concept and of its relevance to the pathogenesis of osteoporosis. Metab Bone Dis Relat Res. 1982; 4: 1–6.
- Behl Y, Siqueira M, Ortiz J, Li J, Desta T, Faibish D, et al.. Activation of the acquired immune response reduces coupled bone formation in response to a periodontal pathogen. J Immunol. 2008; 181: 8711–8.
- Liu R, Bal HS, Desta T, Krothapalli N, Alyassi M, Luan Q, et al.. Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. J Dent Res. 2006; 85: 510–4.