Abstract
Recent evidence suggests potential, but unproven, links between dietary, metabolic, infective, and gastrointestinal factors and the behavioral exacerbations and remissions of autism spectrum disorders (ASDs). Propionic acid (PPA) and its related short-chain fatty acids (SCFAs) are fermentation products of ASD-associated bacteria (Clostridia, Bacteriodetes, Desulfovibrio). SCFAs represent a group of compounds derived from the host microbiome that are plausibly linked to ASDs and can induce widespread effects on gut, brain, and behavior. Intraventricular administration of PPA and SCFAs in rats induces abnormal motor movements, repetitive interests, electrographic changes, cognitive deficits, perseveration, and impaired social interactions. The brain tissue of PPA-treated rats shows a number of ASD-linked neurochemical changes, including innate neuroinflammation, increased oxidative stress, glutathione depletion, and altered phospholipid/acylcarnitine profiles. These directly or indirectly contribute to acquired mitochondrial dysfunction via impairment in carnitine-dependent pathways, consistent with findings in patients with ASDs. Of note, common antibiotics may impair carnitine-dependent processes by altering gut flora favoring PPA-producing bacteria and by directly inhibiting carnitine transport across the gut. Human populations that are partial metabolizers of PPA are more common than previously thought. PPA has further bioactive effects on neurotransmitter systems, intracellular acidification/calcium release, fatty acid metabolism, gap junction gating, immune function, and alteration of gene expression that warrant further exploration. These findings are consistent with the symptoms and proposed underlying mechanisms of ASDs and support the use of PPA infusions in rats as a valid animal model of the condition. Collectively, this offers further support that gut-derived factors, such as dietary or enteric bacterially produced SCFAs, may be plausible environmental agents that can trigger ASDs or ASD-related behaviors and deserve further exploration in basic science, agriculture, and clinical medicine.
Autism spectrum disorders (ASDs) are a family of neurodevelopmental disorders of rapidly increasing incidence Citation1 that are characterized by impairments in communication and social interaction along with restrictive and repetitive behaviors.
The brain tissue of patients with autism shows subtle developmental abnormalities, specifically in those areas concerned with language, facial expression, movement, and social behavior Citation2. Individuals with autism may show enlarged brain size in the first few years of life, with altered migration of cortical, amygdalar, and cranial nerve motor neurons, as well as cerebellar neurons Citation3. Cell counts have shown that compared with controls, brain samples of patients with autism contain smaller neurons with increased cell density in cortical, limbic, and cerebellar regions Citation4, possibly because of altered neurogenesis Citation5, apoptosis Citation6, neural cytoarchitecture Citation7 Citation8, or a combination of these factors. There is a growing interest in examining ASDs as a disorder of glial cell function. Glial cells are of great importance in both the developing and mature nervous system, particularly with respect to cell–cell interactions during neural migration, and synaptic plasticity Citation9. Glia form a functional syncytium necessary for the maintenance of a stable neural microenvironment, especially during periods of increased metabolic stress Citation10. Glial abnormalities may manifest as an overall increase in white matter thickness Citation11, increased thickness of the external capsule and increased water content in the white matter Citation12. These observed abnormalities in brain morphology are accompanied by increased CNS immune activity, including increases in reactive astrocytes and activated microglia in brains of patients with ASD, as well as the elevation of proinflammatory cytokines in the cerebral spinal fluid. These findings have been demonstrated in both young and older patients, suggesting that an inflammatory process may be present throughout the life span of individuals with autism Citation13. Similar findings of heightened immune activity (i.e. increased Th2 cytokine levels) have also been demonstrated in peripheral blood monocytes from patients with ASDs Citation14.
To date, the majority of research has focused on genetic causes of ASD, mainly on developmental abnormalities in synaptic organization, neuromigration, and neurotransmission Citation15. However, the findings that genetic syndromes appear to account for only 6–15% of ASD cases, coupled with the observation of discordance of severity among monozygotic twins Citation16, indicate a solely genetic cause to be unlikely. An alternative approach is to examine ASDs as a whole body condition, with many comorbidities involving immune, metabolic, and gastrointestinal abnormalities, that may have a variable symptomatic course Citation17–Citation22. Recent studies show widespread dysfunction in immune regulation, detoxification, environmental exposures, redox regulation/oxidative stress, and energy generation Citation19–Citation22 that affect many organ systems, including the brain. Furthermore, the recent findings of genetic mutations, such as the MET receptor tyrosine kinase and protein kinase Cβ genes, involved in brain development, immune function, and ability to recover from gastrointestinal insults Citation23 Citation24, and the X-linked 6-N-trimethyllysine dioxygenase (TMLHE) gene, involved in carnitine/fatty acid metabolism Citation25, raise the strong possibility that genetic sensitivities to environmental factors may underlie the pathophysiology of the disorder in at least a subset of patients.
Is autism an acquired disorder of mitochondrial dysfunction?
Disorders of mitochondrial function, and their heterogeneous expression in different tissues or within families, fulfill most of the criteria for widespread multiorgan dysfunction and sensitivity to many diseases Citation26 Citation27. Classic mitochondrial disease is overrepresented in patients with ASDs, encompassing 5% of the ASD population Citation28. More intriguingly, an extensive meta-analysis by Rossignol and Frye also found that about 30% of children in the general ASD population exhibit biomarkers consistent with mitochondrial disease Citation22, including a relative carnitine deficiency and altered lactate/pyruvate ratios. Furthermore, a recent study reported that 80% of the children with ASDs showed altered electron transport chain in lymphocytes compared with neurotypic controls Citation29. Mitochondria are central to this theme as polymorphisms in mitochondrial genes, which can be inherited or acquired from early prenatal environmental insults, can result in altered susceptibility to many diseases Citation26 Citation27. Similarly, abnormalities associated with ASDs such as glutathione deficiency Citation30, increased oxidative stress, elevated concentrations of TNF-α Citation31–Citation33, and methylation abnormalities could further impair mitochondrial function. The fact that classic mitochondrial disorder is overrepresented but does not account for the high occurrence of mitochondrial biomarker alterations in this population may indicate that mitochondrial dysfunction in ASDs may be at least partly environmentally acquired. This is important in light of a rapidly growing incidence of ASDs Citation1 and the growing consensus that the systemic abnormalities seen in ASDs may arise from environmental triggers Citation34 in genetically sensitive subpopulations Citation35 Citation36.
Mitochondrial dysfunction can result from a broad assortment of environmental exposures that have been implicated in the development of ASDs. Possible agents, which are not mutually exclusive, include heavy metals Citation37–Citation40, chemicals Citation41, polychlorinated biphenyls Citation42, pesticides Citation43–Citation45, and finally enteric metabolic products of ASD-associated intestinal bacteria Citation46–Citation53.
The ‘inner’ outer environment, the enteric microbiome as a source of environmental triggers of ASDs
The human digestive tract is host to a complex array of intestinal bacterial florae, coined the microbiome, which outnumber host cells at least 10 to 1. The microbiome produces an array of bioactive metabolic products capable of entering systemic circulation. It is important to note that the enteric microbiome and its metabolic products are not static and can be altered throughout the life cycle of the individual, particularly throughout the first 18 months of life Citation54. The metabolic products from the gut microbiome can have profound and dynamic effects on host metabolism, immune function, and gene expression in many organ systems, including the CNS Citation46 Citation55–Citation60. In addition, it is also important to consider the recent impact of the alteration of the human microbiome and its metabolites through the introduction of the high calorie Western diet, coupled with the high exposure of antibiotics and disinfectants to human beings, animals, and plants, as a possible source of environmental triggers of many diseases of increasing incidence Citation59, including ASDs. This is particularly evident from human populations migrating to Western societies, such as the Somali diaspora, who appear to have a much higher incidence of ASDs than in their country of origin Citation61.
Given the high number of reports of antibiotic exposure, hospitalization, and gastrointestinal disturbances Citation62–Citation64 in many patients with ASDs, our laboratory has been examining the neurobiological effects of microbiota-produced short-chain fatty acids (SCFAs), such as propionic acid (PPA) Citation46–Citation53. PPA is a fermentation product of many bacteria, including some enteric species (i.e. Clostridia, Desulfovibrio, and Bacteroidetes) overexpressed in stool samples from patients with ASDs Citation65 Citation66. PPA levels are also elevated in the stool samples of patients with ASDs Citation67. This has led us to propose PPA as a possible environmental trigger of ASDs. We have found that when administered to rodents, brief intracerebroventricular (ICV) infusions of PPA and its related enteric SCFAs produce many behavioral, electrographic, neuropathological, and biochemical changes, consistent with an animal model of ASDs.
Neurobiological effects of enteric SCFAs
PPA is found in the gut, along with other SCFAs, such as acetate and butyrate, each of which are major metabolic products of enteric bacteria, following fermentation of dietary carbohydrates and some amino acids Citation58 Citation68 Citation69. PPA is also produced by Propionibacteria present on the skin, known to cause acne Citation70, and many bacteria present in the oral mucosa, responsible for gingival inflammation Citation71 Citation72. PPA is well known in animal husbandry, where it is the main metabolic product in ruminant cellulose digestion Citation73, may be a sex pheromone Citation74, and is involved in lactation Citation75. It is also naturally present in a variety of foodstuffs (i.e. cheese) Citation73 and is commonly used as a preservative (antifungal) in many processed foods, particularly in refined wheat and dairy products Citation76. However, the majority of PPA is produced in the gut lumen by intestinal bacteria. PPA, being a weak organic acid, exists in ionized and non-ionized forms at physiological pH, thus allowing it to readily cross the gut–blood barrier, and is principally metabolized in liver. It also crosses the blood–brain barrier and enters the CNS Citation77. In addition, PPA gains access to the CNS via monocarboxylate transporter uptake in the gut lumen and cerebrovascular endothelium, which actively transport many carboxylic acids, particularly PPA and ketones. PPA is also a specific ligand of many G-coupled SCFA receptors (GPR41, Citation43) Citation78–Citation80. PPA and other SCFAs are taken up by glia and, to a lesser extent, neurons once they enter the CNS Citation81 Citation82, where they are thought to comprise a major energy source in cellular metabolism, particularly during early brain development Citation81–Citation83. PPA and other SCFAs (i.e. butyrate) affect diverse physiological processes such as cell signaling Citation84, neurotransmitter synthesis and release Citation85, free radical production, mitochondrial function, Citation86 lipid metabolism Citation87, immune function Citation88, gap junction gating, intracellular pH maintenance Citation89, and modulation of gene expression through phosphorylation and histone acetylation Citation90.
Although PPA may be beneficial at appropriate levels, such as improving insulin sensitivity, lowering cholesterol, and reducing food intake Citation69, excessive PPA may have many negative effects on health and behavior. For example, a number of inherited and acquired conditions, such as propionic/methylmalonic acidemia, biotinidase/holocarboxylase deficiency, ethanol/valproate exposure, and, as discussed, mitochondrial disorders, are all known to result from elevations of PPA and other SCFAs, partly through the formation of propionyl coenzyme A (CoA) and sequestration of carnitine Citation20 Citation46 Citation51 Citation91. Collectively, these conditions present at varying ages with developmental delay and regression, seizure/movement disorder, metabolic acidosis, and gastrointestinal symptoms, which also change in severity with fluctuating PPA levels and markers of mitochondrial dysfunction that are somewhat reminiscent of ASDs Citation86 Citation92 Citation93.
Propionic acidemia is caused by deficient activity of either one of two non-identical subunits of the biotin-dependent enzyme propionyl CoA carboxylase Citation94. This mitochondrial enzyme is responsible for the breakdown of PPA and other SCFAs, as well as a number of amino acids. The disorder may be 10 times more common than previously reported, and there are multiple mutations and variable metabolizers in many populations Citation95 Citation96.
Patients with propionic acidemia clinically present with life-threatening illness during the neonatal period, characterized by vomiting, severe metabolic acidosis, and hyperammonemia. Neurological symptoms include developmental delay, seizure, choreathetoid movements, and dystonia. Interestingly, some other patients, including identical twins of more severely affected siblings, may present later in life with varying severities of the disorder, often without measurable increases in blood or urine PPA or metabolites. Treatment includes reduction of carbohydrate and protein contents in the diet, eradication of PPA-producing bacteria, and carnitine supplementation to improve PPA clearance, which have some benefit Citation46 Citation86 Citation92 Citation93.
Thus, PPA and its related SCFAs have broad effects on cellular systems, providing a potential mechanism where metabolic end products of the enteric microbiome can alter host physiology and behavior Citation46 Citation60. This and other evidence led us to propose that increased PPA exposure at key neurodevelopmental periods is a major environmental trigger of the brain and behavioral changes observed in ASDs.
How are dietary and gastrointestinal factors related to autism? – microbiome production of enteric fatty acids as environmental triggers
There is a growing interest suggesting a link between dietary and/or gastrointestinal factors and the worsening and, in some cases, improvement of ASD symptoms, but the mechanisms responsible for this remain elusive. As indicated, SCFAs, such as PPA, are produced by many gut bacteria by the breakdown of dietary carbohydrates and amino acids, particularly from wheat products Citation97. Of particular relevance are the Clostridia and Desulfovibrio, which have been proposed as infectious causes of ASDs Citation64 Citation66. Clostridial species, a family of heterogeneous anaerobic, spore-forming, gram-positive rods Citation98, are major gut colonizers in early life and are producers of PPA.
Clostridia as spore formers are particularly resistant to most antibiotics used for routine perinatal and early childhood infections, and some species are a cause of major hospital- and, recently, community-acquired communicable disease (i.e. C. difficile-induced colitis) Citation98. Interestingly, spore-forming anaerobes and microerophilic bacteria, particularly from clostridial species, have been shown to be elevated in patients with ASDs Citation66 Citation99 Citation100. Recently, Finegold isolated distinct species of Desulfovibrio, a gram-negative, aerotolerant, non-spore former, from the stool of patients with ASDs, and, to a lesser extent, non-affected siblings. Interestingly, in addition to producing PPA following fermentation of peptones, Desulfovibrio is resistant to most common antibiotics and produces the gasotransmitter and potential mitochondrial toxin, hydrogen sulfide Citation101–Citation103. He has suggested eradication of these organisms with oritavancin and aztreonam as a possible treatment of ASDs Citation64. Furthermore, ASDs may show comorbidity with a variety of gastrointestinal disorders, such as alterations in gut motility, intestinal lesions and increased intestinal permeability, bacterial dysbiosis, impaired carbohydrate digestion/absorption, reflux esophagitis, and ileal hyperplasia Citation46 Citation104 Citation105. An association between long-term antibiotic use, hospitalization, abdominal discomfort and the onset of ASD symptoms after normal or near-normal development has also been reported Citation62 Citation63 Citation100 Citation106. These findings raise the possibility that gut-born factors secondary to alteration of the gut microbiome by antibiotics or diet may influence brain function and symptomology in patients with ASDs. Moreover, a compromised gut–blood barrier (i.e. an acquired colitis) or impaired colonocyte energy metabolism Citation102, which use SCFAs as an energy substrate and act as a metabolic ‘sink’, may allow for greater systemic and CNS access for such compounds Citation59 Citation107.
PPA is also known to have a number of direct effects on gastrointestinal physiology. As reviewed in the study by MacFabe et al. Citation46, PPA increases contraction of colonic smooth muscle, dilates colonic arteries, activates mast cells, increases the release of serotonin from gut enterochromaffin cells, and reduces gastric motility and increases the frequency of contractions, reminiscent of the clinical observations of gut dysmotility in patients with ASD. PPA levels increase in infant colonic contents postnatally and following formula feeding opposed to breast feeding Citation54. Interestingly, intracolonic infusions of PPA, mimicking bacterial overgrowth, can induce an experimental colitis in infant rats but not later in life Citation108. However, ascertaining PPA production in vivo is difficult, as secondary to both passive and active diffusion of SCFAs from the gut lumen, and intracellular concentration levels in stool are often a poor indicator of SCFA production by gut bacteria and may change rapidly with diet Citation58.
Taken together, PPA appears to circumstantially possess the necessary properties to interfere with gastrointestinal activity in a manner similar to the abnormalities observed in ASDs. In support of this, reports from parents of children with ASDs suggest that behavioral and gastrointestinal symptoms increase when their children ingest refined food products that provide high carbohydrates for bacterial fermentation to produce PPA or also contain PPA as a preservative. Furthermore, behaviors and gut symptoms improve following the elimination of these products from the diet Citation17 Citation106 or the eradication of PPA-producing bacteria by broad-spectrum antibiotics Citation109. Moreover, symptom exacerbation associated with propionic acidemia and its related conditions bears some resemblance to that reported in ASDs Citation86 Citation92 Citation93. Stool Citation67 and serum studies from patients with ASDs provide further evidence linking PPA to the condition, as patients with ASDs have metabolic dysfunction, including impairments in B12, glutathione, or carnitine metabolism Citation110 Citation111 and mitochondrial disorder/dysfunction Citation20, which are consistent with the effects of PPA on cellular metabolism Citation46 Citation51 Citation53.
The PPA rat model of ASDs – behavioral and electrographic findings
Animal models allow for the examination of factors involved in human disorders, such as ASDs, using experiments that cannot be conducted in humans. The development of such models is essential and permits the experimental examination of the effects of suspected environmental agents on the pathophysiology and core symptoms and comorbidities of ASDs. There have been a number of valid animal models of ASDs, concentrating on genetic knockouts of key neurodevelopmental processes Citation112 Citation113 and also on exposures to environmental factors such as metals, drugs (valproic acid, terbutaline) Citation114 Citation115, viruses Citation116 and inflammatory agents (i.e. lipopolysaccharide, interleukin-6) Citation117. Each of these models causes some, but not all, behavioral or pathological changes reminiscent of ASDs. Therefore, to investigate the hypothesis that that elevated levels of PPA can induce bouts of behavioral, electrophysiological, neuropathological, and biochemical effects similar to those observed in ASDs, our initial research was concentrated on exposure of adult rats to brief pulsed ICV infusions of PPA and its related enteric SCFAs (i.e. acetate, butyrate), through chronic indwelling brain cannula. CNS exposures were initially done to test for a central, and not peripheral, action of these compounds, as it has been argued that the behaviors, dietary/gut symptoms, and metabolic findings in ASDs are solely due to responses to gastrointestinal pain, poor diet, or obsessional eating behavior and not a direct effect of enteric compounds on the function of CNS.
Impairments in social behavior, including abnormal play behaviors and other forms of social contact, are among the most prominent symptoms of ASDs Citation49 Citation118. Likewise, individuals with ASDs commonly suffer from various forms and degrees of cognitive impairment, including learning disabilities, restricted interests favoring objects vs. social interactions, and ‘rigid’ perseverative behavior, and insistence on ‘sameness’, and rituals Citation18 Citation119. Patients with ASDs often experience motor-related symptoms, such as hyperactivity, gait disturbances, and stereotyped movements, possibly grouping ASDs with the movement disorders Citation120 Citation121. There is increased incidence of seizure disorder in ASDs and other conditions associated with elevated levels of SCFAs Citation92 Citation122 Citation123. We thus tested the validity of the PPA model based on the above behavioral and electrographic criteria.
Pulsed ICV infusions of PPA (4 µl of 0.052–0.26 M solution at pH 7.5 over a 1 min period) or control compounds (i.e. isomolar, acetate, butryate and propanol - the non-acidic alcohol analogue of PPA) were performed over a number of time courses (once weekly×5 weeks, once a day×5 day, twice daily for 7–14 days) into the cerebrospinal fluid of adult rats via chronic brain indwelling canullae ().
Fig. 1. Behavioral videos of propionic acid infusions in rats (click headings to view videos). Single intracerebroventricular (ICV) infusions (4 µl of 0.26 M solution over 4 min) of propionic acid (PPA), a metabolic end product of autism-associated enteric bacteria, produce bouts of reversible hyperactive and repetitive behavior (A) in adult rats, compared with phosphate-buffered saline (PBS) vehicle infused control rat (B). Rat pairs infused with PPA show markedly reduced social interaction and play behavior (C), compared with pairs of rats infused with PBS vehicle (D), which show typical social behavior. Ethovision behavioral tracking of control and PPA-treated rat pairs (E), showing further evidence of PPA-induced hyperactive, repetitive and antisocial behavior. PPA-treated rat displays fixation on objects (F) and a specific object preferences (i.e. block vs. sphere). PPA-infused rats also show turning, tics, dystonia, and retropulsion and electrographic evidence of complex partial seizures and basal ganglial spiking, consistent with findings in patients with autism spectrum disorders.
A – PPA repetitive behavior
B – control rat
C – PPA social
D – control pair social
E – Ethovision pair
F –PPA object fixation

PPA-infused rats showed bouts of increased repetitive locomotor activity, turning, retropulsion, tics, social impairment, perseveration, and restrictive preference for objects versus novel rats Citation46–Citation53 (). These behaviors were rapidly induced within 1–2 min after single infusions, but were transient, lasting approximately 20–30 min, consistent with levels in bloods of propionic acidemia patients, and the half-life of PPA Citation124.
Fig. 2. (A) Intracebroventricular (ICV) infusions of propionic acid (PPA) in adult rats increase repetitive locomotor activity (Versamax automated behavioral assay). Group mean values (±SEM) total distance, number of movements, and movement time in rats given 4 µl ICV infusions of phosphate-buffered saline (PBS) vehicle or PPA (0.26 M) twice daily for 7 days. BL = baseline session with no infusion; T1–T7 consecutive treatment days. *p<0.05. (B) Single ICV infusions (4 µl of 0.26 M solution over min) of PPA and isomolar acetic acid (SA) in adult rat pairs reduce social behavior (Ethovision automated behavioral assay). Behavior is measured as mean distance apart (cm) and time spent (%) in 5 cm proximity during dark hours. Data points represent group means of data collected during 10 min periods. Animals were placed in a large open field in same-drug pairs. Both PPA and SA pairs displayed a significantly greater mean distance apart and spent significantly less time in close proximity to each other, consistent with impaired social behavior found in autism, compared with phosphate-buffered saline (PBS) vehicle controls. Propanol the non-acidic alcohol analogue of PPA was without effect. *=different from PBS control group at p<0.05 or better. (C) Tracks representing object and socially related behavioral movement of adolescent rats receiving ICV infusions of PPA (4 µl of 0.26 M solution over 4 min) or PBS. More locomotion near the caged novel rat (NR) occurred in PBS rats than by PPA rats. Graphic representation of duration of time (s) spent in close proximity (within 18 cm) of the novel rat or novel object (NO). PPA rats showed less approach behavior and remain close to the novel rat less than PBS rats, indicative of object preference over social behavior, consistent with findings in patients with autism. Graphic representation of group mean (±SEM) percent correct turns in the T-maze task during acquisition (Days 1 and 2) and reversal (Days 3 and 4). + = significant difference from Day 2 performance; * = significant different from PBS control group. Figures modified with permission from MacFabe et al. (2008) and (2011), and Shultz et al. (2008). A reproduced with permission from Science (American Association for the Advancement of Science). B and reproduced with permission from Elsevier Ltd.
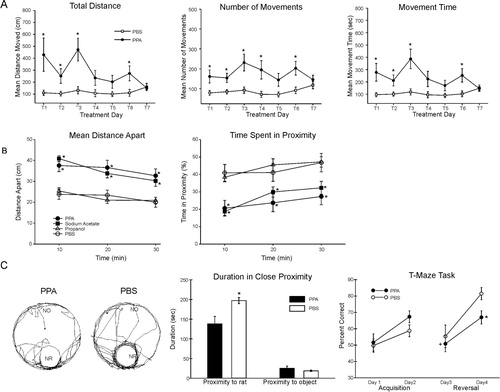
Interestingly, it was found that during the acquisition session in the Morris water maze and T maze, tests for visuospatial learning, the PPA-treated rats displayed mild or no impairment in maze acquisition but marked impairments following maze reversal Citation49 Citation50. In the case of the Morris water maze, when the location of the target platform was moved for the reversal session, the PPA-treated rats displayed marked cognitive impairments, often swimming toward the former location of the platform, consistent with ‘rigid’ perseverative behavior, which could be considered a deficit in ‘un-learning’.
Using rats with stereotactically implanted indwelling cortical, hippocampal, and subcortical brain electrodes, repeated PPA exposures (once weekly×5 weeks) produced a kindling response in hippocampal/neocortical leads and complex-partial-seizure-like behavior, while tics, retropulsion, and dystonia were accompanied by sharp spiking in the basal ganglia. Infusions of butyrate and acetate produced similar, but less pronounced effects, while infusions of propanol were without effect Citation46 (). Current studies have found similar behaviors at PPA doses 1/20th of initial investigations (unpublished observations).
Fig. 3. (I) Intracerebroventricular (ICV) propionic acid (PPA) infusions in rats induce electrographic and behavioral elicitation of kindled seizures and movement disorder. Kindled seizure manifestation in response to repeated weekly 4 µl ICV infusions of low (0.052 M) or high (0.26 M) PPA, isomolar acetic acid (SA), propanol, or phosphate-buffered saline vehicle (PBS) in adult rats: group mean (±SEM) data summed across the five initial testing sessions. Latency to epileptiform spiking was measured from the end of the ICV infusion to the start of seizure. Maximum convulsion stage was rated using the Racine kindling scale. Duration of the longest convulsion was of the longest continuous convulsion during a session. PPA and, to a lesser extent, SA induce a kindling response, while propanol, the non-acidic analogue of PPA, was without effect. * = different from propanol and PBS controls; # = different from low PPA, propanol control, and PBS control; + = different from propanol control; ** = different from all other groups. (II) Abnormal behaviors in response to ICV infusions: group mean frequency (±SEM) of behaviors per baseline or initial test session. Either one or both doses of PPA increased the abnormal behaviors relative to control treatments. Only PPA produced dystonic (snake like), retropulsive behaviors. With the exception of sodium acetate, which increased turning, no control treatments increased abnormal behavior. Black bars indicate treated animals; white bars indicate PBS (vehicle)-treated animals. * = p < 0.05 or better vs. all control groups; # = p < 0.05 or better vs. low PPA, propanol, and PBS control. (III) Representative electrographic seizure records from rat in the high PPA group. (A) Session 2, short bout of epileptiform spiking accompanied by contralateral hindlimb dystonia coincident with spiking (event marker). Note spiking in frontal cortex and caudate but not dorsal hippocampus. (B) Session 3, single epileptiform spikes in caudate and frontal cortex but not hippocampus. Only the caudate spikes were accompanied by brief contralateral hindlimb dystonia (event markers), which led the frontal cortex spikes by approximately 500 ms. A prominent frontal cortex spike (arrow) is not accompanied by a spike in the caudate or by limb dystonia. (C) Session 5, bout of spiking in dorsal hippocampus not accompanied by corresponding bouts of spiking in frontal cortex or caudate or by limb dystonia. (D) Session 5, single epileptiform spikes occur first in the caudate and lead spikes in other traces. These are followed by a short bout of epileptiform spiking that is accompanied by brief retropulsion, followed immediately by contralateral hindlimb dystonia, which ends coincident with the end of the bout of spiking (event marker). (E) Session 5 at approximately 1 min after the records in D, single epileptiform spikes in all three traces, with the caudate spikes leading the spikes in the other traces. Each spike is accompanied by brief contralateral hindlimb adduction and immediate dystonia (event markers). (F) Session 5 at approximately 6 min after the records in E, short bout of epileptiform spiking with hindlimb dystonia beginning coincident with caudate spiking (event marker), with frontal cortex and hippocampal spiking beginning after onset of caudate spiking and hindlimb dystonia. This is followed 2 s later by the beginning of the first sustained kindled seizure displayed by this rat (duration = 35s), which was accompanied by the first conventional kindled convulsion (Stage 2), which did not include limb dystonia. C = caudate; FC = frontal cortex; H = dorsal hippocampus; HD = hindlimb dystonia. Amplitude calibration = 50 µv; time marker = 5 s. Figures modified with permission from MacFabe et al. (2007). Reproduced with permission from Elsevier Ltd.
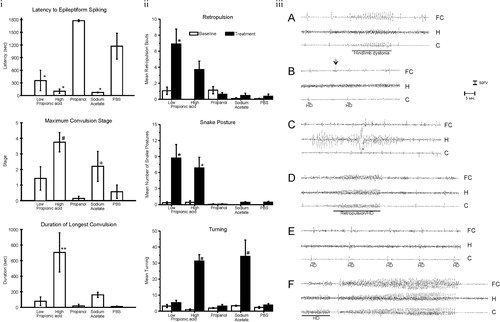
Thus, ICV infusions of PPA and, to a lesser extent, other SCFAs rapidly produce a number of striking behaviors that resemble those seen in patients with ASDs. In addition, many of these behaviors appear to reverse, consistent with the metabolic breakdown of PPA.
Potential underlying mechanisms of PPA and their relation to autism – neurotransmitters
PPA and its related SCFAs are capable of gaining access to the brain and inducing widespread effects on CNS function Citation77, including neurotransmitter synthesis and release, calcium influx, intracellular pH maintenance, lipid metabolism, mitochondrial function, gap-junction-dependent intercellular gating, immune activation, and gene expression, which we have proposed may contribute to the behaviors and biochemical findings observed in the PPA animal model and ASDs Citation46.
Of interest, PPA is capable of altering dopamine, serotonin, and glutamate systems in a manner similar to that observed in ASDs Citation125–Citation130, partly via potentiating intracellular calcium release Citation77 Citation131 Citation132. Furthermore, and of importance to the symptoms observed in both ASDs and the PPA model, similar alterations to the serotonin and dopamine systems have been implicated in abnormal social and motor behaviors Citation127–Citation129. SCFAs, including PPA, may increase synthesis of dopamine and its related catecholamines through induction of tyrosine hydroxylase, a key enzyme in the synthesis of catecholamines Citation133. This may also occur peripherally in the adrenals and sympathetic ganglia, where they may contribute to the enhanced anxiety-like behavior and increased sympathetic tone and cardiovascular instability found in ASDs Citation134.
PPA and other SCFAs are also known to potentiate glutamatergic transmission, inhibit GABAergic transmission and increase the production of enkephalin Citation46 Citation130 Citation135, supportive of a potential mechanism for the enhanced excitation/reduced inhibition theory of ASDs Citation136. PPA is also known to modulate neurofilament phosphorylation/dephosporylation, important in neurodevelopment and neuroplasticity Citation137 Citation138.
PPA-induced intracellular acidification, increased oxidative stress, impaired antioxidant capacity, and gap junction closure
The findings that PPA and its related fatty acids often induce similar behavioral effects, whereas isomolar infusions of propanol do not Citation46 Citation49 Citation50, make it tempting to speculate that the carboxylate functional group and/or acidic properties of PPA may be involved in its effects on behavior. Once low-molecular-weight organic acids are protonated in acidic conditions, they become more lipid soluble and, thus, gain access to the CNS, either by passive diffusion or active transport Citation77 Citation78 Citation80. Once they enter the CNS, they may concentrate within cells and induce intracellular acidification. Previous studies have found that PPA- and acetate-related acidosis in rats can alter social behavior Citation139. Furthermore, clinical metabolic acidosis from varied etiologies in humans often involves bouts of confusion and movement disorder similar in some respects to those found in ASDs Citation140. The effects of varying intracellular pH on cellular physiology are broad. Notably, PPA-induced intracellular acidification can inhibit mitochondrial function, which is vital for normal metabolism of other fatty acids Citation141. Intracellular accumulation of organic acids creates a buildup of acyl-coenzyme, which interrupts metabolism Citation141. PPA sequesters carnitine function and inhibits CoA function and, thus, impairs mitochondrial metabolism and energy production, leading to further decrease in cytoplasmic pH via the accumulation of other organic acids Citation131 Citation141. This is consistent with the impaired mitochondrial metabolism in ASDs, including carnitine deficiency, mitochondrial dysfunction, and systemic elevations of nitric oxide metabolites Citation20 Citation46 Citation142. In addition, valproate, an antiepileptic drug, structurally related to PPA, and a known prenatal risk factor for ASDs Citation143, similarly alters mitochondrial metabolism and causes the depletion of carnitine stores and encephalopathy Citation144 Citation145. Nitropropionic acid, another PPA derivative, is a potent inhibitor of mitochondrial function and produces a model of Huntington chorea when administered to rodents Citation146. The basal ganglia may be particularly sensitive to PPA and its derivatives, because of the region's high metabolic demand and damage in many movement disorders, including organic acidemias and mitochondrial disorders, thus offering an explanation of the tics, dystonias, and repetitive behaviors in these conditions and the PPA rodent model Citation46 Citation86.
An impaired mitochondrial metabolic process similar to the one described could lead to a range of negative effects on the CNS. Of relevance, similar encephalopathic processes associated with increased oxidative stress, such as those found in organic acidemias, are known to produce symptoms consistent with findings in the PPA model and ASDs Citation86 Citation93 Citation147. Consistent with this hypothesis, biochemical analyses of homogenates of the brain sample from PPA-treated rats (4 µl of 0.26 M solution BID×7 days, ICV) demonstrated an increase in oxidative stress markers (protein carbonylation and lipoperoxidation), as well as abnormalities in glutathione-associated pathways, particularly in brain regions implicated in ASDs Citation46 Citation47 Citation148 ().
Fig. 4. (A) Propionic acid (PPA) infusions in rats induces a significant increase in lipid and protein oxidation, supportive of increased oxidative stress via increased oxidant production and decreased antioxidant defenses in homogenates of discrete regions of rodent brain consistent with findings in patients with autism. Interestingly, brain stem appears relatively unaffected. (B) PPA-treated rats showed significantly decreased total GSH, a decreased activity of GPx, but an increase in the activity of GST. Conversely, the activity of GR was unchanged. These findings suggest that GSH may be involved in the metabolic clearance of PPA, or alternatively, the synthesis of GSH was impaired by PPA. The observed reduction of the activity of GPx may be due to the decreased availability of GSH, a cofactor of GPx or may reflect an overall reduction in antioxidant defenses induced by PPA. Black bars indicate PPA-treated animals; white bars indicate PBS (vehicle)-treated animals. Figures modified with permission from MacFabe et al. (2008). Reproduced with permission from Science (American Association for the Advancement of Science).
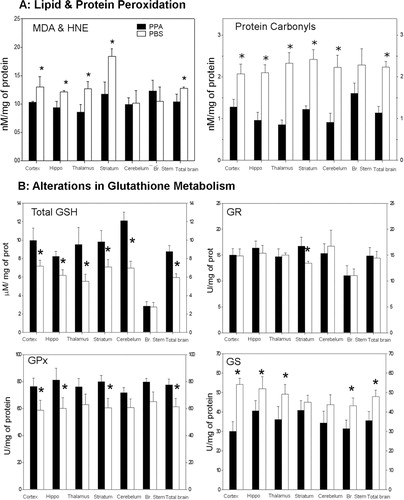
Glutathione participates in both antioxidant defense and xenobiotic detoxification over a broad range of environmental organic compounds and metals Citation149 Citation150, including those implicated in ASDs. Impairments in glutathione-associated pathways suggest reduced cellular defense and are considered markers of increased oxidative stress Citation149. Of particular interest is the evidence of genetic and acquired impairments in glutathione-associated pathways in patients with ASDs Citation150–Citation152, suggesting a plausible mechanism for altered sensitivity to a wide range of environmental agents (metals, pesticides, drugs) proposed in ASDs Citation46 Citation47. Of note, acetaminophen, when given for common pediatric illnesses, may overwhelm glutathione metabolism and has been proposed as a possible trigger for ASDs Citation153–Citation155. Furthermore, even brief exposures to agents that alter redox levels in cells early in development may change cellular developmental trajectory and ultimate cell fate, which may provide a plausible mechanism for neurodevelopmental alterations in ASDs Citation156. Overall, PPA-induced metabolic abnormalities and oxidative stress are consistent with findings from ASDs and ASD-related disorders.
Further to its capability to decrease intracellular pH, both directly via intracellular concentration and indirectly via inhibition of mitochondrial functions, we have proposed that a major effect of PPA is through the closure of gap junctions via intracellular acidification Citation46 Citation50 Citation157. As reviewed in MacFabe et al. Citation46, gap junctions are intercellular channels composed of connexin proteins that are gated by a number of factors influenced upon by PPA, including dopamine, calcium, and cytokines. Gap junctions play a major role in cellular differentiation, and in particular, peripheral nerve, cardiac, uterine, and gastrointestinal function. However, in the CNS, gap junction coupling is vital for the synchronization of neural electrical activity within discrete functional cell groups and is more extensive during early brain development and neuronal migration. Astrocytes are electrotonically connected by gap junctions, forming a syncytium to spatially buffer calcium, glutamate, and potassium Citation158, and apoptotic factors are capable of passing through these glial gap junctions Citation159 Citation160. Thus, closed glial gap junctions may render neurons hyperexcitable to rising extracellular potassium and glutamate Citation160, while closed neuronal gap junctions would be neuroprotective Citation159. In turn, this decrease in gap junction coupling may lead to inhibited cortical pruning in development, consistent with the larger brain size found in ASDs Citation46. Gap junction communication is involved in neurotransmission in areas that are implicated in seizure and movement disorders, such as the basal ganglia, prefrontal cortex, nucleus accumbens, and hippocampus. Intrastriatal injections of gap junction blockers produce stereotypical movements, hyperlocomotion, and disruption of motor sequencing in rodents Citation127 Citation161. Furthermore, gap junction knockout mice show abnormal brain development, exaggerated responses to neurotoxic insults, seizure disorder, and abnormal behaviors Citation162. Interestingly, gap junction blockers also inhibit tight junctions in many cellular systems Citation163, thereby possibly contributing to altered barrier function in vascular endothelium and gut in ASDs Citation107. Given these findings, it seems possible that PPA-induced alterations to gap junction function may contribute to electrophysiologic and motor impairments observed in both the PPA model and ASDs, but also in neural development, as well as systemic effects (i.e. gastrointestinal motility). These potential mechanisms are the subject of further study in our group.
However it is unclear at this stage whether these effects on pH and oxidative stress are causal to behavioral induction or the results of PPA dependent mechanisms such as neuroinflammation or altered lipid metabolism. Quite possibly they are not necessarily exclusive and may be mutually reinforcing, leading to a vicious cycle of these processes.
Neuropathological effects of PPA infusions on inflammation/CREB induction/epigenetics/lipid transport
We have performed a number of studies examining the neurohistological effects of PPA and its related fatty acids on innate neuroinflammatory processes Citation46–Citation50. Immunohistochemical examinations of the brain tissue from PPA-treated rats (1–14 days), demonstrated by DAB immunostaining and semiquantitative image densitometry, show a neuroinflammatory response, characterized by increased activated microglia and reactive astrogliosis in some brain areas, including the hippocampus, cingulate, neocortex, and white matter. Interestingly these effects occur in the absence of apoptotic neuronal loss, as measured by the staining of cleaved caspase 3 ().
Fig. 5. Neuropathology (avidin–biotin complex immunohistochemistry) and semiquantitative image densitometry of coronal brain sections of dorsal hippocampus (CA2) and external capsule of adult rats with 14 day BID ICV infusions of Propionic acid (PPA) or phosphate-buffered saline (PBS). PPA induced significant reactive astrogliosis (anti-GFAP) and microglial activation (anti-CD68), without apoptotic neuronal cell loss (anit-cleaved caspase 3) in rat hippocampus, similar to finding in autopsy brain from patients with autism. Nuclear translocation of anti-CREB and an increase of anti phosphoCREB immunoreactivity are observed in neural, glial, and endovascular epithelium by PPA treatment, suggestive of gene induction. PPA increases monocarboxylate transporter 1 immunoreactivity, primarily in white matter external capsule, suggestive of alterations in brain shot-chain fatty acid transport/metabolism. Black bars indicate PPA-treated animals; white bars indicate PBS (vehicle)-treated animals. Horizontal measurement bar = 100µ. Figures modified with permission from MacFabe et al. (2007); see publication for details of immunostaining procedures. Reproduced with permission from Elsevier Ltd.
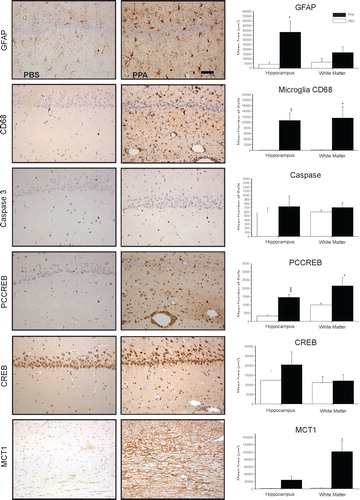
These findings are consistent with those from autopsy cases from patients with ASDs, regardless of age of death, indicating that an ongoing inflammatory process may be present throughout the life of the individual Citation13 Citation164. Microglia are a heterogeneous family of cells of myeloid lineage, which migrate from the bone marrow to the CNS both pre- and postnatally, and play a currently underappreciated role in neurodevelopmental disorders Citation165. When activated, they produce high levels of proinflammatory cytokines and reactive oxygen species such as hydrogen peroxide and nitric oxide. Interestingly, microglia also may be involved in neuroplastic responses, such as synaptic reorganization, and cortical pruning Citation166. Such effects are conceivable in both ASDs and the PPA model, as specific SCFA receptors exist on immune cells Citation88, and increased levels of cytokines such as tumor necrosis factor and macrophage chemoattractant protein are found in ASDs Citation13.
In addition, immunohistochemical analysis of PPA-treated brain sections showed evidence of increased immunoreactivity of the activated, phosphorylated form of cyclic-AMP responsive element binding protein (pCREB). Likewise, the results of in vitro studies show that SCFAs can induce pCREB in PC12 cells, leading to increased catecholamine synthesis Citation46 Citation133. CREB is interesting as this important neuroregulatory protein known to play a key role in the epigenetic expression of a number of genes implicated in neuroplasticity, addiction, movement and mood disorders, and memory acquisition Citation167. Thus, it is possible that increased activation of CREB-dependent epigenetic modulation of memory- or movement-related pathways following PPA administration could result in normal memory acquisition with perseverative behavior, repetitive behavior, or seizure similar to those observed in the PPA model and ASDs Citation46. Further to these findings, the fact that some other SCFAs and valproate, a compound with some structural similarity to PPA and a risk factor for autism, also are capable of further gene regulation via their histone deacetylase inhibition activity Citation90 Citation133 Citation168, makes the epigenetic effects of these compounds on ASD-related genes a subject of further study in our laboratory Citation169.
Preliminary results following immunostaining of brain sections with anti-monocarboxylate transporter 1 antibody (1:1000 dilution, Chemicon AB1286), a major transporter of PPA, related SCFA and ketones (Citation79,Citation80,Citation82,Citation83), reveals significant increases in external capsule white matter immunoreactivity following repeated PPA infusions, suggestive of altered brain fatty acid transport. It is important to note that these neuropathological changes, although consistent with findings in ASD autopsy cases, occur hours or days following PPA administration, which is considerably later than the transient behavioral effects, which are induced within minutes and last approximate 20–30 min. Nonetheless, they may be important in the neuroplastic kindling response and perseverative learning behavior from repeated exposures of PPA and merit further study.
Effects of PPA on lipid metabolism and mitochondrial function
Several reports have indicated that abnormal lipid metabolism may occur in ASDs and ASD-related disorders Citation170–Citation175, including fatty acids, phospholipases A2, and membrane phospholipids. Clinical studies suggest improvements in core symptoms of patients with ASDs following supplementation with polyunsaturated fatty acids (PUFAs) Citation173 Citation176 Citation177 or cholesterol Citation178. PUFAs, the major lipid constituents of neuronal membranes, are absolutely essential for normal brain development and function, modulating membrane fluidity, gene expression, cell signaling, neuronal excitability, and oxidant protection, and provide a source of energy for the cells, particularly during early neurodevelopment Citation110 Citation179. Lipid composition in cellular membranes is particularly important in the formation of lipid rafts, which alter membrane fluidity and the function of membrane-bound proteins, including cell receptors and gap and tight junctions Citation51 Citation53 Citation107 Citation180.
Carnitine is a quaternary ammonium compound synthesized from the amino acids lysine and methionine principally by liver and kidney, and it is also obtained from diet. It is critical for the transport of fatty acids into the inner mitochondrial membrane for β-oxidation and energy production Citation181. Clinical studies have reported a relative carnitine deficiency and abnormal acylcarnitine profile (elevated short and long chain) in erythrocytes of ASDs of uncertain etiology Citation142 Citation182 Citation183 and a common X-linked genetic defect in carnitine synthesis in a subset of patients with ASDs Citation25, suggesting carnitine supplementation as a possible treatment for the disorder Citation20.
We thus used our PPA rodent model to examine whether there is any evidence for alterations in brain phospholipids and acylcarnitines following intraventricular infusions with PPA or butyrate (4 µl of 0.26 M solution BID×7 days) to adult rats Citation51 Citation53.
Brain lipid analysis was performed via GC mass spectroscopy/electrospray ionization mass spectrometry (see Thomas et al. Citation51 Citation53 for technical details). Altered lipid profiles were observed in rat brain phospholipids following infusion with both PPA and, to a lesser extent, butyrate ().
Fig. 6. (A) Changes in phosphatidylethanolamine and phosphatidylcholine in rat brain tissue following repeated 4 µl intracerebroventricular infusions with phosphate-buffered buffer (PBS), butyric acid (BUT), and propionic acid (PPA) 0.26 M twice daily for 7 days are consistent with findings in patients with autism. Values represent means±SE. Arrows indicate significant difference between treatments [PPA and BUT compared with the control (PBS)], and direction of the changes (increase or decrease) at LSD = 0.05, N=6 per treatment. Monounsat = monosaturates; polyunsat = polyunsaturates. (B) Changes in acylcarnitine and ratio of bound to free carnitine in rat brain tissue following intracerebroventricular infusion with PBS, BUT, and PPA are consistent with findings in patients with autism. Values represent means±SE. Means accompanied by different superscript letters (e.g. a, b, and c) indicate significant difference between treatments at LSD = 0.05, N=6 per treatment. Long-chain acylcarnitine = C12–C24, short-chain acylcarnitine = C2–C9. (C) PPA and, to a lesser extent, BUT infusions increase short- and long-chain acylcarnitines, but not medium-chain acylcarnitines, similar to findings in patients with autism. Values (percent by weight) represent means±SE. Means in the same row accompanied by different superscript letters (e.g. a, b, and c) are significantly different between treatments at LSD = 0.05, N=9. Numbers preceding C represents carbon number of the fatty acid moiety attach to carnitine. Long-chain acylcarnitine = C12–C24, short-chain acylcarnitine = C2–C9. Figures modified with permission from Thomas et al. (2010). Reproduced with permission from Wiley Blackwell.
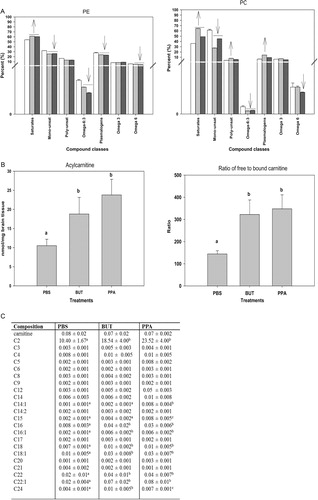
PPA infusion resulted in decreased levels of total monounsaturates and total ω6 fatty acids and elevated levels of total saturates in all of the studied phospholipids. In addition, a decline in total plasmalogen phosphatidylethanolamine and the ratio of ω6:ω3 was also present. Elevated levels of saturated fatty acids have been reported in red blood cells Citation170 Citation176 and plasma Citation184 of several autistic patients. These findings were accompanied by a concomitant decrease in total monounsaturates, particularly, 18:1n9 fatty acid. A decline in the level of this fatty acid along with several other monounsaturates (20:1n9, 22:1n9, 16:1n7, and PE 24:1n9) has also been observed in bloods drawn from patients with autism Citation174 Citation176 Citation185.
Carnitine and acylcarnitine analysis showed a non-significant trend toward lower free carnitine in butyrate- and PPA-treated animals; however, there was a consistent significant (p=0.02) increase in total acylcarnitines, total long-chain (C12 to C24) acylcarnitines, total short-chain (C2 to C9) acylcarnitines, and the ratio of free to bound carnitine following infusions with PPA and butyrate. Increases in the accumulation of short- and long-chain acylcarnitines, but not medium-chain acylcarnitines, were observed. Specifically, when these acylcarnitines were grouped according to chain length, acetyl carnitine (C2) was the major contributor to the increases observed in short-chain acylcarnitines, while C16:0, C18:0, C18:1, C22:0, and C22:1 were the major contributors to the increases observed in long-chain acylcarnitines Citation51 Citation53.
These unique acylcarnitine profiles are similar to those obtained from blood samples of patients with ASDs Citation182 Citation183 which observed elevations in the levels of some long-chain acylcarnitine species (14:1 and 14:2). Elevations in 14:1 and other long-chain acylcarnitines (16:0, 18:0, 18:1, 22:0, and 22:1) consistent with findings in our model. PPA is thought to affect mitochondrial fatty acid metabolism by binding to propionyl CoA and by sequestering carnitine Citation86 Citation186 Citation187. Elevation in the levels of these acylcarnitines indicates a potential metabolic disturbance by PPA infusions to affect mitochondrial metabolism in manners consistent with a unique acylcarnitine profile and a proposed environmental induction of mitochondrial dysfunction and increased oxidative stress in ASDs Citation51 Citation188.
Of particular interest, PPA is a known inhibitor of mitochondrial function, through sequestration of carnitine and the production of propionyl CoA, a potential cytotoxin Citation189 Citation190. PPA also inhibits the incorporation of acetate during lipid synthesis, an effect which is more pronounced in males Citation191 Citation192. This is interesting in light of low cholesterol levels being central to many forms of ASDs Citation178. Since PPA is metabolized through the mitochondrial metabolic pathways and affects mitochondrial cardiolipin levels and membrane stability Citation51 Citation53, we propose that excess exogenous PPA or its related SCFAs overwhelm mitochondrial metabolism, thereby causing mitochondrial dysfunction. Such mitochondrial dysfunction is consistent with an increase in oxidative stress. Evidence for oxidative stress has been demonstrated in the rodent PPA model of ASDs by the increase in nitric oxide-/hydrogen peroxide-producing activated microglia, coupled with the elevation of oxidized proteins and lipids, reduction of glutathione, and alteration of phospholipid/acylcarnitine profiles in brain homogenates, particularly in brain regions affected in ASDs Citation46 Citation51 Citation53. Such findings are all consistent with those obtained from patients with ASDs Citation110 Citation151 Citation152 Citation193 Citation194.
Impairment of carnitine metabolism from a variety of causes may be central to ASD pathogenesis and regression
Although the basis of the reduced blood carnitine levels in ASDs remains unclear, we have made the observation that there are diverse clinical conditions circumstantially linked to ASDs and gastrointestinal dysfunction that show disruptions in carnitine metabolism as a common observation. There has been a growing interest in the role of carnitine in brain physiology and disease Citation181, particularly in brain GABAergic and astrocyte metabolism Citation195. As carnitine is endogenously produced from lysine and methionine, persons with defects in methylation pathways, common in individuals with ASDs Citation196, would have impairments in endogenous carnitine synthesis. A particularly vulnerable subgroup would be the recently discovered X-linked defect in the TMLHE enzyme, responsible for the first step in carnitine biosynthesis, which is a risk factor for non-dysmorphic autism in males Citation25. Collectively, these persons would rely more on dietary sources of carnitine, critical during periods of rapid development, and thus may be more sensitive to factors that impair carnitine uptake from the gut. It is known that carnitine is transported across the gut–blood and blood–brain barriers via the Na+-dependent organic cation/carnitine transporter 2 (OCNT2) Citation197. Carnitine transport deficits have been implicated in colitis Citation198 and also may impair blood–brain barrier integrity, allowing non-neurotropic influenza A virus to enter the CNS and inducing a neonatal encephalopathy Citation199. Interestingly, long-term administration of common antibiotics (i.e. β-lactams) for routine pediatric infections, in addition to altering gut flora favoring PPA-producing species, has been shown to directly inhibit the OCNT2 transporter, affecting carnitine transport across gut–blood and blood–brain barriers Citation197, and, thus, may contribute to a relative systemic carnitine deficiency. Additionally, antibiotics given as forms of pivalyl esters will cause an increased urinary loss of carnitine. Such interferences could be significant considering the reported high incidence of antecedent long-term antibiotic use in some patients with ASDs Citation65 Citation66 Citation200 Citation201 and the finding of unique enteric PPA-producing bacteria and gut carbohydrate malabsorbtion in regressive ASDs Citation65 Citation66 Citation105. This offers a potential explanation for autistic regression and also temporary behavioral improvements in some patients following vancomycin or metronidazole treatment, which transiently eradicates these bacteria Citation65 Citation66 Citation109 Citation201. However, given that Finegold has proposed eradication of ASD-causing bacteria with oritavancin and aztreonam, antibiotics which could further depress carnitine absorption, as a possible treatment of ASDs, warrants the necessity of following patient carnitine levels and providing possible carnitine supplementation in such a study Citation64. Furthermore, removal of refined carbohydrates from the diet, which has been suggested as an empiric treatment to improve the behavioral fluctuations and gastrointestinal symptoms in ASDs, may act by reducing substrate for these bacteria to produce PPA Citation46. Feeding of a high-carbohydrate diet in rats is known to increase SCFA levels and produce anxiety and aggressive behavior Citation139. Interestingly, preeclamptic mothers, who have an increased risk of having offspring affected by ASDs Citation202, have similar short- and long-chain acylcarnitine profiles Citation203 as found in the patients with ASDs and the PPA rodent model. Although the overall relationships remain unproven, it appears that taken collectively, we have proposed the above observations link the decreased carnitine levels in some patients with ASDs with several genetic and environmental factors consistent with regression, gastrointestinal symptomatology, microbiology, and lipid biomarkers and with experimental findings obtained using the PPA model. Furthermore, oral carnitine and its derivative acetyl-l-carnitine have both neuroprotective Citation181 Citation204 Citation205 and coloprotective properties Citation206 and deserve further investigation as therapeutic agents in developmental disorders, including ASDs Citation22 Citation46 Citation51 Citation207. These findings would also warrant more rigorous and possibly repeated carnitine/acylcarnitine screening of ‘patients at risk’, such as infants with clinical evidence of developmental delay and gastrointestinal dysfunction, particularly in the presence of maternal/infant hospital-acquired infection or long-term antibiotic use.
Conclusions/limitations/future directions
It is important to note an animal model is unlikely to completely replicate a human disease. The usefulness of models relates to the various types of validity that can be shown to exist for specific conditions. Face validity, or the degree to which the model is able to capture the phenomenology of the disorder, is usually an essential first step in developing a model Citation208 Citation209, but additional evidence for construct validity, that is, a probable theoretical rationale for the model, possibly based on etiology is equally as important. Crawley Citation120 has provided a list of symptoms of ASDs that would aid in the establishment of face validity for a rodent model, along with suggested behavioral tests for such symptoms.
In the initial development of our model of ASDs, we focused on four broad aspects, such as behavioral, brain electrographic and neuropathological characteristics and biochemical markers, which would provide some face, as well as, construct validity for the model. At a behavioral level, we were interested in determining whether PPA would induce hyperactivity, stereotypies and repetitive behaviors, object preference, perseveration, and social impairment consistent with ASDs. Electroencephalographic recordings allowed us to monitor for cortical and subcortical epileptiform activity and development of seizures along with behavioral assessment of seizure and movement disorder. Finally, we also looked for possible neuropathological effects that might be consistent with those observed in humans, such as innate neuroinflammatory, astroglial and microglial changes, and neuroplastic (i.e. CREB activation, monocarboxylate transport) changes, as well as biochemical markers suggestive of increased oxidative stress, altered phospholipid/acylcarnitine profiles, and mitochondrial dysfunction.
These initial studies provide support for the face validity of the intraventricular PPA administration in adult rat model of autism, as the behavioral and electrographic changes observed resemble those seen in the human condition. The rapid induction and transient nature of these peculiar behavioral and electrographic effects, their potentiation with repeated exposures, and the absence of such behaviors in rats receiving control compounds (1-propanol) suggest the involvement of diverse PPA-activated neural mechanisms and present the first attempt in the field to model the possible fluctuating, as opposed to static behavioral course in ASDs.
Evidence from human studies suggests that ASD is a condition that represents an ongoing neuroinflammatory or neurometabolic disorder possibly resulting from an increased sensitivity to oxidative stress or acquired mitochondrial dysfunction from a variety of environmental risk factors. The neuropathological and biochemical findings of our model support this hypothesis. Interestingly, the observed impairments in the glutathione system, increased oxidative stress, and altered phospholipids/acylcarnitine profiles, suggestive of mitochondrial dysfunction in our model, are consistent with the findings in human autism cases and would provide a plausible mechanism for increased environmental sensitivity to a variety of agents. The similarities in neuropathological and biochemical changes between the animal model and human ASD cases, coupled with the presence of PPA-producing bacteria, could represent similar underlying etiological processes subsequent to central nervous system insult with PPA. However, much additional work needs to be done before these similarities can be taken as support for construct validity of the animal model.
It is important to note, other than reports of increased PPA in stool samples, studies are lacking Citation210 that have systematically examined PPA and its related metabolites in ASDs. The short half-life and rapid metabolism via β oxidation, incorporation into acylcarnitines and other lipids, and intracellular concentration of PPA via specific transporters and pH-dependant mechanisms also make definitive measurement and interpretation difficult. However, there is some indirect evidence in patients with ASDs suggestive of increased PPA, including a relative carnitine deficiency, unique phospholipid and short- and long-chain acylcarnitine profiles, elevations of nitric oxide metabolites, and oxidative stress markers, known to be elevated by PPA in experimental systems, including our rodent model. The underappreciated incidence of complex gene polymorphisms in propionic acidemia in some populations, a potential population of partial metabolizers of PPA, and documented ASD symptoms in some of these patients, is interesting. The antiepileptic drug valproate, a prenatal risk factor for autism, alters both fatty acid and biotin metabolism and causes the depletion of carnitine stores each of which could theoretically increase PPA levels. Prenatal exposures to ethanol, known to produce developmental delay, also increases PPA levels to millimolar levels putatively by depleting intracellular carnitine stores.
Despite the lack of studies on PPA and its related fatty acids in ASDs, abnormalities of lipid metabolism and mitochondrial dysfunction have been postulated as a potential cause of the condition. Patient studies have noted impairments in mitochondrial β-oxidation of fatty acids, reductions in essential PUFAs levels, and anecdotal reports of improvement in some patients with essential fatty acid and carnitine supplementation.
Likewise, the intriguing finding that long-term administration of common antibiotics for routine infections not only may favor PPA-producing bacteria but also may impair colonocyte function and directly inhibits gut carnitine transport, leading to an acquired mitochondrial encephalopathy, should certainly be investigated as a key factor in ASD regression. Notably, screening for unique enteric bacterial populations, plasma phospholipids/acylcarnitines, inherited carnitine transport defects, ‘partial metabolizers’ of PPA (i.e. organic acidemias, mitochondrial disorder), and gut dysfunction deserves further examination. This may be useful not only in patients with ASDs but also in ‘persons at risk’, including individuals in neonatal units, those with immune deficiencies and patients on long-term antibiotic usage. Likewise, to follow these biomarkers in response to proposed treatments (eradication of PPA-producing bacteria, probiotics, dietary carbohydrate restriction, carnitine supplementation) in animal models and ultimately patients would prove valuable. Importantly, the longitudinal examination of the infant gut microbiome, characterizing not only the presence, but also the absence, of specific gut bacterial population in infants who progress neurotypically or subsequently go on to develop ASDs will be critical. Novel bacterial species, such as Desulfovibrio, and their ability to produce H2S, a possible mitochondrial toxin, possessing both colonotoxic and neurotoxic effects synergistic to PPA, and their interaction with Clostridia and other members of the gut microbiome deserve further investigation. Lastly the neurobiological effects of other short chain fatty acids, their derivatives and the array of other metabolites of the microbiome remain largely unexplored.
In a broader context, it is intriguing to postulate that microbiota, through natural selection, have evolved to use their metabolites to modulate the physiology and ultimately behavior of the host, to promote survival. This has been documented in behavioral neurobiology, with such examples as cordyceps fungus producing climbing behavior in ants, and borna and rabies viruses eliciting salivary transmission and biting behavior in mammals Citation211 Citation212. It has recently been established that transplantation of fecal material in germ-free mice can alter brain gene expression and phenotypic behavior of the host Citation213. In light of this, the observation of restrictive eating of carbohydrates, diarrhea and fecal smearing in patients with ASDs which could theoretically promote organism growth and spread, is intriguing. It is also worth noting that many of the effects of lower doses of SCFAs on gut physiology and immune function are indeed beneficial to host and ultimately bacterial survival. Finally, it is important to note the ability of SCFAs to elicit anxiety-like, perseverative, repetitive, ritualistic, and antisocial behaviors that are common to many other neuropsychiatric conditions such as obsessive compulsive, anxiety, attention deficit/hyperactive, mood, and eating disorders, irritable bowel syndrome, pediatric autoimmune neuropsychiatric disorder associated with streptococcal infections (PANDAS), and schizophrenia, where infectious agents have been proposed Citation214–Citation216.
The growing incidence of ASDs and ASD-related conditions, coupled with the observed alterations in the human microbiome secondary to dietary, medical, and agricultural factors, and their potential effect on human and animal behavior, should be further examined. The impact of human migration and urbanization, domestication of plant and animals, and resultant human diseases shaping cultures is not trivial and has been discussed elsewhere Citation217.
In conclusion, these present studies examined the effects of intracerebroventricular PPA in adult rats and their relation to the pathophysiology of ASDs. Given the intriguing multiple effects of PPA on many neurological, gastroenterological, metabolic, and immunological processes, together with our initial findings on behavior, electrophysiology, neuropathology, and biochemistry, the rat intraventricular PPA model maybe a useful paradigm for the examination of the disparate symptoms and pathophysiology of ASDs ().
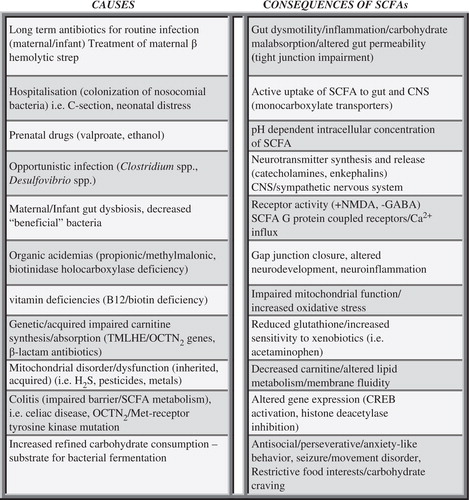
Fig. 7. Potential causes and consequences of increased enteric short-chain fatty acid production and/or decreased breakdown and their relation to autism spectrum disorder. These findings, which are not mutually exclusive, may contribute to the pathophysiology, behavioral symptoms, and comorbidities of autism.
However, notwithstanding of the remarkable effects of these compounds on an ‘intact’ adult brain, autism is, of course, a neurodevelopmental disorder, with evidence of altered nervous system development, involving white matter abnormalities and disorders of neuronal microcircuitry. Treatments of rodents with PPA, other SCFAs and appropriate control compounds, and ultimately infection of ASD-associated bacteria during critical times of pre- and postnatal development are essential steps in extending the validity of this animal model. Such studies using systemic and dietary exposure at these time periods are ongoing in our laboratory and our collaborators Citation52 Citation130 Citation218.
Although compelling, further research examining PPA and its related SCFAs is still needed at both the basic and the clinical levels to better understand the underlying mechanisms of how these and other metabolic products of the host microbiota may modulate host physiology throughout the lifecycle and whether they are directly involved in the disparate brain and behaviors of ASDs and ASD-related conditions. Conceptually, it is the author's opinion that the pathophysiology of ASDs may be more completely understood as being similar to conditions such as ethanol intoxication, or diabetes, and the resultant complex interactions between diet, genetics, metabolism, host microbiome, and behavior, that are well known to exist in these treatable disorders throughout the life cycle. Considering the marked increase, considerable morbidity, and social burden of ASDs, collaboration in the fields of microbiology, clinical and basic neuroscience, immunology, biochemistry, gastroenterology, obstetrics, genetics, and epidemiology is warranted and should be encouraged.
Conflict of interest and funding
The author has not received any funding or benefits from industry or elsewhere to conduct this study.
Acknowledgments
This manuscript was prepared as a summary of two lectures given in 1) honor of the 100th birthday of Dr. Philip Trexler and in memory of the late Dr. Morris Pollard (Notre Dame University, South Bend, IN) and 2) as a Nobel Symposium lecture in ‘The Gut and the Brain – with Focus on Autism Spectrum Disorder’ (Karolinska Institute, Stockholm, Sweden). The author would like to thank all the investigators, collaborators, students, and technologists of the Kilee Patchell-Evans Autism Research Group, University of Western Ontario, Canada. Particular thanks go to Drs. Klaus-Peter Ossenkopp, Martin Kavaliers, Donald Peter Cain (behavior), and Fred Possmayer (biochemistry); graduate students/postdoctoral fellows Kelly Foley, Jennifer Hoffman, Melissa Meeking, Jennifer Mepham; and Drs. Sandy Shultz (behavior), Karina Rodriguez-Capote (oxidative stress), and Raymond Thomas (lipid biochemistry). Expert technical assistance was provided by Roy Taylor (pathology), Francis Boon (behavior, surgery, EEG), and Lisa Tichenoff (Project Manager and generous assistance with manuscript preparation). We would also like to express our utmost thanks to David Patchell-Evans, for his tireless devotion to persons with autism, and his daughter, Kilee Patchell-Evans. Our heartfelt thanks go out to countless parents and caregivers of persons with autism who have shared their stories. This research was supported by contributions from GoodLife Children's Charities, Autism Canada, and Autism Research Institute to DFM.
Related Research Data
References
- Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators. Prevalence of autism spectrum disorders – autism and developmental disabilities monitoring network, 14 sites, United States, 2008. MMWR Surveill Summ. 2012; 61: 1–19.
- Cody H, Pelphrey K, Piven J. Structural and functional magnetic resonance imaging of autism. Int J Dev Neurosci. 2002; 20: 421–38. 10.3402/mehd.v23i0.19260.
- Courchesne E, Pierce K. Brain overgrowth in autism during a critical time in development: implications for frontal pyramidal neuron and interneuron development and connectivity. Int J Dev Neurosci. 2005; 23: 153–70. 10.3402/mehd.v23i0.19260.
- Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci. 2005; 23: 183–7. 10.3402/mehd.v23i0.19260.
- Connolly AM, Chez M, Streif EM, Keeling RM, Golumbek PT, Kwon JM, et al. Brain-derived neurotrophic factor and autoantibodies to neural antigens in sera of children with autistic spectrum disorders, Landau-Kleffner syndrome, and epilepsy. Biol Psychiatry. 2005; 59: 354–63. 10.3402/mehd.v23i0.19260.
- Araghi-Niknam M, Fatemi SH. Levels of Bcl-2 and P53 are altered in superior frontal and cerebellar cortices of autistic subjects. Cell Mol Neurobiol. 2003; 23: 945–52. 10.3402/mehd.v23i0.19260.
- Palomo T, Beninger RJ, Kostrzewa RM, Archer T. Brain sites of movement disorder: genetic and environmental agents in neurodevelopmental perturbations. Neurotox Res. 2003; 5: 1–26. 10.3402/mehd.v23i0.19260.
- Raymond GV, Bauman ML, Kemper TL. Hippocampus in autism: a Golgi analysis. Acta Neuropathol (Berl). 1996; 91: 117–19. 10.3402/mehd.v23i0.19260.
- Bittman K, Becker DL, Cicirata F, Parnavelas JG. Connexin expression in homotypic and heterotypic cell coupling in the developing cerebral cortex. J Comp Neurol. 2002; 443: 201–12. 10.3402/mehd.v23i0.19260.
- Kirchhoff F, Dringen R, Giaume C. Pathways of neuron-astrocyte interactions and their possible role in neuroprotection. Eur Arch Psychiatry Clin Neurosci. 2001; 251: 159–69. 10.3402/mehd.v23i0.19260.
- Carper RA, Moses P, Tigue ZD, Courchesne E. Cerebral lobes in autism: early hyperplasia and abnormal age effects. Neuroimage. 2002; 16: 1038–51. 10.3402/mehd.v23i0.19260.
- Hendry J, Devito T, Gelman N, Densmore M, Rajakumar N, Pavlosky W, et al. White matter abnormalities in autism detected through transverse relaxation time imaging. Neuroimage. 2006; 29: 1049–57. 10.3402/mehd.v23i0.19260.
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005; 57: 67–81. 10.3402/mehd.v23i0.19260.
- Molloy CA, Morrow AL, Meinzen-Derr J, Schleifer K, Dienger K, Manning-Court P, et al. Elevated cytokine levels in children with autism spectrum disorder. J Neuroimmunol. 2005; 172: 198–205. 10.3402/mehd.v23i0.19260.
- Ameis SH, Szatmari P. Imaging-genetics in autism spectrum disorder: advances, translational impact, and future directions. Front Psychiatry. 2012; 3: 46. 10.3402/mehd.v23i0.19260.
- Hu VW, Frank BC, Heine S, Lee NH, Quackenbush J. Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genomics. 2006; 7: 118. 10.3402/mehd.v23i0.19260.
- Jyonouchi H, Sun S, Itokazu N. Innate immunity associated with inflammatory responses and cytokine production against common dietary proteins in patients with autism spectrum disorder. Neuropsychobiology. 2002; 46: 76–84. 10.3402/mehd.v23i0.19260.
- Arndt TL, Stodgell CJ, Rodier PM. The teratology of autism. Int J Dev Neurosci. 2005; 23: 189–99. 10.3402/mehd.v23i0.19260.
- Herbert MR. Contributions of the environment and environmentally vulnerable physiology to autism spectrum disorders. Curr Opin Neurol. 2010; 23: 103–10. 10.3402/mehd.v23i0.19260.
- Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry. 2012; 17: 389–401. 10.3402/mehd.v23i0.19260.
- Frye RE, Rossignol DA. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr Res. 2011; 69: 41R–7R. 10.3402/mehd.v23i0.19260.
- Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2011; 17: 290–314. 10.3402/mehd.v23i0.19260.
- Campbell DB, Buie TM, Winter H, Bauman M, Sutcliffe JS, Perrin JM, et al. Distinct genetic risk based on association of MET in families with co-occurring autism and gastrointestinal conditions. Pediatrics. 2009; 123: 1018–24. 10.3402/mehd.v23i0.19260.
- Lintas C, Sacco R, Garbett K, Mirnics K, Militerni R, Bravaccio C, et al. Involvement of the PRKCB1 gene in autistic disorder: significant genetic association and reduced neocortical gene expression. Mol Psychiatry. 2008; 14: 705–18. 10.3402/mehd.v23i0.19260.
- Celestino-Soper PB, Violante S, Crawford EL, Luo R, Lionel AC, Delaby E, et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc Natl Acad Sci USA. 2012; 109: 7974–81. 10.3402/mehd.v23i0.19260.
- Bayona-Bafaluy MP, Muller S, Moraes CT. Fast adaptive coevolution of nuclear and mitochondrial subunits of ATP synthetase in orangutan. Mol Biol Evol. 2005; 22: 716–24. 10.3402/mehd.v23i0.19260.
- Nishigaki Y, Fuku N, Tanaka M. Mitochondrial haplogroups associated with lifestyle-related diseases and longevity in the Japanese population. Geriatr Gerontol Int. 2010; 10(Suppl 1): S221–35. 10.3402/mehd.v23i0.19260.
- Weissman JR, Kelley RI, Bauman ML, Cohen BH, Murray KF, Mitchell RL, et al. Mitochondrial disease in autism spectrum disorder patients: a cohort analysis. PLoS One. 2008; 3: e3815. 10.3402/mehd.v23i0.19260.
- Giulivi C, Zhang YF, Omanska-Klusek A, Ross-Inta C, Wong S, Hertz-Picciotto I, et al. Mitochondrial dysfunction in autism. JAMA. 2010; 304: 2389–96. 10.3402/mehd.v23i0.19260.
- Vali S, Mythri RB, Jagatha B, Padiadpu J, Ramanujan KS, Andersen JK, et al. Integrating glutathione metabolism and mitochondrial dysfunction with implications for Parkinson's disease: a dynamic model. Neuroscience. 2007; 149: 917–30. 10.3402/mehd.v23i0.19260.
- Samavati L, Lee I, Mathes I, Lottspeich F, Huttemann M. Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem. 2008; 283: 21134–44. 10.3402/mehd.v23i0.19260.
- Vempati UD, Diaz F, Barrientos A, Narisawa S, Mian AM, Millan JL, et al. Role of cytochrome C in apoptosis: increased sensitivity to tumor necrosis factor alpha is associated with respiratory defects but not with lack of cytochrome C release. Mol Cell Biol. 2007; 27: 1771–83. 10.3402/mehd.v23i0.19260.
- Suematsu N, Tsutsui H, Wen J, Kang D, Ikeuchi M, Ide T, et al. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation. 2003; 107: 1418–23. 10.3402/mehd.v23i0.19260.
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011; 68: 1095–102. 10.3402/mehd.v23i0.19260.
- Herbert MR, Russo JP, Yang S, Roohi J, Blaxill M, Kahler SG, et al. Autism and environmental genomics. Neurotoxicology. 2006; 27: 671–84. 10.3402/mehd.v23i0.19260.
- Ashwood P, Anthony A, Torrente F, Wakefield AJ. Spontaneous mucosal lymphocyte cytokine profiles in children with autism and gastrointestinal symptoms: mucosal immune activation and reduced counter regulatory interleukin-10. J Clin Immunol. 2004; 24: 664–73. 10.3402/mehd.v23i0.19260.
- Fowler BA, Woods JS. Ultrastructural and biochemical changes in renal mitochondria during chronic oral methyl mercury exposure: the relationship to renal function. Exp Mol Pathol. 1977; 27: 403–12. 10.3402/mehd.v23i0.19260.
- Shenker BJ, Guo TL, O I, Shapiro IM. Induction of apoptosis in human T-cells by methyl mercury: temporal relationship between mitochondrial dysfunction and loss of reductive reserve. Toxicol Appl Pharmacol. 1999; 157: 23–35. 10.3402/mehd.v23i0.19260.
- Goyer RA. Toxic and essential metal interactions. Annu Rev Nutr. 1997; 17: 37–50. 10.3402/mehd.v23i0.19260.
- Pourahmad J, Mihajlovic A, O'Brien PJ. Hepatocyte lysis induced by environmental metal toxins may involve apoptotic death signals initiated by mitochondrial injury. Adv Exp Med Biol. 2001; 500: 249–52.
- Hiura TS, Li N, Kaplan R, Horwitz M, Seagrave JC, Nel AE. The role of a mitochondrial pathway in the induction of apoptosis by chemicals extracted from diesel exhaust particles. J Immunol. 2000; 165: 2703–11.
- Wong PW, Garcia EF, Pessah IN. Ortho-substituted PCB95 alters intracellular calcium signaling and causes cellular acidification in PC12 cells by an immunophilin-dependent mechanism. J Neurochem. 2001; 76: 450–63. 10.3402/mehd.v23i0.19260.
- Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, et al. Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson's disease. J Neurochem. 2007; 100: 1469–79.
- Yamano T, Morita S. Effects of pesticides on isolated rat hepatocytes, mitochondria, and microsomes II. Arch Environ Contam Toxicol. 1995; 28: 1–7. 10.3402/mehd.v23i0.19260.
- Astiz M, de Alaniz MJ, Marra CA. Effect of pesticides on cell survival in liver and brain rat tissues. Ecotoxicol Environ Saf. 2009; 72: 2025–32. 10.3402/mehd.v23i0.19260.
- MacFabe DF, Cain DP, Rodriguez-Capote K, Franklin AE, Hoffman JE, Boon F, et al. Neurobiological effects of intraventricular propionic acid in rats: possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav Brain Res. 2007; 176: 149–69. 10.3402/mehd.v23i0.19260.
- MacFabe DF, Rodriguez-Capote K, Hoffman JE, Franklin AE, Mohammad-Asef Y, Taylor A, et al. A novel rodent model of autism: intraventricular infusions of propionic acid increase locomotor activity and induce neuroinflammation and oxidative stress in discrete regions of adult rat brain. Am J Biochem & Biotech. 2008; 4: 146–66. 10.3402/mehd.v23i0.19260.
- MacFabe DF, Cain NE, Boon F, Ossenkopp KP, Cain DP. Effects of the enteric bacterial metabolic product propionic acid on object-directed behavior, social behavior, cognition, and neuroinflammation in adolescent rats: relevance to autism spectrum disorder. Behav Brain Res. 2011; 217: 47–54. 10.3402/mehd.v23i0.19260.
- Shultz SR, MacFabe DF, Ossenkopp KP, Scratch S, Whelan J, Taylor R, et al. Intracerebroventricular injection of propionic acid, an enteric bacterial metabolic end-product, impairs social behavior in the rat: implications for an animal model of autism. Neuropharmacology. 2008; 54: 901–11. 10.3402/mehd.v23i0.19260.
- Shultz SR, MacFabe DF, Martin S, Jackson J, Taylor R, Boon F, et al. Intracerebroventricular injections of the enteric bacterial metabolic product propionic acid impair cognition and sensorimotor ability in the Long-Evans rat: further development of a rodent model of autism. Behav Brain Res. 2009; 200: 33–41. 10.3402/mehd.v23i0.19260.
- Thomas RH, Foley KA, Mepham JR, Tichenoff LJ, Possmayer F, MacFabe DF. Altered brain phospholipid and acylcarnitine profiles in propionic acid infused rodents: further development of a potential model of autism spectrum disorders. J Neurochem. 2010; 113: 515–29. 10.3402/mehd.v23i0.19260.
- Ossenkopp KP, Foley KA, Gibson J, Fudge MA, Kavaliers M, Cain DP, et al. Systemic treatment with the enteric bacterial fermentation product, propionic acid, produces both conditioned taste avoidance and conditioned place avoidance in rats. Behav Brain Res. 2012; 227: 134–41. 10.3402/mehd.v23i0.19260.
- Thomas RH, Meeking MM, Mepham JR, Tichenoff L, Possmayer F, Liu S, et al. The enteric bacterial metabolite propionic acid alters brain and plasma phospholipid molecular species: further development of a rodent model of autism spectrum disorders. J Neuroinflammation. 2012; 9: 153. 10.3402/mehd.v23i0.19260.
- Midtvedt AC, Midtvedt T. Production of short chain fatty acids by the intestinal microflora during the first 2 years of human life. J Pediatr Gastroenterol Nutr. 1992; 15: 395–403. 10.3402/mehd.v23i0.19260.
- Patterson PH. Maternal infection and autism. Brain Behav Immun. 2012; 26: 393. 10.3402/mehd.v23i0.19260.
- Yap IK, Angley M, Veselkov KA, Holmes E, Lindon JC, Nicholson JK. Urinary metabolic phenotyping differentiates children with autism from their unaffected siblings and age-matched controls. J Proteome Res. 2010; 14: 705–18.
- Forsythe P, Sudo N, Dinan T, Taylor VH, Bienenstock J. Mood and gut feelings. Brain Behav Immun. 2010; 24: 9–16. 10.3402/mehd.v23i0.19260.
- Roy CC, Kien CL, Bouthillier L, Levy E. Short-chain fatty acids: ready for prime time?. Nutr Clin Pract. 2006; 21: 351–66. 10.3402/mehd.v23i0.19260.
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science. 2012; 336: 1262–7. 10.3402/mehd.v23i0.19260.
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012; 486: 207–14. 10.3402/mehd.v23i0.19260.
- Barnevik-Olsson M, Gillberg C, Fernell E. Prevalence of autism in children of Somali origin living in Stockholm: brief report of an at-risk population. Dev Med Child Neurol. 2010; 52: 1167–8. 10.3402/mehd.v23i0.19260.
- Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M, et al. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord. 2010; 40: 1423–30. 10.3402/mehd.v23i0.19260.
- Atladottir HO, Thorsen P, Schendel DE, Ostergaard L, Lemcke S, Parner ET. Association of hospitalization for infection in childhood with diagnosis of autism spectrum disorders: a Danish cohort study. Arch Pediatr Adolesc Med. 2010; 164: 470–7. 10.3402/mehd.v23i0.19260.
- Finegold SM. Desulfovibrio species are potentially important in regressive autism. Med Hypotheses. 2011; 77: 270–4. 10.3402/mehd.v23i0.19260.
- Finegold SM, Molitoris D, Song Y, Liu C, Vaisanen ML, Bolte E, et al. Gastrointestinal microflora studies in late-onset autism. Clin Infect Dis. 2002; 35: S6–16. 10.3402/mehd.v23i0.19260.
- Finegold SM, Dowd SE, Gontcharova V, Liu C, Henley KE, Wolcott RD, et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe. 2010; 16: 444–53. 10.3402/mehd.v23i0.19260.
- Wang L, Christophersen CT, Sorich MJ, Gerber JP, Angley MT, Conlon MA. Elevated fecal short chain fatty acid and ammonia concentrations in children with autism spectrum disorder. Dig Dis Sci. 2012; 57: 2096–102. 10.3402/mehd.v23i0.19260.
- Jan G, Belzacq AS, Haouzi D, Rouault A, Metivier D, Kroemer G, et al. Propionibacteria induce apoptosis of colorectal carcinoma cells via short-chain fatty acids acting on mitochondria. Cell Death Differ. 2002; 9: 179–88. 10.3402/mehd.v23i0.19260.
- Al-Lahham SH, Peppelenbosch MP, Roelofsen H, Vonk RJ, Venema K. Biological effects of propionic acid in humans; metabolism, potential applications and underlying mechanisms. Biochim Biophys Acta. 2010; 1801: 1175–83. 10.3402/mehd.v23i0.19260.
- Zouboulis CC, Eady A, Philpott M, Goldsmith LA, Orfanos C, Cunliffe WC, et al. What is the pathogenesis of acne?. Exp Dermatol. 2005; 14: 143–52. 10.3402/mehd.v23i0.19260.
- Borgstrom MK, Edwardsson S, Svensater G, Twetman S. Acid formation in sucrose-exposed dental plaque in relation to caries incidence in schoolchildren. Clin Oral Investig. 2000; 4: 9–12. 10.3402/mehd.v23i0.19260.
- Niederman R, Zhang J, Kashket S. Short-chain carboxylic-acid-stimulated, PMN-mediated gingival inflammation. Crit Rev Oral Biol Med. 1997; 8: 269–90. 10.3402/mehd.v23i0.19260.
- Zarate G, Gonzalez S, Chaia AP. Assessing survival of dairy propionibacteria in gastrointestinal conditions and adherence to intestinal epithelia. Methods Mol Biol. 2004; 268: 423–32.
- Brennan PA, Kendrick KM. Mammalian social odours: attraction and individual recognition. Philos Trans R Soc Lond B Biol Sci. 2006; 361: 2061–78. 10.3402/mehd.v23i0.19260.
- Yonezawa T, Haga S, Kobayashi Y, Katoh K, Obara Y. Short-chain fatty acid signaling pathways in bovine mammary epithelial cells. Regul Pept. 2009; 153: 30–6. 10.3402/mehd.v23i0.19260.
- Brock M, Buckel W. On the mechanism of action of the antifungal agent propionate. Eur J Biochem. 2004; 271: 3227–41. 10.3402/mehd.v23i0.19260.
- Karuri AR, Dobrowsky E, Tannock IF. Selective cellular acidification and toxicity of weak organic acids in an acidic microenvironment. Br J Cancer. 1993; 68: 1080–7. 10.3402/mehd.v23i0.19260.
- Tamai I, Takanaga H, Maeda H, Sai Y, Ogihara T, Higashida H, et al. Participation of a proton-cotransporter, MCT1, in the intestinal transport of monocarboxylic acids. Biochem Biophys Res Commun. 1995; 214: 482–9. 10.3402/mehd.v23i0.19260.
- Bergersen L, Rafiki A, Ottersen OP. Immunogold cytochemistry identifies specialized membrane domains for monocarboxylate transport in the central nervous system. Neurochem Res. 2002; 27: 89–96. 10.3402/mehd.v23i0.19260.
- Conn AR, Fell DI, Steele RD. Characterization of alpha-keto acid transport across blood-brain barrier in rats. Am J Physiol. 1983; 245: E253–60.
- Peinado A, Yuste R, Katz LC. Extensive dye coupling between rat neocortical neurons during the period of circuit formation. Neuron. 1993; 10: 103–14. 10.3402/mehd.v23i0.19260.
- Maurer MH, Canis M, Kuschinsky W, Duelli R. Correlation between local monocarboxylate transporter 1 (MCT1) and glucose transporter 1 (GLUT1) densities in the adult rat brain. Neurosci Lett. 2004; 355: 105–8. 10.3402/mehd.v23i0.19260.
- Rafiki A, Boulland JL, Halestrap AP, Ottersen OP, Bergersen L. Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience. 2003; 122: 677–88. 10.3402/mehd.v23i0.19260.
- Nakao S, Moriya Y, Furuyama S, Niederman R, Sugiya H. Propionic acid stimulates superoxide generation in human neutrophils. Cell Biol Int. 1998; 22: 331–7. 10.3402/mehd.v23i0.19260.
- DeCastro M, Nankova BB, Shah P, Patel P, Mally PV, Mishra R, et al. Short chain fatty acids regulate tyrosine hydroxylase gene expression through a cAMP-dependent signaling pathway. Brain Res Mol Brain Res. 2005; 142: 28–38. 10.3402/mehd.v23i0.19260.
- Wajner M, Latini A, Wyse AT, Dutra-Filho CS. The role of oxidative damage in the neuropathology of organic acidurias: insights from animal studies. J Inherit Metab Dis. 2004; 27: 427–48. 10.3402/mehd.v23i0.19260.
- Hara H, Haga S, Aoyama Y, Kiriyama S. Short-chain fatty acids suppress cholesterol synthesis in rat liver and intestine. J Nutr. 1999; 129: 942–8.
- Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003; 278: 25481–9. 10.3402/mehd.v23i0.19260.
- Rorig B, Klausa G, Sutor B. Intracellular acidification reduced gap junction coupling between immature rat neocortical pyramidal neurones. J Physiol. 1996; 490(pt-1): 31–49.
- Parab S, Nankova BB, La Gamma EF. Differential regulation of the tyrosine hydroxylase and enkephalin neuropeptide transmitter genes in rat PC12 cells by short chain fatty acids: concentration-dependent effects on transcription and RNA stability. Brain Res. 2007; 1132: 42–50. 10.3402/mehd.v23i0.19260.
- Schreiber J, Chapman KA, Summar ML, Ah MN, Sutton VR, MacLeod E, et al. Neurologic considerations in propionic acidemia. Mol Genet Metab. 2012; 105: 10–5. 10.3402/mehd.v23i0.19260.
- Nyhan WL, Bay C, Beyer EW, Mazi M. Neurologic nonmetabolic presentation of propionic acidemia. Arch Neurol. 1999; 56: 1143–7. 10.3402/mehd.v23i0.19260.
- Al-Owain M, Kaya N, Al-Shamrani H, Al-Bakheet A, Qari A, Al-Muaigl S, et al. Autism spectrum disorder in a child with propionic acidemia. J Inherit Metab Dis. 2012. (in press).
- Perez B, Desviat LR, Rodriguez-Pombo P, Clavero S, Navarrete R, Perez-Cerda C, et al. Propionic acidemia: identification of twenty-four novel mutations in Europe and North America. Mol Genet Metab. 2003; 78: 59–67. 10.3402/mehd.v23i0.19260.
- Yorifuji T, Kawai M, Muroi J, Mamada M, Kurokawa K, Shigematsu Y, et al. Unexpectedly high prevalence of the mild form of propionic acidemia in Japan: presence of a common mutation and possible clinical implications. Hum Genet. 2002; 111: 161–5. 10.3402/mehd.v23i0.19260.
- Desviat LR, Perez B, Perez-Cerda C, Rodriguez-Pombo P, Clavero S, Ugarte M. Propionic acidemia: mutation update and functional and structural effects of the variant alleles. Mol Genet Metab. 2004; 83: 28–37. 10.3402/mehd.v23i0.19260.
- Haska L, Andersson R, Nyman M. The effect of dietary fiber from wheat processing streams on the formation of carboxylic acids and microbiota in the hindgut of rats. J Agric Food Chem. 2011; 59: 3406–13. 10.3402/mehd.v23i0.19260.
- Stackebrandt E, Rainey FA. Phylogenetic relationships. The clostridia, molecular biology and pathogenesis. Rood JI, McClane BA, Songer JG, Titball RW. Academic Press: New York NY, 1997; 3–19.
- Song Y, Liu C, Finegold SM. Real-time PCR quantitation of clostridia in feces of autistic children. Appl Environ Microbiol. 2004; 70: 6459–65. 10.3402/mehd.v23i0.19260.
- Finegold SM. State of the art; microbiology in health and disease. Intestinal bacterial flora in autism. Anaerobe. 2011; 17: 367–8. 10.3402/mehd.v23i0.19260.
- Barton LL, Fauque GD. Biochemistry, physiology and biotechnology of sulfate-reducing bacteria. Adv Appl Microbiol. 2009; 68: 41–98.
- Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010; 1797: 1500–11. 10.3402/mehd.v23i0.19260.
- Gadalla MM, Snyder SH. Hydrogen sulfide as a gasotransmitter. J Neurochem. 2010; 113: 14–26. 10.3402/mehd.v23i0.19260.
- White JF. Intestinal pathophysiology in autism. Exp Biol Med. 2003; 228: 639–49.
- Williams BL, Hornig M, Buie T, Bauman ML, Cho PM, Wick I, et al. Impaired carbohydrate digestion and transport and mucosal dysbiosis in the intestines of children with autism and gastrointestinal disturbances. PLoS One. 2011; 6: e24585. 10.3402/mehd.v23i0.19260.
- Horvath K, Papadimitriou JC, Rabsztyn A, Drachenberg C, Tildon JT. Gastrointestinal abnormalities in children with autistic disorder. J Pediatr. 1999; 135: 559–63. 10.3402/mehd.v23i0.19260.
- Liu Z, Li N, Neu J. Tight junctions, leaky intestines, and pediatric diseases. Acta Paediatr. 2005; 94: 386–93. 10.3402/mehd.v23i0.19260.
- Nafday SM, Chen W, Peng L, Babyatsky MW, Holzman IR, Lin J. Short-chain fatty acids induce colonic mucosal injury in rats with various postnatal ages. Pediatr Res. 2005; 57: 201–4. 10.3402/mehd.v23i0.19260.
- Mellon AF, Deshpande SA, Mathers JC, Bartlett K. Effect of oral antibiotics on intestinal production of propionic acid. Arch Dis Child. 2000; 82: 169–72. 10.3402/mehd.v23i0.19260.
- Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006; 13: 171–81. 10.3402/mehd.v23i0.19260.
- James SJ, Rose S, Melnyk S, Jernigan S, Blossom S, Pavliv O, et al. Cellular and mitochondrial glutathione redox imbalance in lymphoblastoid cells derived from children with autism. FASEB J. 2009; 23: 2374–83. 10.3402/mehd.v23i0.19260.
- Moy SS, Nadler JJ, Magnuson TR, Crawley JN. Mouse models of autism spectrum disorders: the challenge for behavioral genetics. Am J Med Genet C Semin Med Genet. 2006; 142: 40–51.
- Crawley JN. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol. 2007; 17: 448–59. 10.3402/mehd.v23i0.19260.
- Hornig M, Chian D, Lipkin WI. Neurotoxic effects of postnatal thimerosal are mouse strain dependent. Mol Psychiatry. 2004; 9: 833–45. 10.3402/mehd.v23i0.19260.
- Zerrate MC, Pletnikov M, Connors SL, Vargas DL, Seidler FJ, Zimmerman AW, et al. Neuroinflammation and behavioral abnormalities after neonatal terbutaline treatment in rats: implications for autism. J Pharmacol Exp Ther. 2007; 322: 16–22. 10.3402/mehd.v23i0.19260.
- Lancaster K, Dietz DM, Moran TH, Pletnikov MV. Abnormal social behaviors in young and adult rats neonatally infected with Borna disease virus. Behav Brain Res. 2006; 176: 141–8. 10.3402/mehd.v23i0.19260.
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007; 27: 10695–702. 10.3402/mehd.v23i0.19260.
- Zwaigenbaum L, Bryson S, Rogers T, Roberts W, Brian J, Szatmari P. Behavioral manifestations of autism in the first year of life. Int J Dev Neurosci. 2005; 23: 143–52. 10.3402/mehd.v23i0.19260.
- DiCicco-Bloom E, Lord C, Zwaigenbaum L, Courchesne E, Dager SR, Schmitz C, et al. The developmental neurobiology of autism spectrum disorder. J Neurosci. 2006; 26: 6897–906. 10.3402/mehd.v23i0.19260.
- Crawley JN. Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment Retard Dev Disabil Res Rev. 2004; 10: 248–58. 10.3402/mehd.v23i0.19260.
- Rinehart NJ, Tonge BJ, Iansek R, McGinley J, Brereton AV, Enticott PG, et al. Gait function in newly diagnosed children with autism: cerebellar and basal ganglia related motor disorder. Dev Med Child Neurol. 2006; 48: 819–24. 10.3402/mehd.v23i0.19260.
- Feliz B, Witt DR, Harris BT. Propionic acidemia: a neuropathology case report and review of prior cases. Arch Pathol Lab Med. 2003; 127: e325–8.
- Besag FM. Behavioral aspects of pediatric epilepsy syndromes. Epilepsy Behav. 2004; 5(Suppl 1): S3–13. 10.3402/mehd.v23i0.19260.
- Brusque AM, Mello CF, Buchanan DN, Terracciano ST, Rocha MP, Vargas CR, et al. Effect of chemically induced propionic acidemia on neurobehavioral development of rats. Pharmacol Biochem Behav. 1999; 64: 529–34. 10.3402/mehd.v23i0.19260.
- Cannizzaro C, Monastero R, Vacca M, Martire M. [3H]-DA release evoked by low pH medium and internal H+ accumulation in rat hypothalamic synaptosomes: involvement of calcium ions. Neurochem Int. 2003; 43: 9–17. 10.3402/mehd.v23i0.19260.
- Mitsui R, Ono S, Karaki S, Kuwahara A. Neural and non-neural mediation of propionate-induced contractile responses in the rat distal colon. Neurogastroenterol Motil. 2005; 17: 585–94. 10.3402/mehd.v23i0.19260.
- Moore H, Grace AA. A role for electrotonic coupling in the striatum in the expression of dopamine receptor-mediated stereotypies. Neuropsychopharmacology. 2002; 27: 980–92. 10.3402/mehd.v23i0.19260.
- Sziray N, Leveleki C, Levay G, Marko B, Harsing LG Jr, Mikics E, et al. Mechanisms underlying the long-term behavioral effects of traumatic experience in rats: the role of serotonin/noradrenaline balance and NMDA receptors. Brain Res Bull. 2007; 71: 376–85. 10.3402/mehd.v23i0.19260.
- Neuhaus E, Beauchaine TP, Bernier R. Neurobiological correlates of social functioning in autism. Clin Psychol Rev. 2010; 30: 733–48. 10.3402/mehd.v23i0.19260.
- El-Ansary AK, Ben BA, Kotb M. Etiology of autistic features: the persisting neurotoxic effects of propionic acid. J Neuroinflammation. 2012; 9: 74. 10.3402/mehd.v23i0.19260.
- Bonnet U, Bingmann D, Wiemann M. Intracellular pH modulates spontaneous and epileptiform bioelectric activity of hippocampal CA3-neurones. Eur Neuropsychopharmacol. 2000; 10: 97–103. 10.3402/mehd.v23i0.19260.
- Severson CA, Wang W, Pieribone VA, Dohle CI, Richerson GB. Midbrain serotonergic neurons are central pH chemoreceptors. Nat Neurosci. 2003; 6: 1139–40. 10.3402/mehd.v23i0.19260.
- Shah P, Nankova BB, Parab S, La Gamma EF. Short chain fatty acids induce TH gene expression via ERK-dependent phosphorylation of CREB protein. Brain Res. 2006; 1107: 13–23. 10.3402/mehd.v23i0.19260.
- Ming X, Julu PO, Brimacombe M, Connor S, Daniels ML. Reduced cardiac parasympathetic activity in children with autism. Brain Dev. 2005; 27: 509–16. 10.3402/mehd.v23i0.19260.
- Rigo FK, Pasquetti L, Malfatti CR, Fighera MR, Coelho RC, Petri CZ, et al. Propionic acid induces convulsions and protein carbonylation in rats. Neurosci Lett. 2006; 408: 151–4. 10.3402/mehd.v23i0.19260.
- Gogolla N, Leblanc JJ, Quast KB, Sudhof T, Fagiolini M, Hensch TK. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord. 2009; 1: 172–81. 10.3402/mehd.v23i0.19260.
- de Almeida LM, Funchal C, Pelaez PL, Pessutto FD, Loureiro SO, Vivian L, et al. Effect of propionic and methylmalonic acids on the in vitro phosphorylation of intermediate filaments from cerebral cortex of rats during development. Metab Brain Dis. 2003; 18: 207–19. 10.3402/mehd.v23i0.19260.
- Clavero S, Perez B, Rincon A, Ugarte M, Desviat LR. Qualitative and quantitative analysis of the effect of splicing mutations in propionic acidemia underlying non-severe phenotypes. Hum Genet. 2004; 115: 239–47. 10.3402/mehd.v23i0.19260.
- Hanstock TL, Clayton EH, Li KM, Mallet PE. Anxiety and aggression associated with the fermentation of carbohydrates in the hindgut of rats. Physiol Behav. 2004; 82: 357–68. 10.3402/mehd.v23i0.19260.
- Puwanant M, Mo-Suwan L, Patrapinyokul S. Recurrent D-lactic acidosis in a child with short bowel syndrome. Asia Pac J Clin Nutr. 2005; 14: 195–8.
- Brass EP, Beyerinck RA. Effects of propionate and carnitine on the hepatic oxidation of short- and medium-chain-length fatty acids. J Biochem. 1988; 250: 819–25.
- Filipek PA, Juranek J, Nguyen MT, Cummings C, Gargus JJ. Relative carnitine deficiency in autism. J Autism Dev Disord. 2004; 34: 615–23. 10.3402/mehd.v23i0.19260.
- Kim KC, Kim P, Go HS, Choi CS, Yang SI, Cheong JH, et al. The critical period of valproate exposure to induce autistic symptoms in Sprague-Dawley rats. Toxicol Lett. 2011; 201: 137–42. 10.3402/mehd.v23i0.19260.
- Coulter DL. Carnitine, valproate, and toxicity. J Child Neurol. 1991; 6: 7–14. 10.3402/mehd.v23i0.19260.
- Schulpis KH, Karikas GA, Tjamouranis J, Regoutas S, Tsakiris S. Low serum biotinidase activity in children with valproic acid monotherapy. Epilepsia. 2001; 42: 1359–62. 10.3402/mehd.v23i0.19260.
- Borlongan CV, Koutouzis TK, Sanberg PR. 3-Nitropropionic acid animal model and Huntington's disease. Neurosci Biobehav Rev. 1997; 21: 289–93. 10.3402/mehd.v23i0.19260.
- Chauhan A, Chauhan V, Brown WT, Cohen I. Oxidative stress in autism: increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin – the antioxidant proteins. Life Sci. 2004; 75: 2539–49. 10.3402/mehd.v23i0.19260.
- Chauhan A, Audhya T, Chauhan V. Brain region-specific glutathione redox imbalance in autism. Neurochem Res. 2012; 37: 1681–9. 10.3402/mehd.v23i0.19260.
- Monks TJ, Ghersi-Egea JF, Philbert M, Cooper AJ, Lock EA. Symposium overview: the role of glutathione in neuroprotection and neurotoxicity. Toxicol Sci. 1999; 51: 161–77. 10.3402/mehd.v23i0.19260.
- James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, et al. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr. 2004; 80: 1611–7.
- El-Ansary A, Al-Daihan S, Al-Dbass A, Al-Ayadhi L. Measurement of selected ions related to oxidative stress and energy metabolism in Saudi autistic children. Clin Biochem. 2009; 43: 63–70. 10.3402/mehd.v23i0.19260.
- Al-Gadani Y, El-Ansary A, Attas O, Al-Ayadhi L. Metabolic biomarkers related to oxidative stress and antioxidant status in Saudi autistic children. Clin Biochem. 2009; 42: 1032–40. 10.3402/mehd.v23i0.19260.
- Torres AR. Is fever suppression involved in the etiology of autism and neurodevelopmental disorders?. BMC Pediatr. 2003; 3: 9. 10.3402/mehd.v23i0.19260.
- Neustadt J, Pieczenik SR. Medication-induced mitochondrial damage and disease. Mol Nutr Food Res. 2008; 52: 780–8. 10.3402/mehd.v23i0.19260.
- Schultz ST, Klonoff-Cohen HS, Wingard DL, Akshoomoff NA, Macera CA, Ji M. Acetaminophen (paracetamol) use, measles-mumps-rubella vaccination, and autistic disorder: the results of a parent survey. Autism. 2008; 12: 293–307. 10.3402/mehd.v23i0.19260.
- Noble M, Proschel C, Mayer-Proschel M. Oxidative-reductionist approaches to stem and progenitor cell function. Cell Stem Cell. 2011; 8: 1–2. 10.3402/mehd.v23i0.19260.
- Rorig B, Sutor B. Serotonin regulates gap junction coupling in the developing rat somatosensory cortex. Eur J Neurosci. 1996; 8: 1685–95. 10.3402/mehd.v23i0.19260.
- Anders JJ. Lactic acid inhibition of gap junctional intercellular communication in in vitro astrocytes as measured by fluorescence recovery after laser photobleaching. GLIA. 1988; 1: 371–9. 10.3402/mehd.v23i0.19260.
- Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci. 2002; 22: 644–53.
- Perez-Velazquez JL, Frantseva MV, Naus CC. Gap junctions and neuronal injury: protectants or executioners?. Neuroscientist. 2003; 9: 5–9. 10.3402/mehd.v23i0.19260.
- Juszczak GR, Swiergiel AH. Properties of gap junction blockers and their behavioural, cognitive and electrophysiological effects: animal and human studies. Prog Neuropsychopharmacol Biol Psychiatry. 2009; 33: 181–98. 10.3402/mehd.v23i0.19260.
- Wiencken-Barger AE, Djukic B, Casper KB, McCarthy KD. A role for Connexin43 during neurodevelopment. GLIA. 2007; 55: 675–86. 10.3402/mehd.v23i0.19260.
- Nagasawa K, Chiba H, Fujita H, Kojima T, Saito T, Endo T, et al. Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J Cell Physiol. 2006; 208: 123–32. 10.3402/mehd.v23i0.19260.
- Pardo CA, Eberhart CG. The neurobiology of autism. Brain Pathol. 2007; 17: 434–47. 10.3402/mehd.v23i0.19260.
- Madhusudan A, Vogel P, Knuesel I. Impact of prenatal immune system disturbances on brain development. J Neuroimmune Pharmacol. in press.
- Dringen R. Oxidative and antioxidative potential of brain microglial cells. Antioxid Redox Signal. 2005; 7: 1223–33. 10.3402/mehd.v23i0.19260.
- Carlezon WA Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005; 28: 436–45. 10.3402/mehd.v23i0.19260.
- Wang Z, Xu L, Zhu X, Cui W, Sun Y, Nishijo H, et al. Demethylation of specific Wnt/beta-catenin pathway genes and its upregulation in rat brain induced by prenatal valproate exposure. Anat Rec (Hoboken). 2010; 293: 1947–53. 10.3402/mehd.v23i0.19260.
- Nankova BB, La Gamma EF, Taylor AR, Tichenoff L, MacFabe DF. Intraventricular enteric short chain fatty acid infusions in rats induce behavioural, neuropathological, lipid and epigenetic changes consistent with Autism. abstr. International Meeting for Autism Research, May 17–19, 2012, Toronto.
- Bell JG, Sargent JR, Tocher DR, Dick JR. Red blood cell fatty acid compositions in a patient with autistic spectrum disorder: a characteristic abnormality in neurodevelopmental disorders?. Prostaglandins Leukot Essent Fatty Acids. 2000; 63: 21–5. 10.3402/mehd.v23i0.19260.
- Vancassel S, Durand G, Barthelemy C, Lejeune B, Martineau J, Guilloteau D, et al. Plasma fatty acid levels in autistic children. Prostaglandins Leukot Essent Fatty Acids. 2001; 65: 1–7. 10.3402/mehd.v23i0.19260.
- Richardson AJ. Clinical trials of fatty acid treatment in ADHD, dyslexia, dyspraxia and the autistic spectrum. Prostaglandins Leukot Essent Fatty Acids. 2004; 70: 383–90. 10.3402/mehd.v23i0.19260.
- Amminger GP, Berger GE, Schafer MR, Klier C, Friedrich MH, Feucht M. Omega-3 fatty acids supplementation in children with autism: a double-blind randomized, placebo-controlled pilot study. Biol Psychiatry. 2007; 61: 551–3. 10.3402/mehd.v23i0.19260.
- Wiest MM, German JB, Harvey DJ, Watkins SM, Hertz-Picciotto I. Plasma fatty acid profiles in autism: a case-control study. Prostaglandins Leukot Essent Fatty Acids. 2009; 80: 221–7. 10.3402/mehd.v23i0.19260.
- Tamiji J, Crawford DA. The neurobiology of lipid metabolism in autism spectrum disorders. Neurosignals. 2010; 18: 98–112. 10.3402/mehd.v23i0.19260.
- Bell JG, MacKinlay EE, Dick JR, MacDonald DJ, Boyle RM, Glen AC. Essential fatty acids and phospholipase A2 in autistic spectrum disorders. Prostaglandins Leukot Essent Fatty Acids. 2004; 71: 201–4. 10.3402/mehd.v23i0.19260.
- Meguid NA, Atta HM, Gouda AS, Khalil RO. Role of polyunsaturated fatty acids in the management of Egyptian children with autism. Clin Biochem. 2008; 41: 1044–8. 10.3402/mehd.v23i0.19260.
- Aneja A, Tierney E. Autism: the role of cholesterol in treatment. Int Rev Psychiatry. 2008; 20: 165–70. 10.3402/mehd.v23i0.19260.
- Champeil-Potokar G, Chaumontet C, Guesnet P, Lavialle M, Denis I. Docosahexaenoic acid (22:6n-3) enrichment of membrane phospholipids increases gap junction coupling capacity in cultured astrocytes. Eur J Neurosci. 2006; 24: 3084–90. 10.3402/mehd.v23i0.19260.
- Zhao S, Jia L, Gao P, Li Q, Lu X, Li J, et al. Study on the effect of eicosapentaenoic acid on phospholipids composition in membrane microdomains of tight junctions of epithelial cells by liquid chromatography/electrospray mass spectrometry. J Pharm Biomed Anal. 2008; 47: 343–50. 10.3402/mehd.v23i0.19260.
- Jones LL, McDonald DA, Borum PR. Acylcarnitines: role in brain. Prog Lipid Res. 2010; 49: 61–75. 10.3402/mehd.v23i0.19260.
- Clarke JT, Clark-Taylor BE. Is autism a disorder of fatty acid metabolism? Possible dysfunction of mitochondrial beta oxidation by long chain acyl-CoA dehydrogenase. Med Hypotheses. 2004; 62: 970–5. 10.3402/mehd.v23i0.19260.
- Frye RE. Biomarkers of abnormal energy metabolism in children with autism spectrum disorder. N Am J Med Sci. in press.
- Pastural E, Ritchie S, Lu Y, Jin W, Kavianpour A, Khine Su-Myat K, et al. Novel plasma phospholipid biomarkers of autism: mitochondrial dysfunction as a putative causative mechanism. Prostaglandins Leukot Essent Fatty Acids. 2009; 81: 253–64. 10.3402/mehd.v23i0.19260.
- Bu B, Ashwood P, Harvey D, King IB, Water JV, Jin LW. Fatty acid compositions of red blood cell phospholipids in children with autism. Prostaglandins Leukot Essent Fatty Acids. 2006; 74: 215–21. 10.3402/mehd.v23i0.19260.
- Brass EP, Fennessey PV, Miller LV. Inhibition of oxidative metabolism by propionic acid and its reversal by carnitine in isolated rat hepatocytes. Biochem J. 1986; 236: 131–6.
- Roe RC, Millington DS, Maltby DA, Bohan TP, Hoppel CL. L-carnitine enhances excretion of propoinyl coenzyme A as propionylcarnitine in propoinic acidemia. J Clin Invest. 1984; 73: 1785–8. 10.3402/mehd.v23i0.19260.
- Tonin AM, Grings M, Knebel LA, Zanatta A, Moura AP, Ribeiro CA, et al. Disruption of redox homeostasis in cerebral cortex of developing rats by acylcarnitines accumulating in medium-chain acyl-CoA dehydrogenase deficiency. Int J Dev Neurosci. 2012; 30: 383–90. 10.3402/mehd.v23i0.19260.
- Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, van den Heuvel LP, et al. Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J. 2006; 398: 107–12. 10.3402/mehd.v23i0.19260.
- Brass EP. Interaction of carnitine and propionate with pyruvate oxidation by hepatocytes from clofibrate-treated rats: importance of coenzyme A availability. J Nutr. 1992; 122: 234–40.
- Wolever TM, Fernandes J, Rao AV. Serum acetate: propionate ratio is related to serum cholesterol in men but not women. J Nutr. 1996; 126: 2790–7.
- Wolever TM, Spadafora PJ, Cunnane SC, Pencharz PB. Propionate inhibits incorporation of colonic [1,2-13C]acetate into plasma lipids in humans. Am J Clin Nutr. 1995; 61: 1241–7.
- James SJ, Melnyk S, Jernigan S, Cleves MA, Halsted CH, Wong DH, et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am J Med Genet B Neuropsychiatr Genet. 2006; 141: 947–56.
- Wegiel J, Kuchna I, Nowicki K, Imaki H, Wegiel J, Marchi E, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010; 119: 755–70. 10.3402/mehd.v23i0.19260.
- Scafidi S, Fiskum G, Lindauer SL, Bamford P, Shi D, Hopkins I, et al. Metabolism of acetyl-L-carnitine for energy and neurotransmitter synthesis in the immature rat brain. J Neurochem. 2010; 114: 820–31. 10.3402/mehd.v23i0.19260.
- James SJ, Melnyk S, Fuchs G, Reid T, Jernigan S, Pavliv O, et al. Efficacy of methylcobalamin and folinic acid treatment on glutathione redox status in children with autism. Am J Clin Nutr. 2009; 89: 425–30. 10.3402/mehd.v23i0.19260.
- Miecz D, Januszewicz E, Czeredys M, Hinton BT, Berezowski V, Cecchelli R, et al. Localization of organic cation/carnitine transporter (OCTN2) in cells forming the blood-brain barrier. J Neurochem. 2008; 104: 113–23.
- Yamamoto-Furusho JK, Mendivil-Rangel EJ, Villeda-Ramirez MA, Fonseca-Camarillo G, Barreto-Zuniga R. Gene expression of carnitine organic cation transporters 1 and 2 (OCTN) is downregulated in patients with ulcerative colitis. Inflamm Bowel Dis. 2011; 17: 2205–6. 10.3402/mehd.v23i0.19260.
- Yao D, Kuwajima M, Chen Y, Shiota M, Okumura Y, Yamada H, et al. Impaired long-chain fatty acid metabolism in mitochondria causes brain vascular invasion by a non-neurotropic epidemic influenza A virus in the newborn/suckling period: implications for influenza-associated encephalopathy. Mol Cell Biochem. 2007; 299: 85–92. 10.3402/mehd.v23i0.19260.
- Fallon J. Could one of the most widely prescribed antibiotics amoxicillin/clavulanate “augmentin” be a risk factor for autism?. Med Hypotheses. 2005; 64: 312–5. 10.3402/mehd.v23i0.19260.
- Sandler RH, Finegold SM, Bolte ER, Buchanan CP, Maxwell AP, Vaisanen ML, et al. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J Child Neurol. 2000; 15: 429–35. 10.3402/mehd.v23i0.19260.
- Mann JR, McDermott S, Bao H, Hardin J, Gregg A. Pre-eclampsia, birth weight, and autism spectrum disorders. J Autism Dev Disord. 2010; 40: 548–54. 10.3402/mehd.v23i0.19260.
- Thiele IG, Niezen-Koning KE, van Gennip AH, Aarnoudse JG. Increased plasma carnitine concentrations in preeclampsia. Obstet Gynecol. 2004; 103: 876–80. 10.3402/mehd.v23i0.19260.
- Scafidi S, Racz J, Hazelton J, McKenna MC, Fiskum G. Neuroprotection by acetyl-L-carnitine after traumatic injury to the immature rat brain. Dev Neurosci. 2010; 32: 480–7.
- Patel SP, Sullivan PG, Lyttle TS, Rabchevsky AG. Acetyl-L-carnitine ameliorates mitochondrial dysfunction following contusion spinal cord injury. J Neurochem. 2010; 114: 291–301.
- Fortin G, Yurchenko K, Collette C, Rubio M, Villani AC, Bitton A, et al. L-carnitine, a diet component and organic cation transporter OCTN ligand, displays immunosuppressive properties and abrogates intestinal inflammation. Clin Exp Immunol. 2009; 156: 161–71. 10.3402/mehd.v23i0.19260.
- Rossignol DA, Rossignol LW, Smith S, Schneider C, Logerquist S, Usman A, et al. Hyperbaric treatment for children with autism: a multicenter, randomized, double-blind, controlled trial. BMC Pediatr. 2009; 9: 21. 10.3402/mehd.v23i0.19260.
- Willner P. The validity of animal models of depression. Psychopharmacology (Berl). 1984; 83: 1–16. 10.3402/mehd.v23i0.19260.
- Willner P. Animal models as simulations of depression. Trends Pharmacol Sci. 1991; 12: 131–6. 10.3402/mehd.v23i0.19260.
- Bresolin N, Freddo L, Vergani L, Angelini C. Carnitine, carnitine acyltransferases, and rat brain function. Exp Neurol. 1982; 78: 285–92. 10.3402/mehd.v23i0.19260.
- Kavaliers M, Choleris E, Agmo A, Pfaff DW. Olfactory-mediated parasite recognition and avoidance: linking genes to behavior. Horm Behav. 2004; 46: 272–83. 10.3402/mehd.v23i0.19260.
- Kaushik M, Lamberton PH, Webster JP. The role of parasites and pathogens in influencing generalised anxiety and predation-related fear in the mammalian central nervous system. Horm Behav. in press.
- Heijtz RD, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, et al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA. 2011; 108: 3047–52. 10.3402/mehd.v23i0.19260.
- Suren P, Bakken IJ, Aase H, Chin R, Gunnes N, Lie KK, et al. Autism spectrum disorder, ADHD, epilepsy, and cerebral palsy in norwegian children. Pediatrics. 2012; 130: e152–8. 10.3402/mehd.v23i0.19260.
- Sokol MS. Infection-triggered anorexia nervosa in children: clinical description of four cases. J Child Adolesc Psychopharmacol. 2000; 10: 133–45. 10.3402/mehd.v23i0.19260.
- Kerbeshian J, Burd L. Is anorexia nervosa a neuropsychiatric developmental disorder? An illustrative case report. World J Biol Psychiatry. 2009; 10: 648–57. 10.3402/mehd.v23i0.19260.
- Diamond J. Guns, germs, and steel. New York: W.W. Norton & Company. 1997.
- Foley KA, Kavaliers M, Ossenkopp K-P, MacFabe DF. Prenatal exposure to propionic acid and lipopolysaccharides produces developmental delay, anxiety-like behaviour, and hyper-sensitivity to accoustic startle in adolescent rats. abstr. International Meeting for Autism Research, May17–19, 2012, Toronto.