Abstract
Next-generation sequencing technologies have revolutionized the analysis of microbial communities in diverse environments, including the human body. This article reviews several aspects of one of these technologies, the pyrosequencing technique, including its principles, applications, and significant contribution to the study of the human microbiome, with especial emphasis on the oral microbiome. The results brought about by pyrosequencing studies have significantly contributed to refining and augmenting the knowledge of the community membership and structure in and on the human body in healthy and diseased conditions. Because most oral infectious diseases are currently regarded as biofilm-related polymicrobial infections, high-throughput sequencing technologies have the potential to disclose specific patterns related to health or disease. Further advances in technology hold the perspective to have important implications in terms of accurate diagnosis and more effective preventive and therapeutic measures for common oral diseases.
Culture has been traditionally used for identification of bacteria in the oral cavity, but over the last two decades culture-independent molecular biology methods have been increasingly used. These methods have substantially refined and redefined the knowledge of the microbial diversity in the different oral habitats and expanded the list of candidate pathogens associated with oral infectious diseases Citation1 Citation2 Citation3 Citation4 .
Specifically, the adoption of 16S rRNA gene amplification by polymerase chain reaction (PCR), followed by cloning and Sanger sequencing, allowed an even more comprehensive broad-range investigation of oral bacterial communities. Studies using this approach have revealed that the oral microbiota is more diverse than previously demonstrated by culture methods and that 40–60% of the bacterial taxa found in the mouth have not yet been cultivated and validly named Citation1 Citation2 Citation3 Citation4 Citation5 Citation6 . Cataloguing the bacterial taxa found in healthy and diseased conditions by broad-range methods has allowed further closed-ended molecular studies to target both cultivable and as-yet-uncultivated taxa in multiple samples, revealing several new candidate pathogens involved as for instance with caries Citation7, periodontal diseases, Citation8 and endodontic infections Citation9. In spite of these recent advances, technical difficulties and high cost associated with the cloning and Sanger sequencing approach make it difficult to analyze a large number of clones from a large number of samples. Consequently, the method is expected to reveal only the dominant members of the bacterial community.
Recently, new methods for high throughput DNA sequencing have been introduced. Because the Sanger method is considered as the ‘first-generation’ technology, these newer approaches have been regarded as next-generation DNA sequencing (NGS) technologies. Of the NGS techniques, the pyrosequencing method has been possibly the most widely used in medical microbiology. This approach has been recently introduced in oral microbiology research. Pyrosequencing technology provides a large number of sequence reads in a single run, resulting in very large sampling depth and allowing detection not only of the most dominant microbial community members but also of the low-abundance (rare) taxa Citation10 Citation11. Pyrosequencing of the 16S rRNA gene from diverse environments has demonstrated that the microbial diversity can be orders of magnitude higher than appreciated by previous technologies Citation10 Citation11.
This review article discusses several aspects of the pyrosequencing technique, including the principles, applications, and significant contribution to the study of the human microbiome, with special emphasis on the oral microbiome.
The Sanger sequencing approach
The Sanger approach has stood as the gold standard DNA sequencing technique over the last three decades. In brief, the automated Sanger approach comprises the following steps: DNA purification, DNA synthesis, and labeling using the chain termination method with dye-labeled dideoxynucleotides (ddNTPs), capillary electrophoresis, and fluorescence detection.
In the last years, the automated Sanger method has evolved to generate longer sequence reads of up to 800 bases. The outstanding contribution of Sanger sequencing to scientific advances in diverse areas is incalculable. In oral microbiology, numerous studies have used broad-range PCR, followed by cloning, and Sanger sequencing to unravel the microbiota associated with diverse oral sites in healthy Citation12 Citation13 and disease conditions, including caries Citation3 Citation14 Citation15 Citation16 , endodontic infections Citation4 Citation6 Citation17 Citation18 Citation19 Citation20 , periodontal diseases Citation1 Citation5 Citation21 Citation22 Citation23 , periimplantitis Citation24, and halitosis Citation2 Citation25. Collectively, these studies complemented the data from previous culture studies to reveal that more than 1,000 different bacterial species-level taxa belonging to 13 phyla colonized the human oral cavity Citation26. Six bacterial phyla (Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Spirochaetes, and Fusobacteria) contained the huge majority of oral representatives. Not all of these species-level taxa were present in the same mouth at any one time, and a particular individual usually harbored about 100–200 taxa Citation27. Whereas some species were common to different oral sites, the majority of species were selective for a particular site Citation12. Regardless of the site, as much as 50% or more of the detected taxa have been demonstrated to be as-yet-uncultivated bacteria Citation28.
NGS technologies
Next-generation DNA sequencing technologies that permit massive sequencing with a much higher throughput than the Sanger method have become recently available. The first commercially available NGS platform was introduced in 2005 and as of then NGS methods have revolutionized the field of genomic analysis Citation29. The five most currently used NGS technologies include the 454 pyrosequencing (Roche Applied Science, Basel, Switzerland), Illumina/Solexa Genome Analyzer (Illumina, San Diego, CA, USA) SOLiD (Applied Biosystems, Foster City, CA, USA), the HeliScope Single Molecule Sequencer (Helicos BioSciences, Cambridge, MA, USA), and the Single Molecule Real Time technology (SMRT, Pacific Biosciences, Menlo Park, CA, USA). These platforms share a common technologic characteristic of performing massively parallel sequencing, either of clonally PCR-amplified products (the first three platforms) or even of single DNA molecule (the last two), based on physical separation in a flow cell surface. This feature diverges from the classic Sanger technique that is based on electrophoretic separation of chain-termination products.
454 Pyrosequencing
The 454 pyrosequencing technology is probably the most used NGS platform to study the human microbiome and because of this the focus of this review relies on it. This technology is a sequencing-by-synthesis method that involves a combination of emulsion PCR and pyrosequencing. Pyrosequencing relies on light generation after nucleotides are incorporated in a growing chain of DNA. This approach requires no gels, fluorescent dyes, or ddNTPs.
In the pyrosequencing approach, DNA is isolated, fragmented, ligated to special adapters, and separated into single strands. Emulsion PCR is then carried out for clonal amplification. In this approach, an oil–water emulsion is formed in which the aqueous phase contains the PCR reagents and the DNA template to be sequenced. Capture beads containing one of the oligonucleotide primers attached to them are also included. This oligonucleotide is complementary to one of the adapter sequences used in library construction. The other PCR primer is placed in the solution. Beads are included in excess of DNA template molecules, but not of the expected number of droplets to be formed in the emulsion. After controlled and vigorous agitation of the oil–water system, emulsification takes place and millions of aqueous droplets are formed, within which PCR amplification takes place. Optimization of the concentration of DNA template, beads, and water droplets guarantees that only one template and one bead occur in each droplet. Then, million copies of a unique DNA template are generated on each bead in a clonal PCR amplification. The possibility of more than one bead or template occurring per droplet is greatly reduced by concentration, and in case it occurs, the mixed DNA sequence that forms is ultimately filtered out by the software Citation30.
Next, the emulsion is broken, the DNA is denatured, and beads carrying single-stranded DNA are transferred to the wells of a picotiter plate in such a way that it permits a single bead to occur in each of the several hundred thousand wells. Because each bead has a fixed location in the plate, each sequencing reaction can be monitored. Beads containing the enzymes used in the pyrosequencing reaction steps are then deposited into each well Citation31 Citation32.
The pyrosequencing reaction takes place using a mixture of the single-stranded DNA template, the sequencing primer, and the enzymes DNA polymerase, ATP sulfurylase, luciferase, and apyrase (). Two substrates are also included in the reaction – adenosine 5‘ phosphosulfate (APS) and luciferin. The first one of the four deoxynucleotides (dNTPs) is added to the sequencing reaction, and the DNA polymerase catalyzes its incorporation into the DNA strand, in case there is complementarity. During each incorporation event, a phosphodiester bond between the dNTPs is formed, releasing pyrophosphate (PPi) in a quantity equivalent to the amount of incorporated nucleotide. In sequence, the enzyme ATP sulfurylase converts PPi to ATP in the presence of APS. ATP is used in the conversion of luciferin to oxyluciferin mediated by the enzyme luciferase. This gives rise to light in intensity that is proportional to the amount of ATP used. Light is detected by a charge coupled device camera and detected as a peak in a pyrogram. The height of each peak is proportional to the number of nucleotides incorporated. The system is regenerated with the enzyme apyrase that degrades ATP and unincorporated dNTPs. Then, the next dNTP is added. Addition of dNTPs is performed one at a time. Generation of a signal indicates which nucleotide is the next one occurring in the sequence. As the process goes on, the complementary DNA strand grows and the nucleotide sequence is determined according to the signal peaks in the pyrogram.
Fig. 1. The 454 pyrosequencing approach.
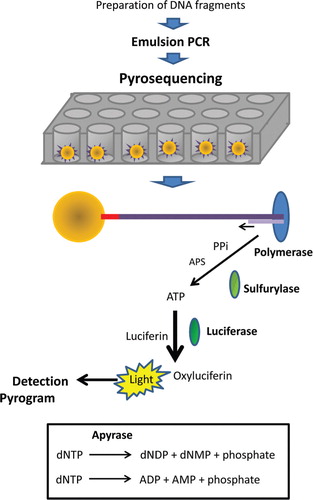
The 454 pyrosequencing technology has evolved over the years, with consequent increase in the length of sequence reads and output. The first-generation instrument (GS 20) yields 100 bp sequence reads and up to 60 Mb per individual run. The second-generation instrument (GS FLX) yields 250 bp sequence reads and up to 150 Mb per run. The third-generation (XLR and now GS FLX Titanium) yields 400 bp sequence reads and about 500 Mb per run. Recently, the GS FLX + was released, which offers read lengths up to 1 kb with a mode read length of 700 bp.
Other NGS platforms
The Applied Biosystems Sequencing by Oligonucleotide Ligation and Detection (SOLiD) system is based on sequencing-by-ligation technology. In the SOLiD approach, sequencing is obtained by measuring serial ligation of an oligonucleotide to the sequencing primer by a DNA ligase enzyme Citation33. Like in pyrosequencing, DNA fragments are ligated to oligonucleotide adapters, attached to beads, and then amplified by emulsion PCR to provide sufficient signal for the sequencing reactions. Beads are deposited on a flow cell surface, and the ligase-mediated sequencing begins by annealing of the sequencing primer to the adapter sequences on each amplified fragment. In a unique approach of sequencing using interrogation probes, each ligation step is accompanied by fluorescence detection, after which a regeneration step prepares the extended primer for the next round of ligation Citation29 Citation31. The newest sequencers using this technology can generate sequence reads that are 50 to 75 bp long.
The Illumina/Solexa sequencing platform is based on the sequencing-by-synthesis principle and is similar to the Sanger method in that it also relies on dye terminator nucleotides incorporated in the sequence by a DNA polymerase. Nonetheless, Illumina/Solexa terminators are reversible, permitting polymerization to proceed even after fluorophore detection. In this method, DNA fragments are immobilized on a flow cell surface and bridge PCR is used for amplification Citation34. The DNA sequencing is initiated with addition of the sequencing primer, DNA polymerase, and four reversible dye terminators. Fluorescence is recorded after incorporation by a four-channel fluorescent scanner Citation33. The newest sequencers using this technology can generate sequence reads that are about 100 bp long.
The HeliScope System by Helicos Biosciences is a single molecule sequencing platform that also utilizes the sequencing-by-synthesis principle. In the HeliScope platform, single DNA molecules are sequenced directly, obviating the need for a previous clonal PCR amplification step. The sample DNA is fragmented and polyadenylated at the 3’ end, with the final adenine being labeled with Cy3 Citation34. The poly-A template molecules are captured by hybridization to poly-T oligonucleotides immobilized on a flow cell surface. Because templates are fluorescently labeled, imaging can identify the array coordinates, where a sequencing read is expected. The label is then cleaved and sequencing proceeds by adding the DNA polymerase and each of four Cy5-labeled nucleotides to the flow cell Citation29. Fluorescent imaging detects incorporation into the individual strands. After chemical cleavage of the label, the cycle is repeated for the next nucleotide. Pacific Biosciences is another company that has developed another single molecule sequencing platform, the SMRT technology Citation35.
depicts many of the features of the most currently used NGS technologies. For a more detailed description of the NGS technologies and chemistries, the reader is referred to some comprehensive reviews Citation29 Citation31 Citation34 Citation36.
Table 1. Next-generation sequencing technologies
Advantages and limitations of pyrosequencing
One of the greatest advantages of the massive parallel pyrosequencing approach over the Sanger sequencing method is that hundreds of thousands of sequence reads can be obtained in a single run, generating sequence information data that are orders of magnitude larger Citation37. Moreover, the cost per base is much lower for pyrosequencing when compared with the Sanger method. This has also helped to widely expand the utilization of DNA-sequencing approaches. Another advantage of the pyrosequencing technique is that it also avoids the biases inherent to the cloning procedure.
In the pyrosequencing method, sequences from different samples can be identified in the same run using the barcoding multiplex approach, in which unique sequences are incorporated into the primers and barcoded amplicons are generatedCitation38. This multiplex approach increases efficiency and throughput, and reduces costs Citation10 Citation39.
The high throughput of the pyrosequencing method impacts on both sampling depth (number of sequences per sample) and breadth (number of samples or individuals analyzed). A greater sampling depth permits a better coverage of one individual sample, increasing the chances of detecting rare species and is of extreme importance for ecological studies. A greater sampling breadth permits to examine more samples or subjects, providing results that are more robust for comparisons. Depending on the goal of the study and the type of samples analyzed, the researcher should decide on which is better suited to his/her purposes. When comparing microbial communities from different sites, it has been argued that NGS platforms can be most wisely utilized to study more samples (breadth) rather than more sequences per sample (depth) Citation40. However, if the samples to be compared come from the same or closely related sites, deeper sequencing may be necessary to reveal minor differences in the community composition Citation41.
As with any other method, pyrosequencing has also its limitations. Most of them are common to other culture-independent molecular methods and were reviewed elsewhere Citation42. A limitation inherent of the pyrosequencing approach refers to detection of long homopolymers (repeated nucleotides), which can result in sequencing errors. Most of the pyrosequences contain none or only a few errors, but a few of them may contain sufficient errors to be interpreted as a rare operational taxonomic unit (OTU), inflating richness estimates. Several strategies have been proposed to deal with these noises Citation43.
The short length of reads generated by the NGS technologies may also represent a limitation when bacterial identification is intended. It is recognized that less phylogenetic information is available from short sequence reads than from near full length of the 16S rRNA gene. Indeed, there are still some bacterial species for which sequencing of the entire 1,500-bp 16S rRNA gene is required for reliable identification Citation44. Furthermore, sequencing of the entire 16S rRNA gene is often required for description of a new species. However, it has been commonly accepted that sequencing the initial 500-bp region of this gene may suffice for distinguishing a large number of human-associated bacteria Citation44.
Most of the currently used NGS platforms generate sequence reads that are too short in length. Because of the short sequence reads, bacterial identification using these methods has focused primarily on hypervariable regions of the 16S rRNA gene. It has been shown that reads spanning these particular regions of the 16S rRNA gene can still be highly informative and that despite the shorter read lengths, the pyrosequencing approach provides a description of the microbiome that is in good agreement with that provided by the cloning and Sanger sequencing approach in terms of higher taxonomic levels and relative abundance values Citation45 Citation46.
Although the first pyrosequencing technologies resulted in short reads, the most recent third generation of pyrosequencing (GS FLX Titanium) produces an average read length of more than 400 bp. This increases the phylogenetic information and permits identification of many taxa to the species level. Nevertheless, for overall community analyses, it has been shown that communities subjected to GS FLX Titanium sequencing after amplification with the V1–V3 and V3–V5 regions resembled communities analyzed with GS FLX sequencing after amplification with the V1–V2 region Citation47. Further advances in technology hold the perspective of generating longer sequence reads.
Pyrosequencing data analysis
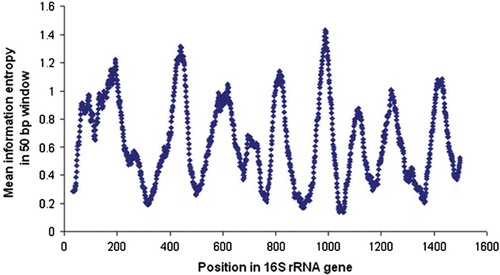
When used to explore the diversity of prokaryotic communities, pyrosequencing analysis relies on the use of segments of the small subunit ribosomal RNA gene or 16S rRNA gene that includes one or more of the hypervariable regions, and that provide the highest discrimination among microorganisms (). Hypervariable regions 1–6 (or V1–V6) have been commonly used.
Fig. 2. Variability within the 16S rRNA gene. From prealigned sequenced >1,200-bp downloaded from RDP, the variability, measured as Shannon information entropy, was calculated at each sequence position, using only positions without a gap in E. coli. The graph shows the Shannon entropy (Y-axis) averaged over 50-bp windows, centered at each position in the gene (X-axis). Shannon entropy at position x was calculated as – Σ p(xi) log2 p(xi), where p(xi) denotes the frequency of nucleotide i. Figure reproduced with permission from Andersson et al. Citation61.
As discussed before, the relatively short reads present some problems in taxonomic assignments of the sequences. Therefore, the technology is generally much more effective in the identification of higher level taxonomic assignments such as phyla, classes, orders, families, and genera, than species or strains. In fact, although there are some attempts to identify actual species using pyrosequencing, most bioinformatics experts agree that this area still needs additional efforts to resolve. A reasonable compromise is to define the number of different OTUs at the common 97, 95, or 90% levels.
Given that there are a large number of sequences generated with pyrosequencing, it is often useful to determine if the number of reads was sufficient to identify most microorganisms in a sample. This is commonly done by an analysis of rarefaction curves of the OTUs Citation48. A rarefaction curve plots the number of sequences analyzed on the Y-axis and the number of OTUs on the X-axis. If the resultant curve shows a plateau, then sufficient richness was present in the sample and was analyzed to define all the component microorganisms. Clearly, the higher the percentage of similarity among OTUs that are selected (i.e. closer to species designation), the more difficult it is to establish a plateau and the more sequences are needed for identification of all microbial taxa present. This is an additional reason for the inability of the technology to define species with precision.
As discussed above, there is a potential for exaggeration of richness and diversity estimates caused by low-quality pyrosequencing reads. Reads with multiple errors can form new OTUs if they are more distant from their true source and commonly occur as singletons or doubletons Citation49. Therefore, bioinformatics tools have been developed to remove low-quality reads, which is now essential to do before analysis is performed. Additionally, sequences that occur less than three times are usually excluded Citation50.
The assignment of taxonomy to sequences, annotations, and functional analysis can be done using a variety of online tools, mostly in the public domain, such as BLAST programs from GenBank at the National Center for Biotechnology Information (NCBI), the Ribosomal Database Project (RDP) Citation51, Greengenes Citation52, GAST Citation45, and the MG-RAST server Citation53. For studies of the oral microbiome, the HOMD Citation26 and CORE Citation54, which are curated 16S rRNA databases, are highly recommended.
Another metric that is used for sequence analysis is UniFrac Citation55 Citation56. Overall, bacterial community composition in different individuals, sites within an individual, or in a site at different time periods can be compared using the unweighted (qualitative) or weighted (quantitative) UniFrac distance metric. This is a phylogenetic tree-based metric ranging from 0 (distance between identical communities) to 1 (distance between totally different communities with no shared ancestry). UniFrac analysis allows the determination of whether the microbial populations overall are likely to be similar or distinct from each other Citation57.
General application of pyrosequencing technology in microbiology
The most important use of pyrosequencing in microbiology is in the efficient analysis of large amounts of sequences, whether from a single genome, single gene, or a community of microorganisms. Single genome analysis is typically done for sequencing of the genome of an organism of interest due to pathogenicity or prevalence. Currently, over 1,500 bacterial, 19 fungal, and 2,700 viral genomes have been fully sequenced Citation58. The speed of completion of genome sequences is increasing with rapid advances in sequencing technology. Genome analyses allow the understanding of virulence, metabolism, and structure of an organism as well as identify targets for antimicrobial agents. They also provide the basis for metagenomic analyses of complex microbiological communities (see below).
The study of microbial communities using pyrosequencing may be limited to the analysis of the 16S rRNA gene of the bacteria and archaea (or 18S rRNA gene for eukaryotes) in the community, or the analysis of random fragments of genomic DNA of the organisms present, commonly known as shotgun sequencing. The generation of massive genomic sequences reveals a large amount of data on the definition of normal microbiomes in different body areas, and the pathogenesis of disease. The massive amount of sequencing that has taken place in the last decade has served in understanding pathogenesis of diseases and developing new therapeutic modalities in multiple ways. Thus, genome sequencing may help in identifying microbial strains responsible for outbreaks of disease, tracking evolutionary history and disease spread, developing new diagnostic tests based on PCR technologies or DNA microarrays, or identify new strategies for drug targets or vaccines Citation58. This analysis provides a powerful tool for understanding gene expression, protein, and metabolite profiles of the microbiome of microorganisms in a community. Even if these microorganisms are cultivable in the laboratory, their genomic, proteomic, and metabolomic profiles are more likely to be different in the artificial environment than they are in the natural habitat Citation59.
Pyrosequencing was initially used to compare microbial communities in samples from distinct environmental ecosystems, in a rapid and relatively inexpensive manner Citation60. It was then used in research questions related to human health, with novel findings. For example, 16S rRNA gene analysis of human digestive tract microbiomes in three locations revealed that the throat and stomach microorganisms were closer to each other, whereas fecal communities clustered separately Citation61. A more extensive 16S rRNA gene pyrosequencing analysis of mothers and newborns born by cesarian or vaginal delivery showed that not only are microbial compositions remarkably different in different body habitats but also that newborn microbial acquisition depended on the mode of delivery and may play a role in affecting their future health Citation62. Other studies using pyrosequencing to study the human microbiome are reviewed in the next sections.
Pyrosequencing analysis of the human microbiome
The resident microbiota has been regarded as integral to the host's health. This is because the resident microbial communities have been shown to assist digestion, provide vitamins, confer resistance against exogenous pathogens, regulate metabolism, and stimulate the immune system Citation63. Therefore, deciphering the human microbiome in health and disease assumes great relevance toward a better understanding of these processes and has the potential to serve as a background for establishment of diagnostic, preventive, or therapeutic measures for numerous conditions.
In 2007, the US National Institutes of Health (NIH) launched an international collaborative project termed Human Microbiome Project (http://commonfund.nih.gov/hmp/) pointing to the need to characterize the diversity of the microbiome of the human body in different sites, including oral, nasal, skin, gastrointestinal, and urogenital regions, and to determine how changes in the human microbiome affect health and disease Citation64. Massively parallel pyrosequencing has been widely used in studies with these purposes. One of the main focuses of these studies has been to define a core microbiome across individuals Citation65. The core microbiome can be understood as those microbial taxa (but also genes) that are shared by all or the great majority of humans. Some authors have even used this term to mean the taxa present in most individuals.
Most studies have focused exclusively on a given site and will be reviewed in the next section. In a comprehensive study that compared the human microbiome from diverse sites, Costello et al. Citation55 used a multiplexed barcoded pyrosequencing approach to evaluate samples from 27 distinct body sites of healthy adults on four occasions. Sampled sites included the oral cavity, gut, and skin surfaces. The V2 region of the 16S rRNA gene was targeted to generate a dataset of about 1 million sequences (average of 1,315 sequences/sample). Overall, the detected taxa comprised representatives of 22 bacterial phyla. Of these, 92% of the sequences belonged to four phyla: Actinobacteria (37%), Firmicutes (34%), Proteobacteria (12%), and Bacteroidetes (9.5%). Each body site was found to harbor a unique microbiota and a group of dominant taxa that remained stable over time. Low-abundance taxa varied significantly. A high interindividual variability was found within sites.
Gut
Bacterial diversity in the gut
The gut contains the largest number of microorganisms associated with the human body and is regarded as one of the most densely populated microbial ecosystems on this planet Citation66 Citation67. In a study by Turnbaugh et al. Citation68, the pyrosequencing approach was used to evaluate the diversity of bacterial communities in fecal samples from a monozygotic twin pair. Findings were also compared to communities from the gut and other body sites of related and unrelated individuals. The V2 region of the 16S rRNA gene was targeted to generate approximately 1.2–1.5 million sequencing reads. The total diversity of species-level bacterial phylotypes in the reads obtained from each twin was about 800. Most species-level phylotypes were present at low abundance. A comparison across 27 body habitats demonstrated high levels of diversity. The combined data from the 27 body sites revealed an estimated 4,949 species-level phylotypes.
A study of rural children in Burkina Faso and in Italy analyzed approximately 440,000 sequences from the V5–V6 regions of the 16S rRNA gene and showed that the Bacteroidetes phylum was far more abundant in microbiomes of African children, with a unique abundance of bacteria from the genus Prevotella and Xylanibacter Citation69. Most Bacteroidetes representatives detected in the African children are known to have genes for cellulose and xylan hydrolysis, indicating that they are possibly involved in obtaining energy from the plant-rich diet. Moreover, Enterobacteriaceae taxa were significantly underrepresented in African children when compared to Italian children. These findings pointed to the fact that microbiomes vary geographically with their hosts, and diet may be one factor involved with such a variation.
Gut microbiome in obese individuals
Community profiling analysis of the human gut microbiome using cloning and Sanger sequencing disclosed a higher Firmicutes/Bacteroidetes ratio in obese individuals than in lean individuals Citation70. Furthermore, a barcode pyrosequencing approach has been successfully proposed and used to determine the Firmicutes/Bacteroidetes ratio in the gut microbiota of obese humans Citation71, with the potential of reducing costs, and increasing depth of coverage.
The microbial community structures of individuals with normal weight, morbidly obese, or postgastric-bypass surgery were investigated in a study using pyrosequencing Citation72. Analysis of approximately 180,000 sequences spanning the V6 region of the 16S rRNA gene demonstrated that members of the Firmicutes phylum were dominant in normal-weight and obese individuals, but significantly decreased in postgastric-bypass individuals. The latter had a proportional increase in Gammaproteobacteria. Prevotellaceae were highly enriched in the obese individuals. Three other families, namely Coriobacteriaceae (phylum Actinobacteria), Erysipelotrichaceae (phylum Firmicutes), and Alcaligenaceae (phylum Proteobacteria), were also more abundant in obese subjects. Verrucomicrobia were generally rare in obeses but abundant in the other individuals. The main conclusions of this study were that despite interindividual differences, obesity and gastric-bypass clearly affected the intestinal microbiome.
In an analysis of the influence of host genotype, environmental exposure, and host adiposity on the gut microbiome, Turnbaugh et al. Citation73 characterized the fecal microbial communities of adult female monozygotic and dizygotic twin pairs matched for leanness or obesity, and their mothers. The authors analyzed about 10,000 near full-length and 2 million partial (V2 and V6 regions) 16S rRNA gene sequences as well as more than 2 Gb from the microbiomes of 154 individuals. The gut microbial community of each individual varied in the specific bacterial taxa detected. One interesting finding from this study is that a core microbiome at the species level was not observed. Instead, a wide array of microbial genes was shared among individuals, comprising a core microbiome at a functional level. Obese individuals were associated with changes in the microbiota at the phylum level and a significant decrease in diversity.
Gut microbiome in diabetics
The composition of the gut microbiota in type-2 diabetic individuals was compared to non-diabetic individuals (controls) by a study using pyrosequencing targeting the V4 region of the 16S rRNA gene Citation74. Analysis of nearly 700,000 sequences showed that the proportion of members of the Firmicutes phylum was significantly higher in the controls when compared with diabetics. Moreover, the Bacteroidetes/Firmicutes ratio correlated positively and significantly with plasma glucose concentration. Class Betaproteobacteria was highly enriched in diabetics compared to non-diabetic subjects and positively correlated with plasma glucose. The authors concluded that type-2 diabetes may be associated with changes in the gut microbiome composition, especially at the phylum and class levels.
Gut microbiome in autistic subjects
Finegold et al. Citation75 used the pyrosequencing approach to examine the fecal microbiota of subjects with various severities of autism, siblings not showing autistic symptoms (sibling controls), and non-sibling control subjects. At the phylum level, members of Bacteroidetes were found at high levels in the severely autistic group, whereas members of Firmicutes were dominant in controls. At the species level, Desulfovibrio species and Bacteroides vulgatus occurred in significantly higher numbers in severely autistic children than in controls. Higher bacterial diversity was disclosed in the feces of autistic individuals when compared with controls. The authors emphasized that it remains uncertain whether autism leads to changes in the gut microbiota or the changed microbiota exerts any influence on the disease or its syndromes.
Gut microbiome as influenced by antibiotics
Some studies have used pyrosequencing to investigate the impact of systemic antibiotic therapy on the gut microbiome. Dethlefsen et al. Citation76 surveyed the distal gut bacterial communities of healthy individuals before and after treatment with ciprofloxacin. Their analyses involved more than 7,000 full-length rRNA sequences and over 900,000 pyrosequencing reads from V6 and V3 variable regions of the 16S rRNA gene. Ciprofloxacin treatment influenced the abundance of about one-third of the bacterial taxa in the gut, decreasing the community diversity. The magnitude of this effect varied among individuals. Despite disturbance, the community membership had largely returned to the pretreatment state within 4 weeks, which is suggestive of community resilience. However, several taxa failed to recover within 6 months.
In a further study, Dethlefsen and Relman Citation63 examined the distal gut microbiota of individuals over 10 months, which included two courses of therapy with ciprofloxacin. Analysis of more than 1.7 million pyrosequencing reads spanning the V1, V2, and V3 regions of the 16S rRNA gene revealed that the effect of ciprofloxacin on the gut microbiome was profound and rapid, with a significant decrease in diversity and a shift in community composition developing within 3–4 days of antibiotic initiation. One week following the end of each antibiotic course, the bacterial communities started to return to their initial state. However, return was frequently incomplete. In all individuals, the composition of the gut microbiota was stabilized by the end of the study, even though it was altered from its initial state.
The short- and long-term effects of clarithromycin and metronidazole treatment on the microbiota of the lower intestine and throat were investigated by Jakobsson et al. Citation77. While the microbial communities of untreated control subjects were relatively stable over time, a decreased diversity of the gut and throat microbiomes was apparent immediately after the antibiotic therapy. A dramatic decline in Actinobacteria in both feces and throat was observed immediately after treatment. Although the bacterial diversity subsequently returned to the pretreatment states, perturbation remained in some cases for up to 4 years following the antibiotic therapy. In conclusion, the authors reported that a common 1-week antibiotic treatment regimen with clarithromycin and metronidazole resulted in a long-term impact in both throat and gut microbiomes.
Skin
Considering the whole area available for colonization, the skin is certainly one of the largest microbial habitats associated with the human body. In the study by Costello et al. Citation55 using pyrosequencing to evaluate the microbiome in different body regions, very interesting results were reported for the skin. Clustering of the sequences obtained revealed different groups: a ‘head’ group, including the forehead, external nose, external ears, and hair, where Propionibacterineae prevailed (60–80%); and an ‘arm’ group, including forearms, palms, and index fingers, where Propionibacterineae were less abundant (20–40%). Separate clusters were evident for sites on the trunk and legs, which were dominated by Staphylococcus species (armpits and soles of the feet) or Corynebacterium species (navel and back of the knees). The highest levels of bacterial diversity were observed in the forearms, palms, index fingers, back of the knees, and soles of the feet.
The bacterial communities on the palm surfaces of both the dominant and non-dominant hands of undergraduate students were assessed using pyrosequencing Citation78. Analysis of more than 330,000 sequences spanning the V1–V2 regions of the 16S rRNA gene revealed a high bacterial diversity that was higher in women than in men. A typical palm hand surface harbored more than 150 unique species-level taxa. A total of 4,742 unique taxa were detected across all of the hands examined. Although representatives of 25 phyla were disclosed, more than 90% of the sequences belonged to three phyla (Actinobacteria, Firmicutes, and Proteobacteria). The most abundant genera on nearly all palm surfaces were Propionibacterium, Streptococcus, Staphylococcus, and Corynebacterium. Even though a core microbiome was observed on the palm surface, pronounced intra- and interindividual variations in the bacterial community composition were reported. Interestingly, hands from the same individual shared only 17% of their taxa, whereas hands from different individuals shared 13%.
Another study from the same group Citation57 revealed the stability and uniqueness of the skin microbiome among a group of individuals tested for their use of computer keyboards and mouse. The discrimination between individuals was stronger with the unweighted UniFrac metric than with the weighted metric, suggesting that differences in community membership (rather than community structure) discriminated best among individuals.
Vagina
The vagina is known to harbor a resident microbiota that is important for maintaining vaginal health and preventing infections such as bacterial vaginosis, yeast infections, urinary tract infections, and sexually transmitted diseases. In the first study to use pyrosequencing for the analysis of the vaginal microbiome, Sundquist et al. Citation79 obtained 100,000–200,000 sequence reads of about 100 bp length and confirmed that Lactobacillus is the dominant genus in the vagina. They also detected a significant presence of other genera, including Psychrobacter, Magnetobacterium, Prevotella, Bifidobacterium, and Veillonella.
Another study compared the diversity of the vaginal microbiome in women with bacterial vaginosis as compared to healthy women from China using several culture-independent methods, including pyrosequencing Citation80. Analysis of ca. 250,000 pyrosequencing reads of the V3 region of the 16S rRNA gene revealed that there was a significant interindividual variability in the bacterial communities. Overall, the most predominant bacterial phyla were Firmicutes, Bacteroidetes, Actinobacteria, and Fusobacteria. Of these, Firmicutes was the most dominant in healthy individuals, whereas Firmicutes, Bacteroidetes, Actinobacteria, and Fusobacteria constituted the complex vaginal microbiota in the vaginosis group. Eight bacterial genera, namely Gardnerella, Atopobium, Megasphaera, Eggerthella, Aerococcus, Leptotrichia/Sneathia, Prevotella, and Papillibacter, were strongly associated with vaginosis. The vaginal bacterial diversity in women with vaginosis was remarkably higher when compared with the healthy controls.
The vaginal microbiome of asymptomatic North American women from four ethnic groups (white, black, Hispanic, and Asian) was evaluated by pyrosequencing of V1–V2 regions of the 16S rRNA gene Citation81. Data consisting of about 900,000 sequences with an average length of 240 bp revealed that the vaginal bacterial communities clustered into five groups. Four were dominated by Lactobacillus iners, Lactobacillus crispatus, Lactobacillus gasseri, or Lactobacillus jensenii. The fifth one had fewer lactobacilli and higher proportions of anaerobic bacteria, including Prevotella, Dialister, Atopobium, Gardnerella, Megasphaera, Peptoniphilus, Sneathia, Eggerthella, and Finegoldia. Interestingly, all communities contained members known to produce lactic acid, including Lactobacillus, Streptococcus, Megasphaera, and Atopobium. This study found no single core microbiome for the human vagina.
Other sites and conditions
Nasopharynx and oropharynx
Charlson et al. Citation82 used pyrosequencing to analyze the microbiome from the nasopharynx and oropharynx of smoking and non-smoking healthy adults. Analysis of nearly 800,000 sequences encompassing the V1–V2 regions demonstrated that the microbiota from smokers was significantly more diverse than non-smokers and clustered separately. The distributions of several genera were systematically altered by smoking in both the nasopharynx and oropharynx, and there was an enrichment of obligate anaerobes in the oropharynx. The authors concluded that distinct regions of the human upper respiratory tract contain typical microbial communities, with altered patterns observed in smokers. These have the potential to influence the prevalence of respiratory tract complications in these individuals.
Chronic wounds
Biofilm infections have been considered as the primary impediment to the healing of chronic wounds. Dowd et al. Citation83 used pyrosequencing to evaluate the bacterial diversity of chronic diabetic foot ulcers and observed that the most prevalent bacterial genus associated with these wounds was Corynebacterium. Obligate anaerobes, including members from the genera Bacteroides, Peptoniphilus, Finegoldia, Anaerococcus, and Peptostreptococcus, also comprised a significant portion of the wound biofilm communities. Other major components of the bacterial communities included Streptococcus, Serratia, Staphylococcus, and Enterococcus.
In another study, the same group Citation84 surveyed samples from three types of chronic wound types: diabetic foot ulcers, venous leg ulcers, and pressure ulcers. Analysis of 129,000 pyrosequencing reads spanning the V4–V6 regions disclosed specific major bacterial communities that were evident in the biofilms of all chronic wound types, including Staphylococcus, Pseudomonas, Peptoniphilus, Enterobacter, Stenotrophomonas, Finegoldia, and Serratia. Specifically, the predominant bacterial taxa in venous leg ulcer biofilms were Enterobacter, Serratia, Stenotrophomonas, and Proteus species. In diabetic foot ulcers, the predominant genera were Staphylococcus, Peptoniphilus, Rhodopseudomonas, Enterococcus, Veillonella, Clostridium, and Finegoldia. Pressure ulcer biofilms were dominated by species from the genera Peptoniphilus, Serratia, Peptococcus, Streptococcus, and Finegoldia. Each of the wound types revealed marked differences in bacterial community structure. In another study evaluating samples from venous leg ulcers, Wolcott et al. Citation85 confirmed that polymicrobial communities commonly colonize the wounds. Predominant taxa included a previously uncharacterized bacteroidales, various anaerobes, Staphylococcus, Corynebacterium, and Serratia.
Smith et al. Citation86 used pyrosequencing to identify bacterial populations in decubitus ulcers. Analysis of nearly 220,000 sequences longer than 350 bp and including the V1–V3 regions demonstrated that the biofilm associated with decubitus ulcers was polymicrobial in nature and characterized by a great interindividual variability. A total of 228 genera and 487 predicted species were identified from 49 wounds. Several genera and species were found in high percentage in individual wounds. Species that predominated in several cases were Corynebacterium striatum, Streptococcus agalactiae, Pseudomonas aeruginosa, Anaerococcus species, Serratia marcescens, Staphylococcus aureus, and Enterococcus faecalis. Several anaerobic species were detected.
Other infectious diseases
Several studies have used the pyrosequencing technology to successfully detect pathogens associated with neonatal sepsis Citation87, brain abscess Citation88, and diarrhea Citation89. A study Citation90 described the incorporation of pyrosequencing for routine pathogen identification of atypical isolates in a children's hospital. Using pyrosequencing, isolates that lacked a definitive identification by culture and biochemical testing yielded genus- or species-level identifications in approximately 90% of cases. The authors concluded that coupled with culture, pyrosequencing-based bacterial identification can be a valuable tool for improved bacterial identification in a pediatric hospital setting.
Pyrosequencing analyses of the oral microbiome
A number of laboratories are currently working on analyzing the oral microbiome in health and disease. Traditional research questions such as the comparison between the oral microbiomes in healthy and diseased sites in the same oral cavity, in healthy and diseased patients, in diseased patients following treatment procedures, and in patients with normal health and those with systemic diseases such as diabetes mellitus and atherosclerosis are being explored using the pyrosequencing technology. The intent is to gain better understanding of the pathogenesis of oral microbial disease, of virulence determinants that lead to extensive disease, and lack of response to therapy or the interrelationship of oral and systemic diseases.
Using 454 GS FLX pyrosequencing, an analysis of 71 saliva and 98 supragingival samples revealed a total of 28,978 unique V6 tag sequences from 22 known phyla Citation91. However, 99.6% of these sequences belonged to one of the seven major phyla: Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria, Proteobacteria, Spirochetes, or candidate division TM7. There was much more diversity in plaque bacteria versus those in saliva, with 267 versus 185 different genera, and 10,000 versus 5,600 OTUs at the 3% difference, respectively. Despite this high number of OTUs, the bacterial richness estimated from rarefaction curves indicated that the richness is incomplete, that is, these numbers did not reveal all the bacterial taxa present. Nevertheless, relative abundance of the phylotypes indicates that the majority of OTUs present had low abundance and that the 1,000 most abundant OTUs account for 90–95% of all sequences. Further analysis by the same group of children of various ages revealed that in deciduous teeth dentition, Proteobacteria were prominent, but with a change to permanent dentition, Bacteroidetes, the Veillonellaceae family, Spirochaetes, and candidate division TM7 increased Citation92. Further extensive analyses of the oral microbiome in multiple sites from three individuals revealed remarkable consistency among them in that 47% of OTUs (at 3%) and 72% of higher taxa were common among them Citation49. Within an individual oral cavity, over 3,600 sequences comprising over 500 species-level phylotypes and 88–104 higher taxa (genus level or above) were found. The abundant phylotypes and relative abundance in the three oral microbiomes are shown in .
Fig. 3. Shared abundant phylotypes in three oral microbiomes and their relative abundance. Relative abundance of shared phylotypes within an individual microbiome. Only abundant phylotypes that contributed to at least 0.1% of the individual microbiome are shown. The most abundant phylotypes (≥0.5% of the microbiome) are grouped separately in the upper panel. Phylotypes were defined as OTUs clustering sequences at a 3% genetic difference. The highest taxon (in most cases, genus) at which the OTU was identified is shown together with the cluster identification number. Different colors indicate three different microbiomes, S1, S2, and S3, respectively. Figure reproduced with permission from Zaura et al. Citation49.

Pyrosequencing combined with UniFrac analysis has also shown that the oral microbiome is relatively stable within the same individual, in three samples collected over 1 month, allowing for subject-specific grouping Citation93. This finding could potentially lead to future uses of salivary samples in forensic identification of individuals, as was noted with skin samples before.
These findings are typical of the characteristics of complex microbial populations, as would be expected in the oral cavity. Examining microbial niches that are more isolated and localized may reveal less complexity and allow more effective comparisons among patients with different presentations of disease. Therefore, a pyrosequencing analysis was performed recently of microorganisms in seven primary and persistent endodontic infections. Endodontic infections would be expected to be less complex than microbiomes in saliva or plaque Citation94. In addition, a comparison was made in this study of the microbial diversity with pyrosequencing using the 16S rRNA tag sequences in the second generation 454 GS FLX platform and traditional Sanger sequencing of cloned 16S rRNA gene. Pyrosequencing yielded a 600-fold increase in depth of coverage compared to Sanger sequencing. Using the RDP-II Classifier, analysis at different taxonomic ranks showed that Sanger sequencing and pyrosequencing yielded 8 versus 13 phyla, 10 versus 22 classes, 11 versus 43 orders, 20 versus 97 families, and 25 versus 179 genera, respectively. These results showed that even in the relatively isolated root canal environment, significant microbial diversity exists, and that pyrosequencing allowed much better characterization of the endodontic microbiome than did traditional Sanger sequencing.
These findings for endodontic infections were confirmed and expanded by another study using pyrosequencing, this one investigating the bacterial diversity of the communities established specifically in the apical root canal Citation95. DNA extracts from cryopulverized apical root segments of 10 extracted teeth with primary apical periodontitis were subjected to multiplex tag-encoded GS FLX Titanium pyrosequencing. The sequences obtained were taxonomically classified into 187 bacterial species-level OTUs (at 97% similarity), 84 genera, and 10 phyla. The most represented, abundant, and prevalent phyla were Proteobacteria, Firmicutes, Bacteroidetes, Fusobacteria, and Actinobacteria. The majority of species-level phylotypes occurred at low levels. The mean number of species-level phylotypes per sample was 37, ranging from 13 to 80. A great interindividual variation in the composition of the apical microbiota was evident.
In another study of the endodontic microbiota using pyrosequencing, Santos et al. Citation96 compared samples from acute (symptomatic) or chronic (asymptomatic) endodontic infections. Overall, about 900 bacterial species-level OTUs (at 97% similarity) belonging to 67 genera and 13 phyla were detected. The most abundant phyla in acute infections were Firmicutes (52%), Fusobacteria (17%), and Bacteroidetes (13%), whereas in chronic infections the dominant phyla were Firmicutes (59%), Bacteroidetes (14%), and Actinobacteria (10%). Members of Fusobacteria were much more prevalent in acute than in chronic cases. The most abundant/prevalent genera in acute infections were Fusobacterium and Parvimonas. Twenty genera were exclusively detected in acute infections and 18 in chronic infections. Only 18% of the OTUs were shared by acute and chronic infections. Acute infections were significantly more diverse than chronic infections. Although a high interindividual variation in bacterial communities was observed, many samples tended to group together according to the type of infection (acute or chronic), which may suggest the existence of patterns related to disease severity.
The root canal environment is normally sterile, and therefore, the use of this highly sensitive and powerful technology to identify microbial resistance to endodontic treatment modalities is likely to be very important in future, particularly with the growing need to reach a high degree of root canal disinfection in preparation for regenerative endodontic procedures Citation97.
A comparison was recently reported between microbial analysis of oral samples using 454 pyrosequencing and targeted identification by DNA microarray with the human oral microbe identification microarray (HOMIM) Citation98. Oral lavage samples were collected from 20 individuals. Correlations and relative abundance were compared at phylum and genus levels, between 16S rRNA sequence read ratio and HOMIM hybridization intensity. The major phyla present were identified with high correlation by the two methods (r=0.70–0.86). 16S rRNA gene pyrosequencing identified 77 genera and HOMIM identified 49. There were 37 genera detected by both methods, and contained more than 98% of the classified bacteria. Concordance by the two methods and correlations were high for common genera (Streptococcus, Veillonella, Leptotrichia, Prevotella, and Haemophilus). This again shows that the data revealed by pyrosequencing add depth of coverage and better definition of diversity of the sample compared to other microbial analysis methods. It is salient to point out that although pyrosequencing revealed more taxa, HOMIM identifies taxa at the species level and specifically those for which there are probes.
In the realm of the relationship of oral and systemic disease, a recent study used 454 pyrosequencing to examine the relationship between oral (periodontal swab), gut, and atherosclerotic plaque samples in 15 atherosclerosis and 15 control patients Citation99. Unweighted UniFrac analysis revealed strong clustering by body site indicating distinct overall microbial communities in the three locations. Although the OTU analysis did not show differences between the oral samples in atherosclerotic and control individuals, OTUs from three genera were found to be in the same consensus lineage between oral and atheroma samples: Veillonella (7 OTUs from 11 patients), Streptococcus (17 OTUs from 6 patients), and Propionibacterium (4 OTUs from 8 patients). In addition, the abundance of Fusobacterium was positively correlated with levels of cholesterol and LDL cholesterol (p=0.028 and 0.005, respectively) Citation99.
As discussed before, the oral metagenome can be analyzed beyond the 16S rRNA gene data. This has been carried out using both the Roche 454 pyrosequencing and Illumina GA iiX platforms to pooled plaque samples of volunteers Citation100. This analysis required a significant amount of DNA and was done in a way to avoid sampling of human DNA. The authors concluded that using the 31 phylogenetic marker genes for community profiling may yield more accurate estimates than 16S rRNA-based assays because of the presence of a single copy for each marker per microbial genome.
Finally, the oral fungal microbiome (mycobiome) was investigated in 20 healthy individuals by pyrosequencing Citation101. The oral fungal microbiome was found to be diverse as revealed by the presence of 74 culturable and 11 non-culturable fungal genera. Fifteen genera (which included four known pathogenic fungi and non-culturable organisms) were present in 20% or more of the tested samples; Candida species were the most frequently obtained genera, isolated from 75% of all study participants, followed by Cladosporium (65%), Aureobasidium (50%), Saccharomycetales (50%), Aspergillus (35%), Fusarium (30%), and Cryptococcus (20%).
Future perspectives
The advent of massively parallel pyrosequencing has allowed the development of large population-based studies of the microbiome of diverse human body sites. The results brought about by these studies have significantly contributed to refining and augmenting the knowledge of the community membership and structure in and on the human body in healthy and diseased conditions. As most of the oral infectious diseases are now regarded as biofilm-related polymicrobial infections, these high-throughput technologies have the potential to revolutionize our knowledge of the oral microbiome in the sense that specific patterns related to health or disease may be identified. This can have important implications in terms of accurate diagnosis and more effective preventive and therapeutic measures. Additionally, these methods can be used for metagenomic analyses, permitting not only to identify the microbial species present but also to screen their genomes for functional roles and pathogenic potential in the communities.
Despite the limitations of pyrosequencing, namely the cost, need for large amounts of DNA, complexity of analysis, and relatively short reads, the increasing availability of massive computing power and the efficiency of data generation and analysis are likely to render this technology a major player in the field of microbiology in the near future. There is an obvious tendency for still greater advances and decreased costs. Therefore, one may envision the possibility of NGS technologies being established as the main diagnostic means for microbial infections in clinical laboratories.
Conflict of interest and funding
There is no conflict of interest in the present study for any of the authors.
References
- Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, et al.. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001; 183: 3770–83.
- Kazor CE, Mitchell PM, Lee AM, Stokes LN, Loesche WJ, Dewhirst FE, et al.. Diversity of bacterial populations on the tongue dorsa of patients with halitosis and healthy patients. J Clin Microbiol. 2003; 41: 558–63.
- Munson MA, Banerjee A, Watson TF, Wade WG. Molecular analysis of the microflora associated with dental caries. J Clin Microbiol. 2004; 42: 3023–9.
- Munson MA, Pitt-Ford T, Chong B, Weightman A, Wade WG. Molecular and cultural analysis of the microflora associated with endodontic infections. J Dent Res. 2002; 81: 761–6.
- Kumar PS, Griffen AL, Moeschberger ML, Leys EJ. Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis. J Clin Microbiol. 2005; 43: 3944–55.
- Sakamoto M, Rôças IN, Siqueira JFJr, Benno Y. Molecular analysis of bacteria in asymptomatic and symptomatic endodontic infections. Oral Microbiol Immunol. 2006; 21: 112–22.
- Corby PM, Lyons-Weiler J, Bretz WA, Hart TC, Aas JA, Boumenna T, et al.. Microbial risk indicators of early childhood caries. J Clin Microbiol. 2005; 43: 5753–9.
- Kumar PS, Griffen AL, Barton JA, Paster BJ, Moeschberger ML, Leys EJ. New bacterial species associated with chronic periodontitis. J Dent Res. 2003; 82: 338–44.
- Siqueira JFJr, Rôças IN. The microbiota of acute apical abscesses. J Dent Res. 2009; 88: 61–5.
- Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, et al.. Microbial diversity in the deep sea and the underexplored rare biosphere. Proc Natl Acad Sci U S A. 2006; 103: 12115–20.
- Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol. 2010; 12: 118–23.
- Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005; 43: 5721–32.
- Bik EM, Long CD, Armitage GC, Loomer P, Emerson J, Mongodin EF, et al.. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 2010; 4: 962–74.
- Kanasi E, Dewhirst FE, Chalmers NI, Kent RJr, Moore A, Hughes CV, et al.. Clonal analysis of the microbiota of severe early childhood caries. Caries Res. 2010; 44: 485–97.
- Aas JA, Griffen AL, Dardis SR, Lee AM, Olsen I, Dewhirst FE, et al.. Bacteria of dental caries in primary and permanent teeth in children and young adults. J Clin Microbiol. 2008; 46: 1407–17.
- Becker MR, Paster BJ, Leys EJ, Moeschberger ML, Kenyon SG, Galvin JL, et al.. Molecular analysis of bacterial species associated with childhood caries. J Clin Microbiol. 2002; 40: 1001–9.
- Sakamoto M, Siqueira JFJr, Rôças IN, Benno Y. Molecular analysis of the root canal microbiota associated with endodontic treatment failures. Oral Microbiol Immunol. 2008; 23: 275–81.
- Handal T, Caugant DA, Olsen I, Sunde PT. Bacterial diversity in persistent periapical lesions on root-filled teeth. J Oral Microbiol. 2009; 1: 1946.
- Ribeiro AC, Matarazzo F, Faveri M, Zezell DM, Mayer MP. Exploring bacterial diversity of endodontic microbiota by cloning and sequencing 16S rRNA. J Endod. 2011; 37: 922–6.
- Saito D, de Toledo Leonardo R, Rodrigues JLM, Tsai SM, Hofling JF, Gonçalves RB. Identification of bacteria in endodontic infections by sequence analysis of 16S rDNA clone libraries. J Med Microbiol. 2006; 55: 101–7.
- Faveri M, Mayer MP, Feres M, de Figueiredo LC, Dewhirst FE, Paster BJ. Microbiological diversity of generalized aggressive periodontitis by 16S rRNA clonal analysis. Oral Microbiol Immunol. 2008; 23: 112–8.
- Paster BJ, Russell MK, Alpagot T, Lee AM, Boches SK, Galvin JL, et al.. Bacterial diversity in necrotizing ulcerative periodontitis in HIV-positive subjects. Ann Periodontol. 2002; 7: 8–16.
- De Lillo A, Ashley FP, Palmer RM, Munson MA, Kyriacou L, Weightman AJ, et al.. Novel subgingival bacterial phylotypes detected using multiple universal polymerase chain reaction primer sets. Oral Microbiol Immunol. 2006; 21: 61–8.
- Koyanagi T, Sakamoto M, Takeuchi Y, Ohkuma M, Izumi Y. Analysis of microbiota associated with peri-implantitis using 16S rRNA gene clone library. J Oral Microbiol. 2010; 2: 5104.
- Haraszthy VI, Zambon JJ, Sreenivasan PK, Zambon MM, Gerber D, Rego R, et al.. Identification of oral bacterial species associated with halitosis. J Am Dent Assoc. 2007; 138: 1113–20.
- Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, et al.. The human oral microbiome. J Bacteriol. 2010; 192: 5002–17.
- Paster BJ, Olsen I, Aas JA, Dewhirst FE. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000. 2006; 42: 80–7.
- Olsen I, Preza D, Aas JA, Paster BJ. Cultivated and not-yet-cultivated bacteria in oral biofilms. Microb Ecol Health Dis. 2009; 21: 65–71.
- Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. Clin Chem. 2009; 55: 641–58.
- Higuchi R, Gyllensten U, Persing DH. Next-generation DNA sequencing and microbiology. Molecular microbiology. diagnostic, principles and practice. Persing DH, Tenover FC, Tang Y-W, Nolte FS, Hayden RT, van Belkum AASM Press. Washington DC, 2011; 301–12.
- Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008; 9: 387–402.
- Rothberg JM, Leamon JH. The development and impact of 454 sequencing. Nat Biotechnol. 2008; 26: 1117–24.
- Varshney RK, Nayak SN, May GD, Jackson SA. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009; 27: 522–30.
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008; 26: 1135–45.
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al.. Real-time DNA sequencing from single polymerase molecules. Science. 2009; 323: 133–8.
- Metzker ML. Sequencing technologies–the next generation. Nat Rev Genet. 2010; 11: 31–46.
- Engelbrektson A, Kunin V, Wrighton KC, Zvenigorodsky N, Chen F, Ochman H, et al.. Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J. 2010; 4: 642–7.
- Tringe SG, Hugenholtz P. A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol. 2008; 11: 442–6.
- Parameswaran P, Jalili R, Tao L, Shokralla S, Gharizadeh B, Ronaghi M, et al.. A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res. 2007; 35: e130.
- Kuczynski J, Costello EK, Nemergut DR, Zaneveld J, Lauber CL, Knights D, et al.. Direct sequencing of the human microbiome readily reveals community differences. Genome Biol. 2010; 11: 210.
- Lemos LN, Fulthorpe RR, Triplett EW, Roesch LF. Rethinking microbial diversity analysis in the high throughput sequencing era. J Microbiol Methods. 2011; 86: 42–51.
- Siqueira JFJr, Rôças IN. Molecular analysis of endodontic infections. Endodontic microbiology. Fouad AFWiley-Blackwell. Ames Iowa, 2009; 68–107.
- Reeder J, Knight R. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat Methods. 2010; 7: 668–9.
- Clarridge JE. 3rd. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev. 2004; 17: 840–62.
- Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, Sogin ML. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008; 4: e1000255.
- Nasidze I, Quinque D, Li J, Li M, Tang K, Stoneking M. Comparative analysis of human saliva microbiome diversity by barcoded pyrosequencing and cloning approaches. Anal Biochem. 2009; 391: 64–8.
- Wu GD, Lewis JD, Hoffmann C, Chen YY, Knight R, Bittinger K, et al.. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 2010; 10: 206.
- Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol. 2005; 71: 1501–6.
- Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the healthy core microbiome of oral microbial communities. BMC Microbiol. 2009; 9: 259.
- Lazarevic V, Whiteson K, Huse S, Hernandez D, Farinelli L, Osteras M, et al.. Metagenomic study of the oral microbiota by Illumina high-throughput sequencing. J Microbiol Methods. 2009; 79: 266–71.
- Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, et al.. The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 2005; 33: D294–D296.
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al.. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006; 72: 5069–72.
- Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, et al.. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008; 9: 386.
- Griffen AL, Beall CJ, Firestone ND, Gross EL, Difranco JM, Hardman JH, et al.. CORE: a phylogenetically-curated 16S rDNA database of the core oral microbiome. PLoS One. 2011; 6: e19051.
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009; 326: 1694–7.
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005; 71: 8228–35.
- Fierer N, Lauber CL, Zhou N, McDonald D, Costello EK, Knight R. Forensic identification using skin bacterial communities. Proc Natl Acad Sci U S A. 2010; 107: 6477–81.
- Relman DA. Microbial genomics and infectious diseases. N Engl J Med. 2011; 365: 347–57.
- Xu J. Metagenomics and Ecosystems Biology: conceptual frameworks, tools and methods. Metagenomics. Macro DCaister Academic Press. NorfolkUK, 2010; 1–15.
- Edwards RA, Rodriguez-Brito B, Wegley L, Haynes M, Breitbart M, Peterson DM, et al.. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics. 2006; 7: 57.
- Andersson AF, Lindberg M, Jakobsson H, Backhed F, Nyren P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE. 2008; 3: e2836.
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al.. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010; 107: 11971–5.
- Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011; 108(Suppl 1): 4554–61.
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007; 449: 804–10.
- Gonzalez A, Clemente JC, Shade A, Metcalf JL, Song S, Prithiviraj B, et al.. Our microbial selves: what ecology can teach us. EMBO Rep. 2011; 12: 775–84.
- Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA. 1998; 95: 6578–83.
- Egert M, de Graaf AA, Smidt H, de Vos WM, Venema K. Beyond diversity: functional microbiomics of the human colon. Trends Microbiol. 2006; 14: 86–91.
- Turnbaugh PJ, Quince C, Faith JJ, McHardy AC, Yatsunenko T, Niazi F, et al.. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci U S A. 2010; 107: 7503–8.
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al.. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. 2010; 107: 14691–6.
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006; 444: 1022–3.
- Armougom F, Raoult D. Use of pyrosequencing and DNA barcodes to monitor variations in Firmicutes and Bacteroidetes communities in the gut microbiota of obese humans. BMC Genomics. 2008; 9: 576.
- Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, et al.. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A. 2009; 106: 2365–70.
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al.. A core gut microbiome in obese and lean twins. Nature. 2009; 457: 480–4.
- Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al.. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010; 5: e9085.
- Finegold SM, Dowd SE, Gontcharova V, Liu C, Henley KE, Wolcott RD, et al.. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe. 2010; 16: 444–53.
- Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008; 6: e280.
- Jakobsson HE, Jernberg C, Andersson AF, Sjolund-Karlsson M, Jansson JK, Engstrand L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One. 2010; 5: e9836.
- Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A. 2008; 105: 17994–9.
- Sundquist A, Bigdeli S, Jalili R, Druzin ML, Waller S, Pullen KM, et al.. Bacterial flora-typing with targeted, chip-based pyrosequencing. BMC Microbiol. 2007; 7: 108.
- Ling Z, Kong J, Liu F, Zhu H, Chen X, Wang Y, et al.. Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genomics. 2010; 11: 488.
- Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, et al.. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011; 108(Suppl 1): 4680–7.
- Charlson ES, Chen J, Custers-Allen R, Bittinger K, Li H, Sinha R, et al.. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS One. 2010; 5: e15216.
- Dowd SE, Wolcott RD, Sun Y, McKeehan T, Smith E, Rhoads D. Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS One. 2008; 3: e3326.
- Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA, et al.. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol. 2008; 8: 43.
- Wolcott RD, Gontcharova V, Sun Y, Dowd SE. Evaluation of the bacterial diversity among and within individual venous leg ulcers using bacterial tag-encoded FLX and titanium amplicon pyrosequencing and metagenomic approaches. BMC Microbiol. 2009; 9: 226.
- Smith DM, Snow DE, Rees E, Zischkau AM, Hanson JD, Wolcott RD, et al.. Evaluation of the bacterial diversity of pressure ulcers using bTEFAP pyrosequencing. BMC Med Genomics. 2010; 3: 41.
- Jordan JA, Butchko AR, Durso MB. Use of pyrosequencing of 16S rRNA fragments to differentiate between bacteria responsible for neonatal sepsis. J Mol Diagn. 2005; 7: 105–10.
- Al Masalma M, Armougom F, Scheld WM, Dufour H, Roche PH, Drancourt M, et al.. The expansion of the microbiological spectrum of brain abscesses with use of multiple 16S ribosomal DNA sequencing. Clin Infect Dis. 2009; 48: 1169–78.
- Nakamura S, Maeda N, Miron IM, Yoh M, Izutsu K, Kataoka C, et al.. Metagenomic diagnosis of bacterial infections. Emerg Infect Dis. 2008; 14: 1784–6.
- Luna RA, Fasciano LR, Jones SC, Boyanton BLJr, Ton TT, Versalovic J. DNA pyrosequencing-based bacterial pathogen identification in a pediatric hospital setting. J Clin Microbiol. 2007; 45: 2985–92.
- Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, et al.. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res. 2008; 87: 1016–20.
- Crielaard W, Zaura E, Schuller AA, Huse SM, Montijn RC, Keijser BJ. Exploring the oral microbiota of children at various developmental stages of their dentition in the relation to their oral health. BMC Med Genomics. 2011; 4: 22.
- Lazarevic V, Whiteson K, Hernandez D, Francois P, Schrenzel J. Study of inter- and intra-individual variations in the salivary microbiota. BMC Genomics. 2010; 11: 523.
- Li L, Hsiao WW, Nandakumar R, Barbuto SM, Mongodin EF, Paster BJ, et al.. Analyzing endodontic infections by deep coverage pyrosequencing. J Dent Res. 2010; 89: 980–4.
- Siqueira JFJr, Alves FR, Rôças IN. Pyrosequencing analysis of the apical root canal microbiota. J Endod. 2011; 37: 1499–503.
- Santos AL, Siqueira JFJr, Rôças IN, Jesus EC, Rosado AS, Tiedje JM. Comparing the bacterial diversity of acute and chronic dental root canal infections. PLoS One. 2011; 6: e28088.
- Fouad AF. The microbial challenge to pulp regeneration. Adv Dent Res. 2011; 23: 285–9.
- Ahn J, Yang L, Paster BJ, Ganly I, Morris L, Pei Z, et al.. Oral Microbiome Profiles: 16S rRNA pyrosequencing and microarray assay comparison. PLoS ONE. 2011; 6: e22788.
- Koren O, Spor A, Felin J, Fak F, Stombaugh J, Tremaroli V, et al.. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A. 2011; 108(Suppl 1): 4592–8.
- Xie G, Chain PS, Lo CC, Liu KL, Gans J, Merritt J, et al.. Community and gene composition of a human dental plaque microbiota obtained by metagenomic sequencing. Mol Oral Microbiol. 2010; 25: 391–405.
- Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, Naqvi A, et al.. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010; 6: e1000713.