
Abstract
Thousands of bacterial phylotypes colonise the human body and the host response to this bacterial challenge greatly influences our state of health or disease. The concept of infectogenomics highlights the importance of host genetic factors in determining the composition of human microbial biofilms and the response to this microbial challenge. We hereby introduce the term ‘genetic dysbiosis’ to highlight the role of human genetic variants affecting microbial recognition and host response in creating an environment conducive to changes in the normal microbiota. Such changes can, in turn, predispose to, and influence, diseases such as: cancer, inflammatory bowel disease, rheumatoid arthritis, psoriasis, bacterial vaginosis and periodontitis. This review presents the state of the evidence on host genetic factors affecting dysbiosis and microbial misrecognition (i.e. an aberrant response to the normal microbiota) and highlights the need for further research in this area.
During their evolution, vertebrates and their colonising microbes have evolved mechanisms to live in symbiosis with each other. One of the major paradigm shifts in modern biomedicine is the realisation of how heavily vertebrates are colonised by bacteria (Citation1). It is now recognised that humans are supra-organisms (Citation2) with 90% of the cells in the human body being bacterial (Citation3), termed the normal bacterial microbiota. It is estimated that in the human gut, the microbiome outnumbers the human genome by 150-fold (Citation4). Culture-dependent methods have identified several hundred bacterial species colonising the skin and the mucosal surfaces of the oral cavity, airways, gut and genitourinary tract (Citation5). In these locations, bacteria flourish from the first moments after birth and adhere to each other forming aggregates termed biofilms. Culture-independent methods involving cloning and 16S rRNA gene sequencing (Citation6) or cloning-independent 16S rRNA gene analysis using massively parallel next generation DNA sequencing (Citation7) have increased the numbers of bacterial phylotypes recognised as colonising humans into the thousands (Citation8). However, only a small proportion of these colonising bacteria can be cultivated in the laboratory and have ever been studied (Citation5). The differences in bacterial colonisation between individuals in these studies lend strength to the idea that each individual human will, as a rule, have a subset of his or her own colonising bacteria in different body habitats, which may impact on the individuals state of health and disease (Citation9).
Microbial diseases
The traditional meaning of ‘microbial disease’ includes infections such as smallpox, tuberculosis or AIDS, caused by colonisation and infection by a specific pathogenic microbe, usually transmitted between individuals. Some microbes or even microbial strains may indeed be responsible for more than one disease state, an example being E. coli, associated with gastro-intestinal and urinary infections as well as meningitis. However, microbial-based disease extends well beyond this meaning as many major idiopathic diseases have a history of research involving the hypothesis that one or other infectious agent, bacterium, bacterial L-form, virus and so forth, is the cause of the disease. An example of this is rheumatoid arthritis, which was thought to be caused by a wide variety of microorganisms (Citation10). Notably, there is now increasing interest in the relationship between rheumatoid arthritis and periodontitis with a novel hypothesis that the peptidylarginine deiminase of the oral bacterium Porphyromonas gingivalis can generate citrullinated proteins which are highly immunogenic and may function as key auto-antigens in rheumatoid arthritis (Citation11, Citation12). This raises the question as to whether other enzymes of the microbiota can redirect the attention of immunity to produce autoimmune outcomes or, indeed, unknown consequences as a result of protein decoration.
Over the past 20 years there has been a suggestion that bacteria and viruses play a role in the process of atherogenesis (Citation13). Cancer is also a good example of a complex disease process in which viruses certainly participate (Citation14) and in which growing evidence exists for bacterial involvement in its pathogenesis (Citation15, Citation16). It has been estimated that 16% of cancers are initiated by specific microbial factors (Citation17), a clear example being Helicobacter pylori, which is responsible for gastric adenocarcinoma.
Microbial infections have been shown to lead to cancer either by direct action of viral oncogenes such as for human papillomaviruses (HPV) and cervical cancer (Citation18) or by indirect action such as promotion of chronic inflammation (Citation19), DNA damage (Citation20) and production of bioactive carcinogenic metabolites (Citation21). A key mechanism through which microbes can promote dysplastic changes is illustrated by the up-regulation of inflammatory pathways, such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway and pro-inflammatory cytokines by the bacterial cytolethal distending toxin of Helicobacter hepaticus in H. hepaticus-infected mouse livers (Citation22).
In addition to these diseases, recent research has identified a group of conditions probably resulting from dysbiosis, or alternatively referred to as originating from a misrecognition or aberrant response to the normal microbiota, including inflammatory bowel disease (IBD), psoriasis (PS), bacterial vaginosis (BV) and periodontal diseases (PD) (Citation23–(Citation26)). These four diseases seem to cluster together from an epidemiological, pathogenic, genetic and microbial standpoint, with similarities between them being recently reported (Citation27–(Citation36)).
Genetic dysbiosis
A common feature of the diseases described above is that they are not, like common infections, caused by individual bacterial species. Rather, they appear to be the result of a dysbiosis, which is a change in the normal microbiota, or of misrecognition of the normal microbiota within different body environments. A sensible hypothesis is that genetic defects in the recognition and response pathways that the host uses to identify microbial pathogens predispose to either altered microbial colonisation or to the misrecognition of normal microbiota that lead to these diseases. We could refer to ‘genetic dysbiosis’ to define this mechanism. This underpins the concept of infectogenomics (Citation37), for which two distinct pathways have been recognised:
Bacterial recognition
Mammals have a very wide variety of pattern-recognition receptors (PRRs), which recognise evolutionarily-conserved constituents of microbes called pathogen-associated molecular patterns (PAMPs) (Citation38). These include the toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), C-type-lectin like receptors (CLRs), scavenger receptors (SCs), innate DNA receptor proteins termed AIM2-like receptors (ALRs), members of the complement pathways and peptidoglycan-recognition proteins (PRPs). In addition to these proteins, which are mainly cell-bound, there are also a range of soluble PRRs including collectins, ficolins, pentraxins, galectins, sCD (cluster of differentiation) 14 and natural IgM (immunoglobulin M). Each of these families of proteins contains multiple members (Citation39–(Citation41)). In addition, these various receptor-based and soluble PRRs generally interact with various accessory proteins to allow for selective cell signalling. It is consequent upon binding of bacteria, or other microbes (or constituents of these microbes), to these PRRs that the target cell generates pro- and/or anti-inflammatory proteins such as cytokines. Mutations in the promoter regions and coding segments of the individual PRR genes may result in either altered expression of PRRs or differences in the ability to recognise the microbial constituents that they bind to respectively. In addition to the influence of PRR production and PRR kinetics of binding, changes in the interaction between the PRR and obligatory accessory proteins may be a factor in this ‘binding/recognition’ process. Initial evidence has recently been produced for the effect of microbial recognition genes on microbial presence in periodontal (Citation42) and vaginal biofilms (Citation43, Citation44). In these conditions, a proposed hypothesis is that genetic factors which may determine an aberrant epithelial barrier (through defects in PRRs and innate immune signalling pathways) may induce microbial shifts and an inflammatory cascade which can give rise to chronic diseases and even cancer (Citation27).
Bacterial proliferation
Several human surfaces in contact with the outside environment lend themselves to be colonised by a complex structure consisting of several layers of bacteria and other microorganisms, called a biofilm. As well as affecting bacterial recognition, common human genetic variants are likely to be responsible for creating a favourable environment for fostering the growth of specific pathogenic bacteria within biofilms. The hypothesis is that genetic variants predisposing to an excessive inflammatory response create a favourable environment for the selective growth of specific bacteria within the human biofilms, which, due to specific characteristics in their metabolism, grow well in more inflamed environments. Initial evidence in periodontitis suggests that cytokine gene polymorphisms may select and favour the growth of certain components of the sub-gingival biofilm (Citation42, Citation45). Similarly to periodontitis, other human diseases might be affected by the overgrowth of certain components of the biofilms upon stimulation by a more or less ‘inflamed’ environment (Citation37). It has been hypothesized that mutations in genes involved in immune regulatory mechanisms or pro-inflammatory pathways could lead to unrestrained inflammation in the intestine and that inflammation can influence the composition of the microbiota, skewing it in favour of pathological microorganisms (Citation46). However, doubts still exist on whether the inflammatory deregulation is the cause, or actually the consequence, of a microbial shift in such cases.
Genetic dysbiosis in IBD
IBD describes two conditions, namely Crohn's disease (CD), usually affecting the terminal ileum (but which can affect anywhere in the GI tract), and ulcerative colitis (UC), which is a chronic non-transmural inflammatory condition localised in the colon. Both are characterised by an intermittent course with symptoms including abdominal pain, diarrhoea and vomiting. IBDs result from the interaction between genetic susceptibility and environmental factors. Although there is no definition for ‘healthy’ microbiota, several conditions including IBD along with obesity and metabolic syndrome are associated with changes in intestinal microbial biofilms (Citation47). A metagenomic case-control study revealed significant differences in microbial compositions of a subset of IBD patients compared with healthy controls (Citation48). A process of dysbiosis, for example related to childhood exposure to antibiotics, is implicated in the pathogenesis of IBD (Citation49, Citation50). Whilst there is evidence that gut microbiota can modulate the expression of genes involved in different intestinal activities (Citation51) and that some resident bacteria have the potential to cause disease, no bacterium has so far been singled out as causative for IBD (Citation34). Bacterial type VI secretion systems (T6SSs) of H. hepaticus, a gram-negative bacterium of the intestinal microbiota, have been shown to direct an anti-inflammatory gene expression profile in intestinal epithelial cells, thus limiting colonization and intestinal inflammation. This is in contrast with the aforementioned effect of promotion of dysplastic changes by this bacterium in mice (Citation22) and could be explained with the concept that H. hepaticus acts as an intestinal pathobiont, in other words a symbiont able to promote pathology only when certain host genetic or environmental conditions are altered (Citation52). This example illustrates the importance of intestinal bacteria–host interactions and suggests that disruption of this host–microbial balance could contribute to human disorders such as IBD and colon cancer (Citation52). Genome-wide association studies (GWAS) have now made considerable progress in the identification of genetic variants predisposing to IBD. There is evidence for a role of genetic variants in genes coding for bacterial recognition receptors in disease pathology. The best-known and most researched gene associated with IBD is NOD2, which codes for an intracellular pattern-recognition receptor, able to recognise molecules containing bacterial muramyl dipeptide (Citation53). Genetic variants in this gene are associated with a higher risk of developing CD (Citation54), owing to the pleiotropic role of this receptor in bacterial recognition and in the inflammatory response. In general, genes linked to epithelial barrier function seem to be specifically associated with UC, while bacterial recognition genes such as NOD2 and autophagy genes seem to be associated with CD (Citation50). These findings are consistent with the involvement of the more superficial epithelial layers in UC and the deeper trans-mural inflammation of CD (supposedly caused by defects in cellular innate immunity and bacterial handling in the deeper layers of the lamina propria and beyond) (Citation50). Therefore, while it is now clear that both genetic susceptibility and dysbiosis predispose to IBD, further work is required to understand the effect of genetic variants in determining such dysbiosis (Citation55, Citation56).
Initial evidence for a role of genetic variants in conferring greater vulnerability to inflammation and dysbiosis in CD comes from an experimental ileal inflammation model in mice (Citation57). In this model, when a CD susceptible genotype (NOD2 mutations) was super-imposed on Toxoplasma gondii-induced ileitis, an increase in inflammation and dysbiosis was noticed (measured by pyrosequencing as a shift from mainly gram-positive to gram-negative bacteria, associated with invasive E. coli). The authors proposed that inflammation drives a progressive decrease in the microbial diversity leading to perturbations in the microenvironment such as increased availability of substrates for growth of gram-negative bacteria (e.g. iron and serum, dead or dying cells) and loss of niche and substrates for gram-positive flora (e.g. mucus, goblet cells). Genetic susceptibility could potentially impact the threshold for dysbiosis and the individual ability to resolve the self-perpetuating cycle of dysbiosis and inflammation generated by an acute trigger (Citation57). Admittedly, further research in humans is needed to substantiate the ‘dysbiosis’ evidence in IBD, mainly derived from animal models. However, even if IBD-associated dysbiosis was not an initiator of disease, dysbiosis may be important in perpetuating the disease (Citation4).
Genetic dysbiosis in PS
PS is a chronic condition characterised by the appearance of inflammatory, erythematous scaly lesions on the skin, linked to the development of arthritis. The most common form of PS is ‘plaque PS’, while other forms include guttate, pustular, erythrodermic and inverse (or flexural) PS (Citation58). PS research has a long history of associations with bacterial infection/colonisation. Over 50 years ago, it was noted that two thirds of patients with the variant guttate psoriasis (GP) had a preceding sore throat and serological evidence of a streptococcal infection (Citation59). This was then confirmed by the isolation of beta haemolytic streptococci [BHS] from the throats of patients with GP (Citation60). Furthermore, guttate flares in chronic plaque PS are associated with streptococcal pharyngitis (Citation61). Microbiological analysis of psoriatic skin report conflicting evidence on types of bacteria associated with the lesions: 16S rRNA gene analysis using swabbing of the skin to recover bacteria, has revealed differences in bacterial colonisation compared to healthy skin, with an increase in Firmicutes and a decrease in Actinobacteria (Citation25). Similarly, in a study on biopsy specimens (Citation62) using massive parallel sequencing of the 16S rRNA genes revealed differences in microbial colonization between psoriatic and healthy skin. Biopsies give a more reliable picture of the skin microbiome as it has recently been shown that bacteria are present throughout the skin (Citation63) and thus swabs are not sufficient to study the microbiome of the skin. Psoriatic lesions are highly infiltrated with immune cells including CD3+ T cells and CD11c+ dendritic cells (DCs) (Citation64, Citation65). Family history and increased prevalence in twins have directed PS research towards the search for a genetic susceptibility locus (Citation66) and the PSORS1 (PS susceptibility 1) locus was found on chromosome 6p21.3 (Citation67, Citation68). In a recent GWAS study (Citation69), 36 genetic loci were found to be associated with PS. The majority of these loci involved candidate genes governing the innate immune system including NFkB activation and interleukin (IL)-23 signalling. Genes affecting adaptive immune response and epidermal barrier function have also been suspected to have a role in the pathogenesis of PS (Citation70). Thus it is possible that PS is due to dysbiosis of bacteria colonising the skin (Citation36) and that genetic variants predispose to this dysbiosis. However, further research in this field is needed to substantiate this hypothesis.
Genetic dysbiosis in periodontitis
PD are inflammatory diseases of the gingivae (gums) associated with destruction of the supporting tissues of the teeth (periodontal ligament and alveolar bone) and early loss of the dentition (Citation71). Periodontitis is due to an aberrant response to members of the sub-gingival microbiota (Citation72) and, together with its non-destructive partner condition, gingivitis, is one of the most prevalent chronic inflammatory conditions of humanity. Mild to moderate periodontitis affects 13–57% of the population according to different studies, with severe forms affecting up to 25% of individuals (Citation73). A recent survey in an US adult population of 3,742 individuals revealed a prevalence of 47% for periodontitis (Citation74). Periodontitis has a multifactorial aetiology, where the combination of common genetic variants alters the response to the sub-gingival microbiota, predisposing to disease onset and progression (Citation72). Periodontopathogenic bacteria include gram-negative bacteria such as Aggregatibacter actinomycetemcomitans, Porphyromonas gingivalis and Tannerella forsythia (Citation75, Citation76). These bacteria are thought be able to enter the bloodstream through infected periodontia (Citation77), have been found in atheromatous plaques (Citation78), amniotic fluid of pregnant women (Citation79) and are thought to initiate rheumatoid arthritis in susceptible individuals (Citation80). Similarly, initial evidence suggests the existence of an inflammatory reaction triggered by periodontopathogenic bacteria and inducing systemic inflammatory products which stimulate the production of beta amyloid and tau protein in brain tissue leading to Alzheimer's neuropathology (Citation81, Citation82). Our group and others have shown in recent years that specific genetic variants affecting the inflammatory response (e.g. IL-1 and IL-6 genes) are associated with detection of periodontopathogenic bacteria (such as A. actinomycetemcomitans and P. gingivalis) below the gingival margin (Citation42, Citation83–Citation85)). A recent study using a genome-wide approach in 1,020 subjects (Citation45) identified a moderate association between locus 1q42 and periodontopathogenic bacteria (belonging to the so-called ‘red complex’) in sub-gingival pockets and confirmed a moderate association our group previously reported between IL6 genetic variants and presence of pathogenic bacteria sub-gingivally (Citation42). This adds to the evidence suggesting that periodontal ‘dysbiosis’ or a shift towards a more pathogenic microbiota (including pathogenic bacteria which grow well in inflamed environments, such as A. actinomycetemcomitans) may be due to specific genetic variants in the host (Citation86).
Genetic dysbiosis in BV
BV is the most common cause of abnormal vaginal discharge in women of child-bearing age. The prevalence in the population is around 10–20% (Citation87). The aetiology of BV is complex. Although it is not classified as a sexually transmitted infection (STI), unprotected vaginal sex, change of sex partner, STIs, intrauterine devices and Afro-Caribbean ethnicity are considered risk factors (Citation24). BV is more common around menstruation and has been observed in women with pelvic inflammatory disease (PID). Recurrent BV is common and could lead to psychological distress and to complications such as miscarriage and preterm birth, endometritis and PID in women after an elective termination of pregnancy and post-hysterectomy infections (Citation88, Citation89). The microbial composition of the vaginal biofilm is thought to be modulated by oestrogen levels, environmental and behavioural factors as well as by host genetic predispositions (Citation88, Citation90) (Citation91). In most human populations, the healthy vagina is mainly populated by Lactobacillus species, which have a symbiotic relationship with their female host. Under the influence of female reproductive hormones, the vaginal epithelia lead to the production of glycogen that is metabolised by the lactobacilli leading to lactic acid production, which is largely responsible for the normal acidic vaginal pH of <4.5. Both increasing acidity and the production of antimicrobials (such as bacteriocins and hydrogen peroxide) by different lactobacilli species are associated with greater inhibition of growth of potential pathogens, such as Gardnerella vaginalis, Prevotella bivia, Mobiluncus spp. and Group B Streptococcus (Citation92). Lactobacilli may also competitively exclude pathogens by sheer load or through formation of hostile biofilms. In BV there is a decrease in lactobacilli load and in the vaginal acidity, associated with an overgrowth of vaginal anaerobes. Bacterial species associated with BV include G. vaginalis, Mycoplasma hominis, Mobiluncus species, Prevotella species, Leptotrichia sanguinegens/amnionii and Atopobium vaginae (Citation93) . Clinically, BV is diagnosed by the presence of a thin white, homogeneous discharge coating the vaginal walls (asymptomatic in about 50% of the cases), with a lack of inflammation. The vaginal pH in BV is above 4.5. A clinical diagnosis of BV can be confirmed by performing microscopy on a gram-stained vaginal smear. The smear is interpreted in dedicated sexual health clinics by specific scoring systems such as Nugent's, based on the morphological appearance of bacteria that are present, or Hay/Ison criteria, based on abundance of lactobacilli, as recommended by the Bacterial Special Interest Group of the British Association for Sexual Health and HIV. Genetic factors affecting microbial recognition have been implicated in the predisposition to the carriage of organisms associated with BV and the host immunity may be key to why some women who are affected by BV have recurrent or relapsing infections and complications whilst others do not (Citation44). TLRs are expressed by the cervico-vaginal epithelia and help initiate the innate response. Genetic polymorphisms in genes encoding bacterial receptors such as TLR and inflammatory mediators such as IL-1 receptor antagonist (IL-1RA) and tumour necrosis factor (TNF)-α may account for differences in BV and complications between women (Citation91). Therefore, BV could be considered a dysbiosis, where genetic factors affecting bacterial recognition and host response may predispose to a shift in the vaginal microbial biofilm, predisposing to disease initiation. However, as for PS, further evidence to support this hypothesis is needed.
Similarities between chronic diseases linked with genetic dysbiosis
The diseases hypothesized as ‘genetic dysbioses’ in this review (IBD, PS, periodontitis and BV) share a substantial pathogenic similarity (see ), being immune-mediated diseases due to an imbalance between the integrity of barrier organs and their colonising microorganisms. The common underlying mechanism involves the presence of triggering microbial events acting on a genetically susceptible individual, in the presence of predisposing environmental factors. The epithelia of barrier organs such as skin, oral cavity, gastrointestinal tract and oro-genital mucosa act as a mechanical barrier and as a first line of host defence against invading pathogens, recognising microorganisms and producing cytokines and antimicrobial peptides. These functions give these epithelia the ability to regulate the normal host–microbial homeostasis, with a key role in the dysbiosis process (Citation94) and allow them to be physiologically colonised by a number of opportunistic bacteria, which have the potential to cause disease. Epidemiological surveys have recently suggested a certain overlap between the conditions that are the object of this review. The presence of UC has been associated with periodontitis in a case-control study in Brazil (Citation29). A large epidemiological study in Taiwan has shown an association between the presence of periodontitis and the incidence of PS (Citation95). Furthermore, PS has been shown to increase in prevalence in subjects affected by CD (Citation96). Gingivitis has been shown to have a higher prevalence in women affected by BV compared to women with healthy vaginas (Citation97) and similar health behaviours and socioeconomic risk factors have been detected between the two conditions (Citation30). For PD, BV, IBD and PS, genetic variants with a modifying effect on disease predisposition involve microbial recognition and inflammatory pathways, as mentioned above. For example, TLR-4 genes have been implicated in BV, PD and IBD pathogenesis (Citation98–(Citation100)). Cytokine-producing genes such as IL-1β and IL-1RA have been involved in the pathogenesis of BV, PD, IBD and PS (Citation43, Citation101–Citation104). Levels of pro-inflammatory cytokines such as TNF-α are increased in CD, PD and PS (Citation27, Citation105) (Citation106). Among other functional genes important in the barrier organ–microbe homeostasis, NLR (nucleotide-binding domain and leucine-rich repeat containing) genes, encoding mediators of innate immunity and providing the first line of defence against pathogens, may provide a link between these diseases (Citation94). Despite some shared similarities from an aetio-pathogenic standpoint, differences exist in the development of immune response in these conditions, for example mainly T-lymphocyte mediated for IBD, and more B-cell dependent in periodontitis (Citation34).
Fig. 1 Schematic representation of potential pathogenic pathways of genetic dysbioses.
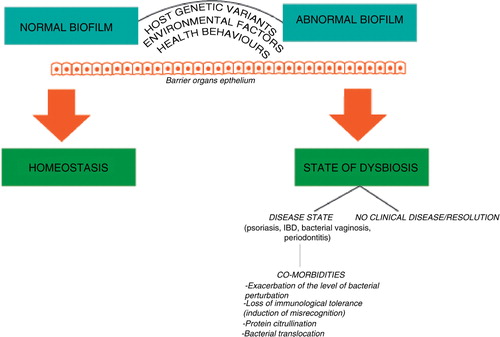
A very intriguing aspect of these genetic dysbioses is the potential co-morbidities. For example, a prevalently anaerobic bacterial environment is associated with both PD and BV. In a recent study in women with BV and gingivitis, the vaginal samples had higher counts of bacteria commonly associated with periodontal disease such as A. actinomycetemcomitans, P. gingivalis, T. forsythia and Prevotella intermedia, in comparison with those with BV but not gingivitis, suggesting that oral diseases may exacerbate the level of bacterial perturbations associated with BV (Citation97). In periodontitis, the ulceration of the pocket epithelium results in migration of periodontal microbes into the bloodstream, even during chewing and tooth brushing (Citation77, Citation107) (Citation108). The survival of these bacteria in the systemic circulation and their final destiny are still unclear. Interestingly, initial evidence showed that out of 50 human specimens obtained during carotid endarterectomy analysed by PCR, 44% of atheromas were found positive for at least one periodontal pathogen (Citation78). Oral pathogens have also been linked with a role in prematurity via haematogenous route (Citation109, Citation110) and with pregnancy complications (Citation111). It is hypothesized that oral pathogens can spread to the vaginal cavity within the female host via the gastrointestinal tract or by transmission between individuals via oro-genital contact (Citation110). A rate of physiological bacterial translocation is thought to occur in the human gut, by the intra-epithelial route and then via the mesenteric lymph nodes (or directly to the portal circulation in case of damage to the epithelium) (Citation112, Citation113). Translocation of indigenous gut microbiota or microbial elements has also been shown in an animal model of sepsis (Citation114) and, in HIV infection, is a major mechanism contributing to generalised immune activation and subsequent disease progression. This is in line with the theory that one of these genetic dysbioses might alter the biodiversity of the microbiota provoking a loss of immunological tolerance to commensal bacteria, hence predisposing to other diseases in the body (Citation94).
Summary and conclusions
This review introduces the concept of genetic dysbiosis, to indicate a host genetically driven deviation from the normal composition of human tissue-distinct microbial biofilms. Inflammatory diseases such as IBD, PS, periodontitis and BV are likely to be initiated through this mechanism. The strongest evidence comes from studies on CD and on periodontitis, showing alterations in the normal microbiota dependent on specific host genetic variants (Citation45, Citation57). An additional pathogenic mechanism for these conditions is represented by a host genome-driven misrecognition of the normal microbiota, which might overlap or in some cases represent a different disease-initiating pathway. Bacterial translocation, protein citrullination and loss of immunological tolerance are mechanisms through which one of these dysbioses could influence and potentially trigger another. Therefore, although the different epithelial and mucosal surfaces of the body have their own distinct microbiotas with little overlap between species in health (Citation9), this may change in the presence of a genetically-driven dysbiosis. Large studies investigating host genetic variants and microbiome analyses in different body locations may substantiate the hypotheses reported here, elucidate the pathogenic process and potentially provide new weapons (such as for example diet or probiotics) in the treating physicians’ prevention and treatment arsenal.
Conflict of interest and funding
The authors declare that they have no conflicts of interest in relation to this paper.
Acknowledgements
This review was undertaken at UCL, which received a proportion of funding from the Department of Health's National Institute of Health Research (NIHR) Biomedical Research Centres funding scheme. The authors thank Prof. Lionel Fry and Prof. Alastair Forbes for their expert review of the psoriasis and inflammatory bowel disease sections, respectively.
References
- McFall-Ngai M, Henderson B, Ruby N. The influence of cooperative bacteria on animal host biology. 2005; New York: Cambridge University Press.
- Ruby E, Henderson B, McFall-Ngai M. Microbiology – we get by with a little help from our (little) friends. Science. 2004; 303: 1305–7.
- Wilson M. Bacterial inhabitants of humans: their ecology and role in health and disease. 2005; New York: Cambridge University Press.
- Wu GD, Lewis JD. Analysis of the human gut microbiome and association with disease. Clin Gastroenterol Hepatol. 2013; 7: 774–7.
- Wilson M. Bacteriology of humans: an ecological perspective. 2008; Oxford: Blackwell.
- Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev. 2004; 68: 669–85.
- Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J. Metagenomic pyrosequencing and microbial identification. Clin Chem. 2009; 55: 856–66.
- Zoetendal EG, Rajilic-Stojanovic M, de Vos WM. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut. 2008; 57: 1605–15.
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012; 486: 207–14.
- Benedek TG. The history of bacteriologic concepts of rheumatic fever and rheumatoid arthritis. Semin Arthritis Rheum. 2006; 36: 109–23.
- Detert J, Pischon N, Burmester GR, Buttgereit F. The association between rheumatoid arthritis and periodontal disease. Arthritis Res Ther. 2010; 12: 218.
- De Pablo P, Dietrich T, Chapple IL, Milward M, Chowdhury M, Charles PJ, etal. The autoantibody repertoire in periodontitis: a role in the induction of autoimmunity to citrullinated proteins in rheumatoid arthritis?. Ann Rheum Dis. 2014; 73(3): 580–6.
- Makris GC, Makris MC, Wilmot VV, Geroulakos G, Falagas ME. The role of infection in carotid plaque pathogenesis and stability: the clinical evidence. Curr Vasc Pharmacol. 2010; 8: 861–72.
- Taylor GS, Blackbourn DJ. Infectious agents in human cancers: lessons in immunity and immunomodulation from gammaherpesviruses EBV and KSHV. Cancer Lett. 2011; 305: 263–78.
- Piazuelo MB, Epplein M, Correa P. Gastric cancer: an infectious disease. Infect Dis Clin North Am. 2010; 24: 853–69.
- Sun J. Enteric bacteria and cancer stem cells. Cancers (Basel). 2010; 3: 285–97.
- de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, etal. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012; 13: F12–23.
- Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007; 370: 890–907.
- Balkwill F, Mantovani A. Cancer and inflammation: implications for pharmacology and therapeutics. Clin Pharmacol Therapeut. 2010; 87: 401–6.
- Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrede JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A. 2010; 107: 11537–42.
- Arthur JC, Jobin C. The struggle within: microbial influences on colorectal cancer. Inflamm Bowel Dis. 2011; 17: 396–409.
- Ge ZM, Rogers AB, Feng Y, Lee A, Xu S, Taylor NS, etal. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell Microbiol. 2007; 9: 2070–80.
- Socransky SS, Haffajee AD, Smith C, Duff GW. Microbiological parameters associated with IL-1 gene polymorphisms in periodontitis patients. J Clin Periodontol. 2000; 27: 810–18.
- Hay P. How important are the newly described bacteria in bacterial vaginosis?. Sex Transm Infect. 2009; 85: 240–1.
- Gao Z, Tseng CH, Strober BE, Pei ZH, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS One. 2008; 3: e2719.
- Man SM, Kaakoush NO, Mitchell HM. The role of bacteria and pattern-recognition receptors in Crohn's disease. Nat Rev Gastroenterol Hepatol. 2011; 8: 152–68.
- Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006; 124: 823–35.
- Floch MH, Madsen KK, Jenkins DJA, Guandalini S, Katz JA, Onderdonk A, etal. Recommendations for probiotic use. J Clin Gastroenterol. 2006; 40: 275–8.
- Brito F, de Barros FC, Zaltman C, Carvalho AT, Carneiro AJ, Fischer RG, etal. Prevalence of periodontitis and DMFT index in patients with Crohn's disease and ulcerative colitis. J Clin Periodontol. 2008; 35: 555–60.
- Srinivasan U, Misra D, Marazita ML, Foxman B. Vaginal and oral microbes, host genotype and preterm birth. Med Hypotheses. 2009; 73: 963–75.
- Preus HR, Khanifam P, Kolltveit K, Mork C, Gjermo P. Periodontitis in psoriasis patients. A blinded, case-controlled study. Acta Odontol Scand. 2010; 68: 165–70.
- Sanu O, Lamont RF. Periodontal disease and bacterial vaginosis as genetic and environmental markers for the risk of spontaneous preterm labor and preterm birth. J Matern Fetal Neonatal Med. 2011; 24: 1476–85.
- Figueredo CM, Brito F, Barros FC, Menegat JS, Pedreira RR, Fischer RG, etal. Expression of cytokines in the gingival crevicular fluid and serum from patients with inflammatory bowel disease and untreated chronic periodontitis. J Periodontal Res. 2011; 46: 141–6.
- Indriolo A, Greco S, Ravelli P, Fagiuoli S. What can we learn about biofilm/host interactions from the study of inflammatory bowel disease. J Clin Periodontol. 2011; 38: 36–43.
- Binus AM, Han J, Qamar AA, Mody EA, Holt EW, Qureshi AA. Associated comorbidities in psoriasis and inflammatory bowel disease. J Eur Acad Dermatol Venereol. 2012; 26: 644–50.
- Fry L, Baker BS, Powles AV, Fahlen A, Engstrand L. Is chronic plaque psoriasis triggered by microbiota in the skin?. Br J Dermatol. 2013; 169: 47–52.
- Kellam P, Weiss RA. Infectogenomics: insights from the host genome into infectious diseases. Cell. 2006; 124: 695–7.
- Janeway CA. Approaching the asymptote – evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989; 54: 1–13.
- Dagenais M, Dupaul-Chicoine J, Saleh M. Function of NOD-like receptors in immunity and disease. Curr Opin Investig Drugs. 2010; 11: 1246–55.
- Hansen JD, Vojtech LN, Laing KJ. Sensing disease and danger: a survey of vertebrate PRRs and their origins. Dev Comp Immunol. 2011; 35: 886–97.
- Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011; 30: 16–34.
- Nibali L, Ready DR, Parkar M, Brett PM, Wilson M, Tonetti MS, etal. Gene polymorphisms and the prevalence of key periodontal pathogens. J Dent Res. 2007; 86: 416–20.
- Genc MR, Vardhana S, Delaney ML, Onderdonk A, Tuomala R, Norwitz E, etal. Relationship between a toll-like receptor-4 gene polymorphism, bacterial vaginosis-related flora and vaginal cytokine responses in pregnant women. Eur J Obstet Gynecol Reprod Biol. 2004; 116: 152–6.
- Verstraelen H, Verhelst R, Nuytinck L, Roelens K, De Meester E, De Vos D, etal. Gene polymorphisms of toll-like and related recognition receptors relation to the vaginal carriage of Gardnerella vaginalis and Atopobium vaginae. J Reprod Immunol. 2009; 79: 163–73.
- Divaris K, Monda KL, North KE, Olshan AF, Lange EM, Moss K, etal. Genome-wide association study of periodontal pathogen colonization. J Dent Res. 2012; 91(7 Suppl): 21S–8S.
- Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, etal. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011; 6: e16393.
- Alonso VR, Guarner F. Linking the gut microbiota to human health. Br J Nutr. 2013; 109: S21–6.
- Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007; 104: 13780–5.
- Isaacs KL, Sartor RB. Treatment of inflammatory bowel disease with antibiotics. Gastroenterol Clin North Am. 2004; 33: 335–45.
- Parkes M. Evidence from genetics for a role of autophagy and innate immunity in IBD pathogenesis. Dig Dis. 2012; 30: 330–3.
- Bibiloni R, Mangold M, Madsen KL, Fedorak RN, Tannock GW. The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn's disease and ulcerative colitis patients. J Med Microbiol. 2006; 55: 1141–9.
- Chow J, Mazmanian SK. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe. 2010; 7: 265–76.
- Inohara N, Chamaillard M, McDonald C, Nunez G. NOD-LRR proteins: role in host–microbial interactions and inflammatory disease. Ann Rev Biochem. 2005; 74: 355–83.
- Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, etal. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001; 411: 603–6.
- Sun LL, Nava GM, Stappenbeck TS. Host genetic susceptibility, dysbiosis, and viral triggers in inflammatory bowel disease. Curr Opin Gastroenterol. 2011; 27: 321–7.
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, etal. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012; 491: 119–24.
- Craven M, Egan CE, Dowd SE, McDonough SP, Dogan B, Denkers EY, etal. Inflammation drives dysbiosis and bacterial invasion in murine models of ileal Crohn's disease. PLoS One. 2012; 7: e41594.
- Roberson EDO, Bowcock AM. Psoriasis genetics: breaking the barrier. Trends Genet. 2010; 26: 415–23.
- Norrlind R. The significance of infections in the origination of psoriasis. Acta Rheumatol Scand. 1955; 1: 135–44.
- Tervaert WC, Esseveld H. A study of incidence of haemolytic streptococci in throat in patients with psoriasis vulgaris, with reference to their role in pathogenesis of this disease. Dermatologica. 1970; 140: 282–90.
- Gudjonsson JE, Thorarinsson AM, Sigurgeirsson B, Kristinsson KG, Valdimarsson H. Streptococcal throat infections and exacerbation of chronic plaque psoriasis: a prospective study. Br J Dermatol. 2003; 149: 530–4.
- Fahlen A, Engstrand L, Baker BS, Powles A, Fry L. Comparison of bacterial microbiota in skin biopsies from normal and psoriatic skin. Arch Dermatol Res. 2012; 304: 15–22.
- Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun. 2013; 4: 1431.
- Chamian F, Lowes MA, Lin SL, Lee E, Kikuchi T, Gilleaudeau P, etal. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proc Natl Acad Sci U S A. 2005; 102: 2075–80.
- Lowes MA, Chamian F, Abello MV, Fuentes-Duculan J, Lin SL, Nussbaum R, etal. Increase in TNF-alpha and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc Natl Acad Sci U S A. 2005; 102: 19057–62.
- Bataille V, Lens M, Spector TD. The use of the twin model to investigate the genetics and epigenetics of skin diseases with genomic, transcriptomic and methylation data. J Eur Acad Dermatol Venereol. 2012; 26: 1067–73.
- Trembath RC, Clough RL, Rosbotham JL, Jones AB, Camp RD, Frodsham A, etal. Identification of a major susceptibility locus on chromosome 6p and evidence for further disease loci revealed by a two stage genome-wide search in psoriasis. Hum Mol Genet. 1997; 6: 813–20.
- Nair RP, Stuart P, Henseler T, Jenisch S, Chia NV, Westphal E, etal. Localization of psoriasis-susceptibility locus PSORS1 to a 60-kb interval telomeric to HLA-C. Am J Hum Genet. 2000; 66: 1833–44.
- Tsoi LC, Spain SL, Knight J, Ellinghaus S, Stuart PE, Capon F, etal. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012; 44: 1341–8.
- Oka A, Mabuchi T, Ozawa A, Inoko H. Current understanding of human genetics and genetic analysis of psoriasis. J Dermatol. 2012; 39: 231–41.
- Armitage GC. Development of a classification system for periodontal diseases and conditions. Ann Periodontol. 1999; 4: 1–6.
- Page RC, Offenbacher S, Schroeder HE, Seymour GJ, Kornman KS. Advances in the pathogenesis of periodontitis: summary of developments, clinical implications and future directions. Periodontology. 2000; 14: 216–48.
- Rylev M, Kilian M. Prevalence and distribution of principal periodontal pathogens worldwide. J Clin Periodontol. 2008; 35: 346–61.
- Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res. 2012; 91: 914–20.
- American Academy of Periodontology. Epidemiology of periodontal diseases. J Periodontol. 1996; 67: 935–45.
- Socransky S, Haffajee A. Lindhe J, Lang N, Karring P. Periodontal infections. Clinical implantology and implant dentistry . 2012; Oxford, UK: Blackwell Munksgaard. 207–267.
- Forner L, Larsen T, Kilian M, Holmstrup P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol. 2006; 33: 401–7.
- Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of periodontal pathogens in atheromatous plaques. J Periodontol. 2000; 71: 1554–60.
- Leon R, Silva N, Ovalle A, Chaparro A, Ahumada A, Gajardo M, etal. Detection of Porphyromonas gingivalis in the amniotic fluid in pregnant women with a diagnosis of threatened premature labor. J Periodontol. 2007; 78: 1249–55.
- Quirke AM, Lugli EB, Wegner N, Hamilton BC, Charles P, Chowdhury M, etal. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: a potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann Rheum Dis. 2014; 73: 263–9.
- Kamer AR, Dasanayake AP, Craig RG, Glodzik-Sobanska L, Bry M, de Leon MJ. Alzheimer's disease and peripheral infections: the possible contribution from periodontal infections, model and hypothesis. J Alzheimers Dis. 2008; 13: 437–49.
- Stein PS, Steffen MJ, Smith C, Jicha G, Ebersole JL, Abner E, etal. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer's disease. Alzheimers Dement. 2012; 8: 196–203.
- Socransky SS, Haffajee AD. Microbial mechanisms in the pathogenesis of destructive periodontal-diseases – a critical assessment. J Periodontal Res. 1991; 26: 195–212.
- Nibali L, Tonetti MS, Ready D, Parkar M, Brett PM, Donos N, etal. Interleukin-6 polymorphisms are associated with pathogenic bacteria in subjects with periodontitis. J Periodontol. 2008; 79: 677–83.
- Nibali L, Madden I, Chillida FF, Heitz-Mayfield LJA, Brett PM, Donos N. IL6-174 genotype associated with Aggregatibacter actinomycetemcomitans in Indians. Oral Dis. 2011; 17: 232–7.
- Nibali L, Donos N, Henderson B. Periodontal infectogenomics. J Med Microbiol. 2009; 58: 1269–74.
- Ugwumadu A, Manyonda I, Reid F, Hay P. Effect of early oral clindamycin on late miscarriage and preterm delivery in asymptomatic women with abnormal vaginal flora and bacterial vaginosis: a randomised controlled trial. Lancet. 2003; 361: 983–8.
- Spiegel CA. Bacterial vaginosis. Clin Microbiol Rev. 1991; 4: 485–502.
- Bilardi JE, Walker S, Temple-Smith M, McNair R, Mooney-Somers J, Bellhouse C, etal. The burden of bacterial vaginosis: women's experience of the physical, emotional, sexual and social impact of living with recurrent bacterial vaginosis. PLoS One. 2013; 8: e74378.
- Forsum U, Holst E, Larsson PG, Vasquez A, Jakobsson T, Mattsby-Baltzer I. Bacterial vaginosis – a microbiological and immunological enigma. Apmis. 2005; 113: 81–90.
- Turovskiy Y, Noll KS, Chikindas ML. The aetiology of bacterial vaginosis. J Appl Microbiol. 2011; 110: 1105–28.
- Matu MN, Orinda GO, Njagi EN, Cohen CR, Bukusi EA. In vitro inhibitory activity of human vaginal lactobacilli against pathogenic bacteria associated with bacterial vaginosis in Kenyan women. Anaerobe. 2010; 16: 210–15.
- Hay P. Life in the littoral zone: lactobacilli losing the plot. Sex Transm Infect. 2005; 81: 100–2.
- Mattozzi C, Richetta AG, Cantisani C, Macaluso L, Calvieri S. Psoriasis: new insight about pathogenesis, role of barrier organ integrity, NLR/CATERPILLER family genes and microbial flora. J Dermatol. 2012; 39: 752–60.
- Keller JJ, Lin HC. The effects of chronic periodontitis and its treatment on the subsequent risk of psoriasis. Br J Dermatol. 2012; 167: 1338–44.
- Li WQ, Han JL, Chan AT, Qureshi AA. Psoriasis, psoriatic arthritis and increased risk of incident Crohn's disease in US women. Ann Rheum Dis. 2013; 72: 1200–5.
- Persson R, Hitti J, Verhelst R, Vaneechoutte M, Persson R, Hirschi R, etal. The vaginal microflora in relation to gingivitis. BMC Infect Dis. 2009; 9: 6.
- Henckaerts L, Pierik M, Joossens M, Ferrante M, Rutgeerts P, Vermeire S. Mutations in pattern recognition receptor genes modulate seroreactivity to microbial antigens in patients with inflammatory bowel disease. Gut. 2007; 56: 1536–42.
- Genc MR, Schantz-Dunn J. The role of gene–environment interaction in predicting adverse pregnancy outcome. Best Pract Res Clin Obstet Gynaecol. 2007; 21: 491–504.
- Schulz S, Zissler N, Altermann W, Klapproth J, Zimmermann U, Glaser C, etal. Impact of genetic variants of CD14 and TLR4 on subgingival periodontopathogens. Int J Immunogenet. 2008; 35: 457–64.
- Cauci S, Di Santolo M, Casabellata G, Ryckman K, Williams SM, Guaschino S. Association of interleukin-1 beta and interleukin-1 receptor antagonist polymorphisms with bacterial vaginosis in non-pregnant Italian women. Mol Hum Reprod. 2007; 13: 243–50.
- Havemose-Poulsen A, Sorensen LK, Bendtzen K, Holmstrup P. Polymorphisms within the IL-1 gene cluster: effects on cytokine profiles in peripheral blood and whole blood cell cultures of patients with aggressive periodontitis, juvenile idiopathic arthritis, and rheumatoid arthritis. J Periodontol. 2007; 78: 475–92.
- Queiroz DMM, Oliveira AG, Saraiva IEB, Rocha GA, Rocha AM, das Gracas Pimenta Sanna M, etal. Immune response and gene polymorphism profiles in Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. 2009; 15: 353–8.
- Julia A, Tortosa R, Hernanz JM, Canete JD, Fonseca E, Ferrandiz C, etal. Risk variants for psoriasis vulgaris in a large case-control collection and association with clinical subphenotypes. Hum Mol Genet. 2012; 21: 4549–57.
- Zhang X, Xia P, Zhang L, Zhang Z. Upregulation of tumor necrosis factor alpha-induced protein 3 mRNA in mild psoriasis vulgaris. Clin Vaccine Immunol. 2013; 20: 1341.
- Gümüş P, Nizam N, Lappin DF, Buduneli N. Saliva and serum levels of B-cell activating factors and tumor necrosis factor-alpha in patients with periodontitis. J Periodontol. 2014; 85: 270–80.
- Conner HD, Haberman S, Collings CK, Winford TE. Bacteremias following periodontal scaling in patients with healthy appearing Gingiva. J Periodontol. 1967; 38: 466–72.
- Daly C, Mitchell D, Grossberg D, Highfield J, Stewart D. Bacteraemia caused by periodontal probing. Aust Dent J. 1997; 42: 77–80.
- Hill GB. Preterm birth: associations with gential and possibly oral microflora. Ann Periodontol. 1998; 3: 222–32.
- Goldenberg RL, Hauth JC, Andrews WW. Mechanisms of disease – intrauterine infection and preterm delivery. N Engl J Med. 2000; 342: 1500–7.
- Gonzales-Marin C, Spratt DA, Allaker RP. Maternal oral origin of Fusobacterium nucleatum in adverse pregnancy outcomes as determined using the 16S-23S rRNA gene intergenic transcribed spacer region. J Med Microbiol. 2013; 62: 133–44.
- O'Boyle CJ, MacFie J, Dave K, Sagar PS, Poon P, Mitchell CJ. Alterations in intestinal barrier function do not predispose to translocation of enteric bacteria in gastroenterologic patients. Nutrition. 1998; 14: 358–62.
- Shanahan F. The host-microbe interface within the gut. Best Pract Res Clin Gastroenterol. 2002; 16: 915–31.
- Naaber P, Smidt I, Tamme K, Liigant A, Tapfer H, Mikelsaar M, etal. Translocation of indigenous microflora in an experimental model of sepsis. J Med Microbiol. 2000; 49: 431–9.