
Abstract
Polymorphonuclear leukocytes (neutrophils) constitute an integrated component of the innate host defense in the gingival sulcus/periodontal pocket. However, the keystone periodontal pathogen Porphyromonas gingivalis has in the course of evolution developed a number of capacities to subvert this defense to its own advantage. The present review describes the major mechanisms that P. gingivalis uses to subvert neutrophil homeostasis, such as impaired recruitment and chemotaxis, resistance to granule-derived antimicrobial agents and to the oxidative burst, inhibition of phagocytic killing while promoting a nutritionally favorable inflammatory response, and delay of neutrophil apoptosis. Studies in animal models have shown that at least some of these mechanisms promote the dysbiotic transformation of the periodontal polymicrobial community, thereby leading to inflammation and bone loss. It is apparent that neutrophil–P. gingivalis interactions and subversion of innate immunity are key contributing factors to the pathogenesis of periodontal disease.
Polymorphonuclear leukocytes (neutrophils) represent the primary cellular defense system in healthy oral tissues (Citation1). They are the most common leukocytes recruited to the gingival crevice or periodontal pocket (Citation2, Citation3) and are needed for periodontal tissue homeostasis (Citation4–Citation6). They attach to and roll along the endothelial cells in the terminal circulation via a weak selectin-mediated binding. As a result of a microbial/pro-inflammatory stimulus, selectins are later removed and surface integrins are up-regulated, creating a firm attachment to the endothelial lining of the blood vessel in the lamina propria of the gingiva (Citation1, Citation7). This mechanism is regulated by adhesion molecules, such as β2- and β1-integrins, chemokines, and cytokines (Citation7, Citation8). In addition, toll-like receptors (TLRs) (especially TLR2, TLR4, and TLR5) can activate integrin-dependent neutrophil adhesion. In fact, the neutrophil adhesion cascade for transmigration as a response to infection or inflammation is a key paradigm in immunity (Citation9).
Although transmigrating neutrophils initially follow the chemokine gradient (e.g. IL-8) deposited by the endothelium, upon extravasation they move towards chemoattractant gradients formed in the infected or inflamed tissue (e.g. chemoattractants derived from bacteria such as N-formyl-methionyl-leucyl-phenylalanine (FMLP) or generated from local complement activation such as the C5a fragment) (Citation9). In the periodontal pocket, migration of polymorphonuclear neutrophils through the junctional epithelium occurs at a rate of 30,000 per min (Citation4, Citation10). In their efforts to localize and kill subgingival, supragingival, and mucosal biofilm and planktonic bacteria, neutrophils phagocytose individual organisms and release superoxide as part of the oxidative burst process. Superoxide is converted to hydrogen peroxide. Neutrophils also release a series of enzymes that have antimicrobial action, such as myeloperoxidase and proteolytic enzymes, from lysosomal granules (Citation1).
The neutrophils form a ‘wall’ against the subgingival biofilm. However, they are not adept at phagocytosing biofilm-associated bacteria, which eventually leads to ‘frustrated phagocytosis’. During this process, neutrophil-derived toxic substances (e.g. reactive oxygen species, ROS) may also be released to the underlying tissue, causing collateral damage to tooth-supportive tissues (Citation11). This mechanism has earned neutrophils the characterization two-edged swords, implying that their presence is not necessarily protective. Collateral damage can be exerted by hyperactive neutrophils or neutrophils in excessive numbers (reviewed in Ref. 7). Recent research has also indicated that neutrophils can be involved in the initiation and progression of periodontitis when their function is subverted by periodontal bacteria (Citation7).
Porphyromonas gingivalis
Porphyromonas gingivalis is a keystone pathogen in human periodontitis (Citation12, Citation13). This description implies for example that it can cause dysbiosis (imbalance in the relative abundance or influence of species within a microbial community) even when present at a low colonization level. Microbiome studies have shown that P. gingivalis is a minor constituent of periodontitis-associated biofilm in man (Citation14–Citation16). In addition, in a mouse model of periodontitis, P. gingivalis exhibited low-level colonization and caused an increase in certain populations of the periodontal microbiota (‘microbial shift’) followed by inflammatory bone loss (Citation17). Accordingly, a keystone pathogen serves a specialized beneficial function for the entire pathogenic community in the same way a differentiated cell serves an entire tissue (Citation4, Citation18). In inducing a microbial shift, P. gingivalis – as a keystone pathogen – disturbs the original biofilm community instead of protecting it (Citation12).
As a Gram-negative, anaerobic, and asaccharolytic rod, P. gingivalis has a number of virulence factors (Citation19), many of which are related to subversion of the innate immune system. This ability is what often characterizes a successful pathogen, as it tends to disable the overall host response while simultaneously enhancing the pathogenicity of a polymicrobial community (Citation20).
The aim of the present review is to characterize the major steps that P. gingivalis can take to subvert neutrophil functions and related immune responses (). We have previously reviewed mechanisms through which P. gingivalis can modulate innate immunity by affecting inflammasome activity (Citation21).
Table 1. Mechanisms and consequences of Porphyromonas gingivalis causing subversion of polymorphonuclear leukocytes
Impaired recruitment
Neutrophils are more than simple foot soldiers of the innate immune system with a restricted set of pro-inflammatory functions. They are in fact complex cells capable of a vast array of specialized functions, contributing not only to acute inflammation but also to chronic inflammatory conditions and adaptive immune responses (Citation22).
P. gingivalis can impair recruitment of neutrophils by interfering with (down-regulating) the expression of chemokines (termed local chemokine paralysis) and cell adhesion molecules such as IL-8, ICAM-1, and E-selectin necessary for leukocyte diapedesis (Citation4, Citation5) (Citation23–Citation27). There was little or no stimulation of E-selectin expression with either P. gingivalis or Helicobacter pylori when whole cells, lipopolysaccharides (LPSs), or cell wall preparations were added to human umbilical cord vein endothelial cells (Citation23). Moreover, P. gingivalis LPS blocked E-selectin expression induced by LPS from other bacteria. This would attenuate the recruitment of neutrophils, which normally have an important role in maintaining oral health and will help bacterial colonization of the periodontal pocket. Deregulation of leukocyte recruitment was suggested to contribute to P. gingivalis–induced periodontitis in a murine model (Citation17). Noteworthy, neutrophil recruitment may also be important for homeostatic functions that are not related to control of infection (Citation5). In a gingival crevice model (epithelial cell–P. gingivalis–neutrophil interaction), P. gingivalis alone induced low levels of IL-1β and IL-8 from epithelial cells, but when neutrophils were added, high levels of both cytokines were produced (Citation28). This finding is consistent with the notion that P. gingivalis subverts the function of neutrophils while promoting their inflammatory response, which not only causes collateral tissue damage but provides the bacterium with nutrients in the form of tissue breakdown products (Citation20).
SerB phosphatase and its role in impaired recruitment
SerB phosphatase has numerous roles in P. gingivalis–host cell interactions. This enzyme, which is secreted by P. gingivalis, is a potent and specific inhibitor of the activation of the NF-κB transcription factor that regulates the production of IL-8. Infection of gingival epithelial cells with P. gingivalis suppressed the production of IL-8 (Citation25, Citation29), which facilitated colonization with this organism. In gingival epithelial cells, SerB phosphatase binds to and dephosphorylates the serine 536 residue of the RelA/p65 subunit of the NF-κB, which prevents nuclear translocation and transcription of the IL-8 gene. By targeting the NF-κB p65 subunit protein, P. gingivalis reduces IL-8 inflammatory responses, promotes survival, and probably establishes a favorable niche not only for itself but for the entire biofilm community. Accordingly, an isogenic mutant deficient in SerB phosphatase induced significantly higher recruitment of neutrophils to the periodontal tissue (Citation25, Citation30). These data testify to the fact that the so-called localized chemokine paralysis (Citation25) occurs in vivo through the action of SerB phosphatase. This implies that the ability to detect and localize colonized bacteria by IL-8 is paralyzed and unable to function in sites invaded by P. gingivalis. Indeed, this could have a destructive effect on innate host defense in the periodontium continuously exposed to bacteria, and the host might no longer be able to direct leukocytes for removal of bacteria (Citation18).
Reduced chemotaxis
P. gingivalis – together with nine other Gram-negative biofilm species – inhibited neutrophil chemotaxis, probably by binding non-chemotactic formylmethionyl peptides to the FMLP receptor, thereby preventing detection of the chemotactic gradient by the neutrophils (Citation31). This suggested that chemotaxis inhibition in vitro was mediated by binding of non-chemotactic bacterial formylmethionyl peptides to the FMLP receptor on neutrophils. Chemotaxis of endotoxin-activated serum, where the active chemotactic component is the complement fragment C5a, was also inhibited. This complement factor has a separate receptor, known as the C5a receptor (C5aR; CD88). The observed chemotactic inhibition might therefore have been due to a more general interference with the neutrophil chemotactic receptors (Citation31). Van Dyke (Citation32) also suggested that P. gingivalis could render the neutrophils incapable of responding normally to chemotactic receptor–mediated events in the periodontal pocket and adjacent connective tissue. The neutrophils could actually be stopped short of the junctional epithelium in connective tissues due to local inhibitors such as soluble bacterial products (Citation32).
Rotstein et al. (Citation33) found that succinic acid, a major metabolic by-product of Bacteroides species, reduced random migration and chemotactic response to FMLP and C5a and virtually obliterated phagocytic killing of Escherichia coli. Butyric acid is produced by P. gingivalis at higher levels than succinate (Citation34, Citation35). Butyrate and propionate reduced the production of CINC-2αβ, which like human IL-8 is a potent neutrophil chemoattractant in the rat (Citation36). In these animals, butyrate also reduced the migration of neutrophils, possibly by inhibition of the production of cytokines and chemokines (Citation35).
Phagocytosis of bacteria by neutrophils is followed by increased neutrophil oxidative metabolism. One of the oxygen metabolites is O2 −, a major component of the bactericidal activity of neutrophils. Many anaerobic bacteria produce enzymes such as superoxide dismutase that neutralize these reactive products (Citation37). Superoxide anion (O2 −) production of human or guinea pig neutrophils stimulated by FMLP was suppressed when the cells were preincubated in culture supernatants of P. gingivalis (Citation38). After 1-h incubation of a P. gingivalis supernatant with these neutrophils, there was a significant reduction in the FMLP receptor on neutrophils (Citation38). However, assessing binding to FMLP is a rather indirect indicator of receptor density on the cell surface. Nevertheless, the subversive functions of P. gingivalis via modulation of neutrophil cell surface receptors could be important in the virulence potential of this bacterium.
FDC 381 and ATCC 33277, outer membrane components of P. gingivalis, were found to have porin-like activity, able to depolarize the electrochemical potential that exists across the neutrophil membrane. Membrane depolarization was associated with immobilization of the neutrophils in response to the chemotactic peptide FMLP (Citation39), but not with cell lysis. The immobilization of neutrophils by P. gingivalis, although it varied between strains, was thought to be a significant virulence factor for this bacterium, combined with the inability of several P. gingivalis strains to activate the microbicidal respiratory burst. The biological relevance of this mechanism for neutrophil immobilization is unclear.
Rotstein et al. (Citation40) found that short-chain fatty acids (SCFAs), particularly succinic acid, inhibited the respiratory burst in neutrophils by reducing the intracellular pH. This might not only inhibit neutrophil chemotaxis but also phagocytic killing (Citation33, Citation41) and could contribute to the virulence of P. gingivalis in polymicrobial infections.
Bielecka et al. (Citation42) discovered that in vitro pretreatment of C5a for 1 h at 37°C with peptidylarginine deiminase (PPAD) from P. gingivalis led to reduced chemotaxis of human neutrophils. Thus a novel pathogenic strategy was revealed for P. gingivalis, which is the sole bacterium expressing PPAD, implying that it can inactivate the pro-inflammatory activity of C5a by deimination (citrullination) of its C-terminal arginine. As discussed above, given enough time, P. gingivalis can eventually inactivate C5a; however, it should be noted that this bacterium also takes advantage of the agonist activity of C5a to subvert neutrophil action (see the section ‘Inhibition of antimicrobial and promotion of inflammatory responses’).
Although the above-discussed in vitro studies have provided important insights into the mechanisms whereby P. gingivalis impairs neutrophil recruitment and chemotaxis, these subversive effects are transient in vivo, and neutrophils eventually transmigrate in great numbers. Nevertheless, even a temporary delay of neutrophil recruitment may in principle allow adequate time for initial colonization and formation of bacterial communities that can subsequently subvert neutrophils, leading to defective killing capacity and dysregulated inflammatory responses (Citation17, Citation19).
Dual TREM-1 regulation
The triggering receptor expressed on myeloid cells 1 (TREM-1) is a cell surface receptor belonging to the immunoglobulin superfamily. It has the capacity to amplify pro-inflammatory cytokine production and regulate apoptosis. Neutrophils are a major source of TREM-1 (Citation43). These authors suggested that P. gingivalis uses its Arg-gingipain to shed off soluble TREM-1 (sTREM-1) from the neutrophils and thereby amplify the inflammatory response. On the other hand, degradation of sTREM-1 by Lys-gingipain may eliminate the ability of neutrophils to promote an inflammatory response or to accomplish its antibacterial action through phagocytosis. The outcome of sTREM-1 regulation of neutrophils by P. gingivalis may be context-dependent. When food resources are limited, P. gingivalis might use its Arg-gingipain to cause an sTREM-1-supported innate immune response, thereby creating an inflammation-rich environment for its survival. If the nature of the inflammatory response tends to compromise its survival, P. gingivalis might use its Lys-gingipain to attenuate inflammation. Dual regulation of sTREM-1 release and degradation by two different gingipains may be a new mechanism whereby P. gingivalis could evade host defenses and establish chronic periodontitis. However, as the temperature of the periodontal pocket increases due to inflammation, a reduction in total arginine- and lysine-specific activities may occur (Citation44).
Resistance to killing
P. gingivalis was shown to be relatively resistant to killing by neutrophils under in vitro anaerobic conditions (Citation45). This resistance was attributed to the ability of P. gingivalis to inactivate cathepsin G, elastase, bacterial-permeability increasing factor, and defensins. Interestingly, crevicular neutrophils from patients with rapidly progressive periodontitis display reduced intracellular killing of P. gingivalis as compared to neutrophils from healthy controls, suggesting that host defects contribute further to the killing resistance of this pathogen (Citation46). The capacity of P. gingivalis to inactivate granular enzymes and antimicrobial peptides and thus evade neutrophil-mediated killing could be an important mechanism for its persistence in the periodontal pocket. In fact, it has been claimed that anaerobes inhibit their own phagocytosis and killing as well as that of aerobic species present in mixed infections, for example by their SCFAs inhibiting chemotactic response, chemiluminescence, and degranulation (Citation41). SCFAs have also been found to increase neutrophil migration to the inflammatory site, thereby contributing to the initiation of the inflammation process (Citation35). The inconsistency in these results may be associated with use of different concentrations or different types of SCFAs, pH, and cell types.
Maekawa et al. (Citation47) recently showed that P. gingivalis induces C5aR–TLR2 co-association in human neutrophils and inhibits its own killing as well as the killing of bystander bacteria. Specifically, P. gingivalis rescued Fusobacterium nucleatum from killing in a C5aR- and TLR2-dependent manner, while not inhibiting the pro-inflammatory activity of neutrophils in vivo. These findings imply that P. gingivalis interacts with and exploits C5aR and TLR2 in human neutrophils to promote the survival of dysbiotic communities where P. gingivalis resides.
For clearance of microbes by neutrophils a concerted action of ROS and microbicidal components within leukocyte secretory granules is required. P. gingivalis produces an unidentified factor that interferes with the bactericidal activity of neutrophils by modulating the generation of ROS (Citation48). More recent publications, revealing the associated intracellular signaling pathways, have indicated that P. gingivalis can stimulate ROS release (Citation49, Citation50). The discrepancy between these results remains unclear.
Although P. gingivalis can induce ROS release (Citation49, Citation50), it should be noted that this bacterium is protected against oxidative stress by the cytoplasmic, non-heme iron protein rubrerythrin (Rbr) (Citation51). Rbr enabled proliferation of P. gingivalis in mice that had a functional oxidative burst response, but not in nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-null mice (Citation52). The in vivo protection provided by Rbr was not associated with the oxidative burst responses of isolated neutrophils. Although the phagocyte-derived oxidative burst response was quite ineffective against infection caused by P. gingivalis, the oxidative response to the Rbr-positive microbe caused host-induced pathology through potent mobilization and systemic activation of neutrophils. Rbr also shielded the organism against reactive nitrogen species thereby ensuring survival of P. gingivalis in the infected host. These findings suggested that the host oxidative burst paradoxically enhances the survival of P. gingivalis by exacerbating local and systemic inflammation. Overall, P. gingivalis has several mechanisms to modulate neutrophil function that seem to be contradictory and are still debated in the literature. It is necessary to keep in mind, however, the contextual relevance of the various mechanisms; a pathogen may inhibit or enhance a given cellular function depending on specific circumstances.
Inhibition of antimicrobial and promotion of inflammatory responses
The Arg-specific gingipains of P. gingivalis have C5 convertase-like activity and generate C5a, which is considered the most potent pro-inflammatory effector of the complement cascade and, in general, promotes host defense (Citation53). It guides neutrophils and other phagocytes to sites of complement activation (Citation54). Paradoxically, P. gingivalis uses C5a to undermine TLR2 immunity. Although P. gingivalis cannot antagonize TLR2 at the receptor level, it can intercept and undermine certain TLR2 signaling events to subvert innate immunity (Citation47, Citation55). In macrophages, for example, P. gingivalis was found to synergize with C5a for cyclic adenosine monophosphate (cAMP) elevation, resulting in inhibition of macrophage production of nitric oxide and enhanced pathogen survival in vitro and in vivo (Citation55). Importantly, nitric oxide has an antibacterial effect against P. gingivalis (Citation56). The reaction mentioned above reflected manipulation of crosstalk interactions between C5aR and TLR2 (Citation53–Citation58). In the context of experimental murine periodontitis, P. gingivalis subverts C5aR in leukocytes to inhibit their antimicrobial effects but not their pro-inflammatory effects, thereby allowing uncontrolled growth and change in the composition of the periodontal microbiota towards a dysbiotic state (Citation17, Citation47) (Citation55).
Maekawa et al. (Citation47) showed that C5aR and TLR2 cooperate in human neutrophils in a subversive crosstalk to prevent P. gingivalis killing without causing a generalized neutrophil immunosuppression. When C5aR or TLR2 – but not other receptors such as complement receptor 3 (CR3) or C-X-C chemokine receptor type 4 (CXCR4) – were blocked, the ability of human neutrophils to kill P. gingivalis was significantly increased. This suggested that intact C5aR and TLR2 signaling is needed for maximal protection of P. gingivalis against human neutrophils. In this regard, the same study also reported that P. gingivalis–induced C5aR–TLR2 crosstalk leads to degradation of MyD88 (). This is a TLR2 signaling adaptor contributing to clearance of P. gingivalis infection but it does not affect TLR2-dependent inflammatory response to P. gingivalis (Citation59), suggesting the operation of a MyD88-independent inflammatory pathway. Noteworthy, F. nucleatum did not cause degradation of MyD88. Degradation of MyD88 was completely reversed by C5aR antagonist or anti-TLR2, thus confirming that it was caused by the C5aR–TLR2 crosstalk (Citation47). In summary, P. gingivalis can evade killing by human neutrophils by using a C5aR/TLR2-mediated mechanism that involves degradation of MyD88 (Citation47).
Fig. 1. Porphyromonas gingivalis subverts neutrophils to evade killing while causing dysbiotic inflammation. P. gingivalis expresses ligands that activate the TLR2–TLR1 receptor complex (TLR2/1) and Arg-specific gingipains (HRgpA and RgpB gingipains), which cleave complement C5 and generate high local concentrations of C5a ligand. The ability of P. gingivalis to co-activate C5aR and TLR2 in human neutrophils results in a subversive crosstalk that leads to ubiquitylation and proteasomal degradation of the TLR2 adaptor MyD88, thereby blocking an antimicrobial response that would otherwise clear the pathogen. The proteolysis of MyD88 requires C5aR/TLR2-dependent release of the cytokine TGF-β1, which mediates ubiquitination of MyD88 via the E3 ubiquitin ligase Smurf1 (enlarged inset). In addition, the C5aR–TLR2 crosstalk activates PI3K, which inhibits phagocytosis by blocking the activity of RhoA GTPase and hence actin polymerization. PI3K signaling, moreover, induces the production of inflammatory cytokines. In contrast to MyD88, a second TLR2 adaptor, Mal, participates in this immune subversion strategy by acting upstream of PI3K. Together, these functionally integrated pathways, as controlled by P. gingivalis, offer ‘bystander’ protection to otherwise susceptible species in polymicrobial communities and promote dysbiotic inflammation and periodontal bone loss in relevant animal models. (From Ref. 20. Used with permission.)
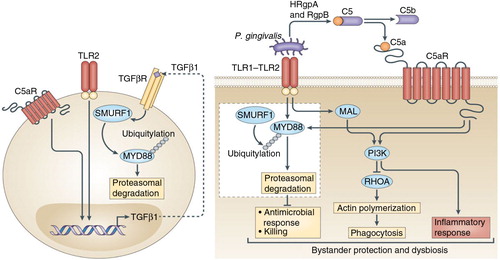
Moreover, the exploitation of C5aR and TLR2 by P. gingivalis helped this bacterium survive via MyD88-independent mechanisms. In this regard, P. gingivalis–instigated C5aR–TLR2 crosstalk in neutrophils also activates an alternative pathway where the TLR2 adaptor MyD88 adaptor-like protein (Mal) induces phosphatidylinositol-3-OH kinase P13K signaling (Citation47). The authors reported that P. gingivalis inhibits phagocytosis by exploiting Ca5aR/TLR2/Mal-dependent PI3K signaling, which suppresses RhoA GTPase activation and hence actin polymerization. Moreover, PI3K was shown to act downstream of C5aR and TLR2 to promote inflammation (). Taken together, P. gingivalis evades phagocytic killing and simultaneously elicits inflammatory responses in neutrophils by activating PI3K in a way that depends on Ca5R and TLR2 but not on MyD88 activation. This mechanism protects P. gingivalis and bystander bacteria and contributes to dysbiotic inflammation and periodontal bone loss (Citation47).
These studies provided a molecular explanation for the inability of human as well as mouse neutrophils to take control of periodontal bacteria and prevent dysbiotic inflammation. Moreover, these studies underscored the ‘inflammophilic’ nature of the periodontal bacteria, which, not surprisingly, do not cause generalized immune suppression in leukocytes. Rather, they target specifically those pathways that can eliminate them while keeping intact or even promoting relatively harmless inflammatory responses that are exploited for procurement of nutrients from tissue breakdown products (Citation60). It should be emphasized that, although harmless for bacteria, these inflammatory responses are detrimental for the host, as they mediate collateral tissue damage.
Pro-adhesive signaling pathway
P. gingivalis induces a pro-adhesive signaling pathway for activation of CR3 in macrophages and neutrophils (Citation61). The pathway is initiated when P. gingivalis fimbriae bind to CD14 and activate TLR2 and PI3K inside-out signaling. This again leads to induction of the high-affinity conformation of CR3 in leukocytes. The TLR2 pro-adhesive signaling pathway is involved in enhancing leukocyte-endothelial cell interactions and transendothelial migration. However, P. gingivalis seems to have co-opted this pathway for enhancing the interaction of its cell surface fimbriae with CR3. Activated CR3 interacts with P. gingivalis fimbriae and induces downregulation of IL-12p70, a key cytokine involved in intracellular bacterial clearance by macrophages. The idea that CR3 is associated with reduced IL-12p70 induction and impaired clearance of P. gingivalis was confirmed using intraperitoneally infected wild-type and CR3-deficient mice (Citation62). Although this immune evasion mechanism was shown in macrophages and has not yet been addressed in neutrophils, exploitation of CR3 by P. gingivalis may represent an effective strategy of P. gingivalis to promote its persistence and virulence.
The pro-adhesive signaling pathway induced by P. gingivalis fimbriae was confirmed to operate also in human monocytes and the pathway was further dissected, involving TLR2, Rac1, PI3K, and CD11b/CD18 (Citation63). Through this pathway, P. gingivalis stimulates monocyte adhesion to endothelial cells and transendothelial migration, potentially providing a mechanistic basis linking P. gingivalis to inflammatory atherosclerotic processes. This appears to be in contrast to the report of Madianos et al. (Citation24), who however studied neutrophil transepithelial migration, whereas Harokopakis et al. (Citation63) focused on monocyte adhesion and transendothelial migration. Because different mechanisms govern transepithelial versus transendothelial migration, these studies may not necessarily reflect a discrepancy.
Delaying neutrophil apoptosis
LPS and lipid A from P. gingivalis delayed neutrophil apoptosis, whereas capsular polysaccharide had no significant effect (Citation64, Citation65). Neutrophils showed a time-dependent increase in the number of apoptotic cells, and apoptosis was delayed in a dose-dependent fashion. In addition, LPS from P. gingivalis W50 prevented apoptosis in HL-60-derived neutrophils (Citation66). The inhibition was probably achieved by signaling through TLR2 but not TLR4. Zaric et al. (Citation67) suggested that impaired induction of immune tolerance to P. gingivalis LPS promoted neutrophil migration and reduced apoptosis. Because neutrophil apoptosis followed by efferocytosis (apoptotic cell phagocytosis) plays a major role in the resolution of inflammation, a delay in apoptosis caused by periodontal bacteria might prolong inflammatory responses with increased potential for tissue damage.
Concluding remarks
Neutrophils are a key factor in the host defense of the gingival sulcus/periodontal pocket. This defense can be subverted by pathogenic bacteria such as P. gingivalis, which in the course of evolution has probably acquired qualities able to facilitate its survival in a hostile environment. The subversion of neutrophil defenses likely contributes to the initiation and progression of periodontitis and hyperactive or supernumerary neutrophils cause tissue damage in the periodontium.
Although P. gingivalis has been considered in this review as a model keystone pathogen in periodontitis, it is probably not the only one, and – most likely – it does not initiate the disease by itself. It should be appreciated that keystone pathogen colonization can be promoted by certain commensals (e.g. Streptococcus gordonii) that are not pathogenic by themselves in the oral cavity. In this way, commensals can be considered accessory pathogens (Citation68). Moreover, whereas keystone pathogens are involved in the breakdown of tissue homeostasis, so-called pathobionts can elicit destructive inflammation when homeostasis is disrupted. The pathogenesis of periodontal disease, therefore, is not mediated by individual pathogens but rather involves polymicrobial synergy and dysbiosis (Citation68). It should be emphasized that a significant strain heterogeneity exists within P. gingivalis, which could affect their virulence potential (Citation69, Citation70). It is also possible that there are individuals who can resist the ability of P. gingivalis to convert a symbiotic microbiota into a dysbiotic one due to their intrinsic immune status. With these limitations in mind, it is apparent that P. gingivalis has an impressive repertoire for subverting innate immunity mediated by neutrophils. However, as alluded to above, this subversion is not in the best interest of P. gingivalis, unless it also causes inflammation to feed on the ‘inflammatory spoils’ generated through degradation of collagen peptides and heme-containing compounds. P. gingivalis seems to stand this test.
Conflict of interest and funding
There is no conflict of interest in the present study for any of the authors.
Acknowledgements
IO acknowledges funding through the European Commission (FP7-HEALTH-306029 ‘TRIGGER’). GH acknowledges funding through the US National Institutes of Health (DE015254, DE017138, DE021685, DE024716, and AI068730).
Related Research Data
References
- Ryder MI. Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontol 2000. 2010; 53: 124–37. [PubMed Abstract].
- Nussbaum G, Shapira L. How has neutrophil research improved our understanding of periodontal pathogenesis. J Clin Periodontol. 2011; 38(Suppl 11): 49–59. doi: http://dx.doi.org/10.1111/j.1600-051X.2010.01678.x [PubMed Abstract].
- Sima C, Glogauer M. Neutrophil dysfunction and host susceptibility to periodontal inflammation: current state of knowledge. Curr Oral Health Rep. 2014; 1: 95–103. doi: http://dx.doi.org/10.1007/s40496-014-0015-x.
- Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010; 8: 481–90. doi: http://dx.doi.org/10.1038/nrmicro2337 [PubMed Abstract].
- Hajishengallis E, Hajishengallis G. Neutrophil homeostasis and periodontal health in children and adults. J Dent Res. 2014; 93: 231–7. doi: http://dx.doi.org/10.1177/0022034513507956 [PubMed Abstract] [PubMed CentralFull Text].
- Moutsopoulos N, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, etal. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med. 2014; 6: 229ra40. doi: http://dx.doi.org/10.1126/scitranslmed.3007696.
- Hajishengallis G, Chavakis T, Hajishengallis E, Lambris JD. Neutrophil haemostasis and inflammation: novel paradigms from studying periodontitis. J Leukoc Biol. 2015; 98: 539–48. doi: http://dx.doi.org/10.1189/jlb.3VMR1014-468R [PubMed Abstract].
- Subramanian P, Mitroulis I, Hajishengallis G, Chavakis T. Regulation of tissue infiltration by neutrophils: role of integrin α3β1 and other factors. Curr Opin Hematol. 2016; 23: 36–43. doi: http://dx.doi.org/10.1097/MOH.0000000000000198 [PubMed Abstract].
- Hajishengallis G, Chavakis T. Endogenous modulators of inflammatory cell recruitment. Trends Immunol. 2013; 34: 1–6. doi: http://dx.doi.org/10.1016/j.it.2012.08.003 [PubMed Abstract] [PubMed CentralFull Text].
- Tonetti MS, Imboden MA, Lang NP. Neutrophil migration into the gingival sulcus is associated with transepithelial gradients of interleukin-8 and ICAM-1. J Periodontol. 1998; 69: 1139–47. [PubMed Abstract].
- Chapple IL, Matthews JB. The role of reactive oxygen and antioxidant species in periodontal tissue destruction. Periodontol 2000. 2007; 43: 160–232. [PubMed Abstract].
- Darveau RP, Hajishengallis G, Curtis MA. Porphyromonas gingivalis as a potential community activist for disease. J Dent Res. 2012; 91: 816–20. doi: http://dx.doi.org/10.1177/0022034512453589 [PubMed Abstract] [PubMed CentralFull Text].
- Hajishengallis G, Darveau RP, Curtis MA. The keystone pathogen hypothesis. Nat Rev Microbiol. 2012; 10: 717–25. doi: http://dx.doi.org/10.1038/nrmicro2873 [PubMed Abstract] [PubMed CentralFull Text].
- Doungudomdacha S, Rawlinson A, Douglas CWI. Enumeration of Porphyromonas gingivalis, Prevotella intermedia and Actinobacillus actinomycetemcomitans in subgingival plaque samples by a quantitative-competitive PCR method. J Med Microbiol. 2000; 49: 861–74. [PubMed Abstract].
- Kumar PS, Leys EJ, Bryk JM, Martinez FJ, Moeschberger ML, Griffen AL. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J Clin Microbiol. 2006; 44: 3665–73. doi: http://dx.doi.org/10.1128/JCM.00317-06 [PubMed Abstract] [PubMed CentralFull Text].
- Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, etal. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013; 7: 1016–25. doi: http://dx.doi.org/10.1038/ismej.2012.174 [PubMed Abstract] [PubMed CentralFull Text].
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, etal. A low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and the complement pathway. Cell Host Microbe. 2011; 10: 497–506. doi: http://dx.doi.org/10.1016/j.chom.2011.10.006 [PubMed Abstract] [PubMed CentralFull Text].
- Darveau RP. The oral microbial consortium's interaction with the periodontal innate defense system. DNA Cell Biol. 2009; 28: 389–95. doi: http://dx.doi.org/10.1089/dna.2009.0864 [PubMed Abstract] [PubMed CentralFull Text].
- Zenobia C, Hajishengallis G. Porphyromonas gingivalis virulence factors involved in subversion of leukocytes and microbial dysbiosis. Virulence. 2015; 6: 236–43. doi: http://dx.doi.org/10.1080/21505594.2014.999567 [PubMed Abstract] [PubMed CentralFull Text].
- Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. 2015; 15: 30–44. doi: http://dx.doi.org/10.1038/nri3785 [PubMed Abstract] [PubMed CentralFull Text].
- Olsen I, Yilmaz ÖSubversion of inflammasome activity by Porphyromonas gingivalis in periodontitis and associated systemic diseases. J Oral Microbiol. 2016; 8: 30385. doi: http://dx.doi.org/10.3402/jom.v8.30385.
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013; 13: 159–75. doi: http://dx.doi.org/10.1038/nri3399 [PubMed Abstract].
- Darveau RP, Cunningham MD, Bailey T, Seachord C, Ratcliffe K, Bainbridge B, etal. Ability of bacteria associated with chronic inflammatory disease to stimulate E-selectin expression and promote neutrophil adhesion. Infect Immun. 1995; 63: 1311–17. [PubMed Abstract] [PubMed CentralFull Text].
- Madianos PN, Papapanou PN, Sandros J. Porphyromonas gingivalis infection of oral epithelium inhibits neutrophil transepithelial migration. Infect Immun. 1997; 65: 3983–90. [PubMed Abstract] [PubMed CentralFull Text].
- Darveau RP, Belton CM, Reife RA, Lamont RJ. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. 1998; 66: 1660–5. [PubMed Abstract] [PubMed CentralFull Text].
- Huang GT, Haake SK, Kim JW, Park NH. Differential expression of interleukin-8 and intercellular adhesion molecule-1 by human gingival epithelial cells in response to Actinobacillus actinomycetemcomitans or Porphyromonas gingivalis infection. Oral Microbiol Immunol. 1998; 13: 301–9. [PubMed Abstract].
- Huang GT-J, Kim D, Lee JK, Lee JK-H, Kuramitsu HK, Haake SK. Interleukin-8 and intercellular adhesion molecule 1 regulation in oral epithelial cells by selected periodontal bacteria: multiple effects of Porphyromonas gingivalis via antagonistic mechanisms. Infect Immun. 2001; 69: 1364–72. [PubMed Abstract] [PubMed CentralFull Text].
- Bondy-Carey JL, Galicia J, Bagaitkar J, Potempa JS, Potempa B, Kinane DF, etal. Neutrophils alter epithelial response to Porphyromonas gingivalis in a gingival crevice model. Mol Oral Microbiol. 2013; 28: 102–13. doi: http://dx.doi.org/10.1111/omi.12008 [PubMed Abstract] [PubMed CentralFull Text].
- Takeuchi H, Hirano T, Whitmore SE, Morisaki I, Amano A, Lamont RJ. The serine phosphatase SerB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-κB RelA/p65. PLoS Pathog. 2013; 9: e1003326. doi: http://dx.doi.org/10.1371/journal.ppat.1003326 [PubMed CentralFull Text].
- Bainbridge B, Verma RK, Eastman C, Yehia B, Rivera M, Moffatt C, etal. Role of Porphyromonas gingivalis phosphoserine phosphatase enzyme SerB in inflammation, immune response, and induction of alveolar bone resorption in rats. Infect Immun. 2010; 78: 4560–9. doi: http://dx.doi.org/10.1128/IAI.00703-10 [PubMed Abstract] [PubMed CentralFull Text].
- Van Dyke TE, Bartholomew E, Genco RJ, Slots J, Levine MJ. Inhibition of neutrophil chemotaxis by soluble bacterial products. J Periodontol. 1982; 53: 502–8. [PubMed Abstract].
- Van Dyke TE. Neutrophil receptor modulation in the pathogenesis of periodontal diseases. J Dent Res. 1984; 63: 452–4. [PubMed Abstract].
- Rotstein OD, Pruett TL, Fiegel VD, Nelson RD, Simmons RL. Succinic acid: a metabolic by-product of Bacteroides species, inhibits polymorphonuclear leukocyte function. Infect Immun. 1985; 48: 402–8. [PubMed Abstract] [PubMed CentralFull Text].
- Saito K, Takahashi N, Horiuchi H, Yamada T. Effects of glucose on formation of cytotoxic end-products and proteolytic activity of Prevotella interemedia, Prevotella nigrescens and Porphyromonas gingivalis. J Periodont Res. 2001; 36: 355–60. [PubMed Abstract].
- Vinolo MAR, Rodrigues HG, Hatanaka E, Sato FT, Campaio SC, Curi R. Suppressive effect of short-chain acids on production of proinflammatory mediators of neutrophils. J Nutr Biochem. 2011; 22: 849–55. doi: http://dx.doi.org/10.1016/j.jnutbio.2010.07.009 [PubMed Abstract].
- Shibata F, Konishi K, Kato H, Komorita N, al-Mokdad M, Fujioka M, etal. Recombinant production and biological properties of rat cytokine-induced neutrophil chemoattractants, GRO/CINC-2 alpha, CINC-2 beta and CINC-3. Eur J Biochem. 1995; 231: 306–11. [PubMed Abstract].
- Amano A, Shizukuishi S, Tsunemitsu A, Tsunasawa S. Identity of amino acid sequences of superoxide dismutase purified from both anaerobically maintained and aerated Porphyromonas gingivalis. Oral Microbiol Immunol. 1992; 7: 368–71. [PubMed Abstract].
- Maeda K, Hirofuji T, Chinju N, Tanigawa K, Iwamoto Y, Hatakeyama T, etal. The modulation of polymorphonuclear leukocyte function by Bacteroides gingivalis. Adv Dent Res. 1988; 2: 315–18. [PubMed Abstract].
- Novak MJ, Cohen HJ. Depolarization of polymorphonuclear leukocytes by Porphyromonas (Bacteroides) gingivalis 381 in the absence of respiratory burst activation. Infect Immun. 1991; 59: 3134–42. [PubMed Abstract] [PubMed CentralFull Text].
- Rotstein OD, Nasmith PE, Grinstein S. The Bacteroides by-product succinic acid inhibits neutrophil respiratory burst by reducing intracellular pH. Infect Immun. 1987; 55: 864–70. [PubMed Abstract] [PubMed CentralFull Text].
- Eftimiadi C, Buzzi E, Tonetti M, Buffa P, Buffa D, van Steenbergen MT, etal. Short-chain fatty acids produced by anaerobic bacteria alter the physiological responses of human neutrophils to chemotactic peptide. J Infect. 1987; 14: 43–53. [PubMed Abstract].
- Bielecka E, Scavenius C, Kantyka T, Jusko M, Mizgalska D, Szmigielski B, etal. Peptidyl arginine deiminase from Porphyromonas gingivalis abolishes anaphylatoxin C5a activity. J Biol Chem. 2014; 289: 32481–7. doi: http://dx.doi.org/10.1074/jbc.C114.617142 [PubMed Abstract] [PubMed CentralFull Text].
- Bostanci N, Thurnheer T, Aduse-Opoku J, Curtis MA, Zinkernagel AS, Belibasakis GN. Porphyromonas gingivalis regulates TREM-1 in human polymorphonuclear neutrophils via its gingipains. PLoS One. 2013; 8: e75784. doi: http://dx.doi.org/10.1371/journal.pone.0075784.
- Percival RS, Marsh PD, Devine DA, Rangarajan M, Aduse-Opoku J, Shepherd P, etal. Effect of temperature on growth, hemagglutination, and protease activity of Porphyromonas gingivalis. Infect Immun. 1999; 67: 1917–21. [PubMed Abstract] [PubMed CentralFull Text].
- Odell EW, Wu PJ. Susceptibility of Porphyromonas gingivalis and P. asaccharolytica to the non-oxidative killing mechanisms of human neutrophils. Archs Oral Biol. 1992; 37: 597–601.
- Eick S, Pfister W, Sigusch B, Straube E. Phagocytosis of periodontopathogenic bacteria by crevicular granulocytes is depressed in progressive periodontitis. Infection. 2000; 28: 301–4. [PubMed Abstract].
- Maekawa T, Krauss JL, Abe T, Jotwani R, Triantafilou M, Triantafilou K, etal. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe. 2014; 15: 768–78. doi: http://dx.doi.org/10.1016/j.chom.2014.05.012 [PubMed Abstract] [PubMed CentralFull Text].
- Yoneda M, Maeda K, Aono M. Suppression of bactericidal activity of human polymorphonuclear leukocytes by Bacteroides gingivalis. Infect Immun. 1990; 58: 406–11. [PubMed Abstract] [PubMed CentralFull Text].
- Wang H, Zhou H, Duan X, Jotwani R, Vuddaraju H, Liang S, etal. Porphyromonas gingivalis-induced reactive oxygen species activate JAK2 and regulate production of inflammatory cytokines through c-Jun. Infect Immun. 2014; 82: 4118–26. doi: http://dx.doi.org/10.1128/IAI.02000-14 [PubMed Abstract] [PubMed CentralFull Text].
- Guentsch A, Puklo M, Preshaw PM, Glockmann E, Pfister W, Potempa J, etal. Neutrophils in chronic and aggressive periodontitis in interaction with Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans. J Periodontal Res. 2009; 44: 368–77. doi: http://dx.doi.org/10.1111/j.1600-0765.2008.01113.x [PubMed Abstract] [PubMed CentralFull Text].
- Sztukowska M, Bugno M, Potempa J, Travis J, Kurtz DMJr.. Role of rubrerythrin in the oxidative stress response of Porphyromonas gingivalis. Mol Microbiol. 2002; 44: 479–88. [PubMed Abstract].
- Mydel P, Takahashi Y, Yumoto H, Sztukowska M, Kubica M, Gibson FCIII, etal. Roles of the host oxidative immune response and bacterial antioxidant rubrerythrin during Porphyromonas gingivalis infection. PLoS Pathog. 2006; 2: e76. doi: http://dx.doi.org/10.1371/journal.ppat.002007653.
- Hajishengallis G. Immune evasion strategies of Porphyromonas gingivalis. J Oral Biosci. 2011; 53: 233–40. doi: http://dx.doi.org/10.2330/joralbiosci.53.233 [PubMed Abstract] [PubMed CentralFull Text].
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement – a key system for immune surveillance and homeostasis. Nat Immunol. 2010; 11: 785–97. doi: http://dx.doi.org/10.1038/ni.1923 [PubMed Abstract] [PubMed CentralFull Text].
- Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P, etal. Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal. 2010; 3: ra11. doi: http://dx.doi.org/10.1126/scisignal.2000697 [PubMed CentralFull Text].
- Backlund CJ, Sergesketter AR, Offenbacher S, Schoenfisch MH. Antibacterial efficacy of exogenous nitric oxide on periodontal pathogens. J Dent Res. 2014; 93: 1089–94. doi: http://dx.doi.org/10.1177/0022034514529974 [PubMed Abstract] [PubMed CentralFull Text].
- Hajishengallis G, Lambris JD. Microbial manipulation of receptor crosstalk in innate immunity. Nat Rev Immunol. 2011; 11: 187–200. doi: http://dx.doi.org/10.1038/nri2918 [PubMed Abstract] [PubMed CentralFull Text].
- Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, etal. The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol. 2011; 186: 869–77. doi: http://dx.doi.org/10.4049/jimmunol.100325 [PubMed Abstract] [PubMed CentralFull Text].
- Burns E, Eliyahu T, Uematsu S, Akira S, Nussbaum G. TLR2-dependent inflammatory response to Porphyromonas gingivalis is MyD88 independent, whereas MyD88 is required to clear infection. J Immunol. 2010; 184: 1455–62. doi: http://dx.doi.org/10.4049/jimmunol.0900378 [PubMed Abstract].
- Hajishengallis G. The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol. 2014; 29: 248–57. [PubMed Abstract] [PubMed CentralFull Text].
- Hajishengallis G, Harokopakis E. Porphyromonas gingivalis interactions with complement receptor 3 (CR3): innate immunity or immune evasion?. Front Biosci. 2007; 12: 4547–57. [PubMed Abstract].
- Hajishengallis G, Shakhatreh M-AK, Wang M, Liang S. Complement receptor 3 blockade promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J Immunol. 2007; 179: 2359–67. [PubMed Abstract].
- Harokopakis E, Albzreh MH, Martin MH, Hajishengallis G. TLR2 transmodulates monocyte adhesion and transmigration via Rac1- and PI3K- mediated inside-out signaling in response to Porphyromonas gingivalis fimbriae. J Immunol. 2006; 176: 7645–56. [PubMed Abstract].
- Hiroi M, Shimojima T, Kashimata M, Miyata T, Takano H, Takahama M, etal. Inhibition by Porphyromonas gingivalis LPS of apoptosis induction in human peripheral blood polymorphonuclear leukocytes. Anticancer Res. 1998; 18: 3475–9. [PubMed Abstract].
- Preshaw PM, Schifferle RE, Walters JD. Porphyromonas gingivalis lipopolysaccharide delays human polymorphonuclear leukocyte apoptosis in vitro. J Periodontal Res. 1999; 34: 197–202. [PubMed Abstract].
- Murray DA, Wilton JMA. Lipopolysaccharide from the periodontal pathogen Porphyromonas gingivalis prevents apoptosis of HL60-derived neutrophils in vitro. Infect Immun. 2003; 71: 7232–5. [PubMed Abstract] [PubMed CentralFull Text].
- Zaric S, Shelburne C, Darveau R, Quinn DJ, Weldon S, Taggart CC, etal. Impaired immune tolerance to Porphyromonas gingivalis lipopolysaccharide promotes neutrophil migration and decreased apoptosis. Infect Immun. 2010; 78: 4151–6. doi: http://dx.doi.org/10.1128/IAI.00600-10 [PubMed Abstract] [PubMed CentralFull Text].
- Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med. 2015; 21: 172–83. doi: http://dx.doi.org/10.1016/j.molmed.2014.11.004 [PubMed Abstract] [PubMed CentralFull Text].
- Griffen AL, Lyons SR, Becker MR, Moeschberger ML, Leys EJ. Porphyromonas gingivalis strain variability and periodontitis. J Clin Microbiol. 1999; 37: 4028–33. [PubMed Abstract] [PubMed CentralFull Text].
- Amano A, Kuboniwa M, Nakagawa I, Akiyama S, Morisaki I, Hamada S. Prevalence of specific genotypes of Porphyromonas gingivalis fimA and periodontal health status. J Dent Res. 2000; 79: 1664–8. [PubMed Abstract].