Abstract
Obesity is a significant risk factor for developing osteoarthritis in weight-bearing and non-weight-bearing joints. Although the pathogenesis of obesity-associated osteoarthritis is not completely understood, recent studies indicate that pro-inflammatory metabolic factors contribute to an increase in osteoarthritis risk. Adipose tissue, and in particular infrapatellar fat, is a local source of pro-inflammatory mediators that are increased with obesity and have been shown to increase cartilage degradation in cell and tissue culture models. One adipokine in particular, leptin, may be a critical mediator of obesity-associated osteoarthritis via synergistic actions with other inflammatory cytokines. Biomechanical factors may also increase the risk of osteoarthritis by activating cellular inflammation and promoting oxidative stress. However, some types of biomechanical stimulation, such as physiologic cyclic loading, inhibit inflammation and protect against cartilage degradation. A high percentage of obese individuals with knee osteoarthritis are sedentary, suggesting that a lack of physical activity may increase the susceptibility to inflammation. A more comprehensive approach to understanding how obesity alters daily biomechanical exposures within joint tissues may provide new insight into the protective and damaging effects of biomechanical factors on inflammation in osteoarthritis.
Osteoarthritis is a painful diarthrodial joint disease that develops as a result of an insufficient and often aberrant repair of damaged synovial joint tissue. Common features of osteoarthritis include progressive focal lesions in the articular cartilage, abnormal bone remodeling, meniscal tears, mild synovitis, and muscle weakness. Aging is the primary predictor of osteoarthritis, with most people older than 70 years of age showing some signs of disease in at least one joint Citation1. The etiology of aging-associated osteoarthritis is not well understood, although cellular senescence may contribute to disease pathology. Many of the pro-inflammatory mediators and proteases attributed to a senescence-associated secretory phenotype in fibroblasts, such as Interluekin-1β (IL-1β), IL-6, IL-8, matrix metalloproteinase 3 (MMP-3), and MMP-13, are commonly increased in osteoarthritic tissues Citation2 Citation3. These pro-inflammatory mediators, along with nitric oxide and prostaglandin E2, promote an imbalance in the synthesis and degradation of the cartilage extracellular matrix that eventually lead to cartilage degradation Citation4. Identifying the age-associated changes in chondrocyte biology that promote the cellular senescence phenotype is an active area of research, with current studies indicating a role for DNA damage, mitochondrial dysfunction, oxidative stress, and cellular stress protective mechanisms Citation3.
The extent to which circulating inflammatory factors contribute to osteoarthritis pathology is uncertain. Synovial fluid and serum inflammatory cytokines, such as IL-1, IL-6, IL-8 and Tumor Necrosis Factor-α (TNF-α), are elevated in osteoarthritis patient cohorts and animal models, suggesting that systemic inflammation initiates osteoarthritis pathology Citation5. A recent study provided additional evidence linking systemic inflammation and osteoarthritis, but from a different point of view. Rather than focusing on factors associated with the presence of osteoarthritis, the authors investigated inflammatory factors associated with the absence of osteoarthritis in an aged cohort. When older adults with and without osteoarthritis were compared, those individuals without osteoarthritis showed a reduced production of IL-1β and IL-6 in whole blood samples stimulated with lipopolysaccharide Citation6. Thus, resistance to a systemic inflammatory stress may protect against osteoarthritis. Recent genetic studies of inflammatory marker polymorphisms and knee osteoarthritis indicate that IL-1 receptor antagonist (IL-1Ra) gene polymorphisms may be associated with differences in the severity of age-dependent knee osteoarthritis Citation7. The ability of inflammatory genetic markers or other systemic inflammatory mediators to predict osteoarthritis risk remains an important area of investigation, especially in the study of obesity-associated osteoarthritis.
Obesity and inflammation in osteoarthritis
Obesity is one of the most significant preventable risk factors for developing knee osteoarthritis Citation8. One way in which obesity may increase the risk of osteoarthritis is by altering knee joint loading patterns, such as occurs with varus/valgus laxity and knee joint malalignment Citation9 Citation10. However, mechanical factors alone do not seem to be sufficient to explain the relationship between osteoarthritis incidence and obesity Citation11 Citation12. Obesity also increases the risk of developing osteoarthritis in non-weight-bearing joints such as the hand, and several recent studies, summarized below, suggest that systemic inflammatory mediators contribute to the increased risk of osteoarthritis with obesity. Adipose tissue, especially from the abdomen, is a rich source of pro-inflammatory cytokines, which are often referred to as adipokines. Many of the adipokines elevated with obesity and metabolic syndrome have also been shown to mediate synovial tissue inflammation and upregulate cartilage matrix synthesis and degradation Citation5 Citation13. In a recent study, cardiometabolic disease clustering increased the risk of osteoarthritis even when controlling for BMI Citation14, suggesting that a common set of metabolic factors underlie both cardiovascular and osteoarthritis disease conditions. Osteoarthritis, like cardiovascular disease, involves altered lipid metabolism coupled with increased systemic and cellular expression of pro-inflammatory mediators Citation15 Citation16. Establishing a causal relationship between osteoarthritis and metabolic inflammation is difficult because both factors are correlated with changes in joint loading biomechanics that occur with obesity. Nevertheless, a growing number of studies, although not all Citation17, support a role for metabolic factors in osteoarthritis Citation3 Citation11 Citation15 Citation18–Citation23.
One of the central mediators being investigated for a potential metabolic-osteoarthritis link is leptin. Leptin is a 16-kd polypeptide hormone encoded by the obese (ob) gene Citation24. Leptin is primarily secreted by adipocytes, functioning as an afferent signal in a hypothalamic negative-feedback loop to regulate adipose tissue mass and body weight. We have previously reported that female mice with impaired leptin signaling are protected from obesity-induced osteoarthritis Citation19. These findings indicate that elevated body fat in the absence of leptin signaling is insufficient to induce systemic inflammation and osteoarthritis in C57BL/6J mice. Moreover, in a 45-week diet-induced obesity study in female C57BL/6J mice, we found that serum leptin concentrations predicted knee osteoarthritis severity even when statistically controlling for the effect of diet and percentage body fat Citation18.
The extent to which leptin mediates the onset versus the progression of osteoarthritis is uncertain. The primary pro-inflammatory and catabolic effects of leptin in chondrocytes occur through co-stimulation with other inflammatory mediators, such as IL-1, TNF-α, and IFN-γ Citation21 Citation25. Furthermore, there is some evidence that tissue disease status and patient BMI are important determinants of leptin-mediated cartilage degeneration. We previously showed that physiologic doses of leptin were not sufficient to induce or synergistically increase cartilage nitric oxide production or proteoglycan release in healthy porcine cartilage explants Citation18. Moreover, when osteoarthritic chondrocytes are obtained from patients with a range of body mass indexes, MMP13 gene expression was only altered by physiologic concentrations of leptin in cells from obese patients Citation23. It is not known if leptin receptor expression is increased in chondrocytes from obese patients independent from osteoarthritis disease status Citation26. As leptin has also been shown to increase the expression of anabolic growth factors, such as IGF-1 and TGF-β Citation27, increased sensitivity to leptin within osteoarthritic cells appears to mediate an overall increase in cartilage extracellular matrix turnover, a characteristic of early to middle stages of disease.
Recent studies indicate a potentially important role for the infrapatellar fat pad in mediating intra-articular knee inflammation Citation21 Citation28–Citation31. In particular, Hui et al. Citation21 used conditioned medium from ex vivo cultures of infrapatellar fat to show that leptin secretion from infrapatellar fat is sufficient to induce MMP1 and MMP13 gene expression in osteoarthritic chondrocytes. Leptin is unique among other adipokines analyzed to date in that only leptin has been found to exist at higher concentrations in the synovial fluid compared to serum, suggesting an important role for infrapatellar fat in intra-articular leptin production Citation32. Moreover, the infrapatellar fat pad also secretes IL-6, adiponectin, and visfatin at greater levels than subcutaneous fat in ex vivo culture Citation31. An important question that remains to be addressed is to what extent cartilage and synovial inflammation drive infrapatellar fat inflammation. Most studies investigating inflammation in the infrapatellar fat pad harvest fat tissue from severely osteoarthritic joints during joint replacement surgery. Bastiaansen-Jenniskens et al. Citation28 reported increased levels of immuno-regulatory CD206+ macrophages in infrapatellar fat harvested from osteoarthritic joints compared to joints undergoing ACL repair. The increased population of immuno-regulatory macrophages may explain the unexpected finding that infrapatellar fat-conditioned media from osteoarthritic joints inhibits the function of catabolic mediators in healthy cartilage Citation28. Future studies are needed to characterize the inflammatory phenotype of infrapatellar fat pads prior to the development of osteoarthritis. Testing this question in lean and obese animal models may provide much needed insight into the role of infrapatellar fat and inflammation in obesity-associated osteoarthritis.
Biomechanical modulation of cartilage inflammation
Numerous factors, such as physical activity levels, body weight, and skeletal anatomy, affect the range of mechanical forces acting on synovial joint tissues. Mechanical signals are not only generally well-tolerated by cells embedded within load-bearing tissues such as articular cartilage, they are essential for normal function and maintenance of the extracellular matrix Citation33. Cartilage is subjected to mechanical forces in the form of compression, shear stress, and hydrostatic pressure Citation34. These mechanical signals are sensed by chondrocytes and activate signaling pathways that promote tissue homeostasis by balancing the production of anabolic growth factors and catabolic cytokines and inflammatory mediators, including IGF-1, TGF-β, IL-10, IL-1, IL-6, TNF-α, prostaglandin E2, and nitric oxide Citation33 Citation35. Pro-inflammatory mediators are highly expressed under conditions of acute or chronic high intensity loading Citation12 Citation36 Citation37. These catabolic loads also significantly increase the production of reactive oxygen species, which stimulate a pro-oxidative cellular environment leading to proteolytic enzyme activation and matrix degradation Citation38 Citation39. Understanding how different forms, magnitudes, and frequencies of mechanical stimulation activate pro-anabolic and pro-catabolic pathways may lead to new biomechanical-based therapeutic approaches for treating obesity-associated osteoarthritis.
Anti-inflammatory mechanisms
Exercise is generally considered anti-inflammatory and therefore may be an important treatment for reducing osteoarthritis risk, especially in obese individuals with metabolic inflammation Citation40. However, current evidence on how increased physical activity affects the risk of developing osteoarthritis in obese individuals is inconclusive, perhaps due to the reliance on self-report recall of physical activity. Therefore, we sought to study the interaction of obesity and exercise on the development of osteoarthritis using an animal model Citation20. After 4 weeks of feeding mice a very high-fat diet, mice were divided into exercise and sedentary groups for an additional 4 weeks. We found that voluntary wheel running was protective or neutral rather than damaging, indicating that increased joint loading per se is not sufficient to explain the increased incidence of knee osteoarthritis in diet-induced obese mice. Furthermore, exercise improved glucose tolerance and disrupted the co-expression of systemic pro-inflammatory cytokines. Both lean and obese mice responded to the exercise stimulus with an adaptive increase in heart mass relative to lean tissue mass and without any change in body mass or percent body fat. Thus, like the benefits of being ‘fit and fat’ rather than ‘thin and sedentary’ for cardiovascular disease Citation41, these findings suggest that increased aerobic exercise may act independent of weight loss in promoting joint health.
Exercise may also be protective against inflammation and matrix catabolism through direct biomechanical stimulation of articular cartilage. In a rabbit model of antigen-induced arthritis, cyclic passive joint motion reduced the expression of pro-inflammatory mediators IL-1β and cyclooxygenase-2 and increased the expression of the anti-inflammatory cytokine IL-10 Citation42 Citation43. Additional work by Agarwal et al. has shown that NF-κB is a critical mechano-sensitive transcription factor that can either be inhibited or activated depending on the type of biomechanical stimulation. Cyclic biomechanical loading suppresses NF-κB activation and down-regulates IL-1α and TNF-α dependent gene transcription Citation44 Citation45. Cyclic loading also reduces the expression of inflammatory-induced cartilage catabolic mediators, matrix metalloproteinases and aggrecanases, as well as cartilage matrix degradation Citation44–Citation48. In contrast, static and high-strain loads activate NF-κB signaling and downstream inflammatory mediators, including inducible nitric oxide synthase, prostaglandin E2, reactive oxygen species, and pro-inflammatory cytokines Citation34 Citation43 Citation49 Citation50. Damaging biomechanical loads also lead to cellular ATP depletion Citation51 Citation52 and apoptosis Citation53. Thus, identifying how different magnitudes, durations, and frequencies of mechanical stimulation regulate the transition between anti-inflammatory and pro-inflammatory mechanisms is critical for understanding disease pathogenesis and developing effective therapeutic targets.
Redox regulation
As with obesity, a central cellular characteristic of osteoarthritis is an increase in the production of reactive oxygen species, cellular oxidation Citation35 Citation54, and apoptosis Citation55 Citation56. Maintaining oxidative homeostasis requires a sufficient activity of endogenous antioxidant enzymes, such as superoxide dismutase (SOD), catalase, and glutathione, which appear to be impaired with aging Citation57–Citation59. Biomechanical stress resulting from injurious or static loads can also be a source of pro-oxidant stress in chondrocytes by inducing the production of reactive oxygen species Citation60. Superoxide anion is thought to be the primary reactive oxygen species that is biomechanically induced through the activation of NADPH oxidase or mitochondrial stress Citation61 Citation62. Superoxide anion is not stable and is readily transformed to hydrogen peroxide (H2O2) by SOD. H2O2 inhibits protein and DNA synthesis in cartilage tissue through the depletion of ATP Citation63. Supplementing chondrocytes with antioxidants prior to stressful loading can protect against cell death and matrix degradation, suggesting that biomechanically-induced oxidative stress could be reduced with an increase in endogenous antioxidant activities Citation39 Citation64–Citation66. In fact, treatment with low repetitive doses of H2O2 preconditions cartilage against biomechanical stress Citation67.
Cyclic biomechanical loading may precondition cartilage with a regulated production of pro-oxidants leading to enhanced antioxidant activities and improved cellular redox control. Low to moderate levels of pro-oxidants act as signaling mediators in pathways involving transcriptional regulation of cytokines, proteinases, adhesion molecules, and extracellular matrix components Citation68–Citation70. One potential mechanism for biomechanically-induced upregulation of endogenous antioxidant capacity is through stabilization of the transcription factor NF-E2-related factor-2 (Nrf2). Nrf2 protects cells and tissues from oxidative stress by activating a family of antioxidant proteins containing the antioxidant responsive element enhancer sequence Citation71. Enhanced antioxidant capacity would improve cellular redox regulation and may also explain how cyclic biomechanical loading downregulates the NF-κB signaling pathway, which involves several redox-sensitive signaling elements. Thus, the regulated production of reactive oxygen species via physiologic biomechanical stimulation may increase endogenous anti-oxidant capacity, thereby providing an adaptive response that may protect against joint inflammation and cellular oxidative stress Citation72 Citation73.
Conclusion
Obesity and osteoarthritis: treatment challenges and opportunities
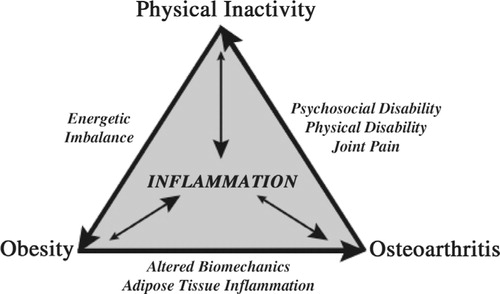
Inflammation is a central component of a progressive disease cycle involving obesity, osteoarthritis, and physical inactivity (). Metabolic inflammation associated with obesity is believed to exacerbate the condition by contributing to metabolic inflexibility and the sustained production of pro-inflammatory mediators Citation74. Metabolic inflammation also appears to increase osteoarthritis risk, although the mechanisms for this association are not yet well understood. Several recent studies suggest that metabolic inflammation and hyperlipidemia increase the susceptibility of chondrocytes to biomechanically-induced cellular stress, as occurs following joint injury Citation11 Citation22. Cyclic physiologic loading of the limbs, as occurs with a physically active lifestyle and exercise, is an effective means of reducing inflammation systemically as well as locally in adipose tissue and cartilage. Obesity is in part attributed to a reduction in physical activity levels, and recent studies indicate that knee osteoarthritis greatly exacerbates this problem. Approximately 10% of obese adults with knee osteoarthritis meet the recommended levels of daily physical activity and nearly 50% are physically inactive, with no moderate-to-vigorous activity ≥10 min during the previous week Citation75. These and other data indicate that osteoarthritis is a major barrier to being physically active Citation76. Physical inactivity, like obesity, is considered an independent risk factor for inflammation Citation77. Thus, a decrease in regular joint loading may increase the susceptibility of joint tissues to inflammatory stress. An important challenge for the future is to integrate quantitative measures of systemic and local inflammation with biomechanical exposure data. Does obesity cause local knee inflammation by altering load distribution and increasing joint stress? Or does a lack of regular cyclic joint loading increase metabolic inflammation and the susceptibility of synovial joint tissues to cellular oxidative stress and extracellular matrix degradation? The answers to these questions are not mutually exclusive, and they highlight the challenges ahead to understand the pathogenesis of obesity-associated osteoarthritis.
Fig. 1. Progressive relationship among obesity, osteoarthritis, and physical inactivity. Obesity is an independent risk factor for osteoarthritis. The mechanisms responsible for this link are not completely understood but are thought to involve altered biomechanical loading and metabolic inflammation associated with excess adipose tissue and lipids. Osteoarthritis pain increases sedentary behavior and is associated with psychosocial and physical disability. Physical inactivity is an independent risk factor for inflammation due to the reduced expression of systemic and cellular anti-inflammatory mediators. Physiologic cyclic loading of cartilage tissue has been shown to reduce the expression of pro-inflammatory mediators and decrease cytokine-induced extracellular matrix degradation. Physical inactivity reduces daily energy expenditure thereby promoting weight gain and continuation of the cycle. Emerging evidence indicates that osteoarthritis likely impedes the management of chronic metabolic diseases associated with aging, such as obesity, diabetes, and heart disease, because of its negative impact on physical activity.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
Acknowledgements
Supported by grants from the National Institutes of Health (National Center for Research Resources, 5P20RR018758-08, the National Institute of General Medical Sciences, 8P20GM103441-08, and the National Institute of Arthritis and Musculoskeletal and Skin Diseases, AR-060155), the Oklahoma Center for the Advancement of Science and Technology, and the Arthritis Foundation. The authors thank Drs. Wan-Pin Chang, Farshid Guilak, Ken Humphries, Mike Kinter, Virginia Kraus, Paul Rindler, and Luke Szweda for many insightful discussions.
Related Research Data
References
- Loeser RF. Age-related changes in the musculoskeletal system and the development of osteoarthritis. 2010; 263371-86. 10.3402/pba.v2i0.17470.
- Goldring SR, Goldring MB. The role of cytokines in cartilage matrix degeneration in osteoarthritis. 2004; 427SupplS27-36. 10.3402/pba.v2i0.17470.
- Loeser RF. Aging and osteoarthritis. 2011; 235492-6. 10.3402/pba.v2i0.17470.
- Goldring MB, Goldring SR. Osteoarthritis. 2007; 2133626-34. 10.3402/pba.v2i0.17470.
- Gomez R, Conde J, Scotece M, Gomez-Reino JJ, Lago F, Gualillo O. Whats new in our understanding of the role of adipokines in rheumatic diseases?. 2011; 79528-36. 10.3402/pba.v2i0.17470.
- Goekoop RJ, Kloppenburg M, Kroon HM, Frolich M, Huizinga TW, Westendorp RG et al. Low innate production of interleukin-1beta and interleukin-6 is associated with the absence of osteoarthritis in old age. 2010; 187942-7. 10.3402/pba.v2i0.17470.
- Kerkhof HJ, Doherty M, Arden NK, Abramson SB, Attur M, Bos SD et al. Large-scale meta-analysis of interleukin-1 beta and interleukin-1 receptor antagonist polymorphisms on risk of radiographic hip and knee osteoarthritis and severity of knee osteoarthritis. 2011; 193265-71. 10.3402/pba.v2i0.17470.
- Felson DT, Chaisson CE. Understanding the relationship between body weight and osteoarthritis. 1997; 114671-81. 10.3402/pba.v2i0.17470.
- Felson DT, Goggins J, Niu J, Zhang Y, Hunter DJ. The effect of body weight on progression of knee osteoarthritis is dependent on alignment. 2004; 50123904-9. 10.3402/pba.v2i0.17470.
- Sharma L, Lou C, Cahue S, Dunlop DD. The mechanism of the effect of obesity in knee osteoarthritis: the mediating role of malalignment. 2000; 433568-75. 10.3402/pba.v2i0.17470.
- Aspden RM. Obesity punches above its weight in osteoarthritis. 2011; 7165-8. 10.3402/pba.v2i0.17470.
- Griffin TM, Guilak F. The role of mechanical loading in the onset and progression of osteoarthritis. 2005; 334195-200. 10.3402/pba.v2i0.17470.
- Lago F, Dieguez C, Gomez-Reino J, Gualillo O. The emerging role of adipokines as mediators of inflammation and immune responses. 2007; 183–4313-25. 10.3402/pba.v2i0.17470.
- Sowers M, Karvonen-Gutierrez CA, Palmieri-Smith R, Jacobson JA, Jiang Y, Ashton-Miller JA. Knee osteoarthritis in obese women with cardiometabolic clustering. 2009; 61101328-36. 10.3402/pba.v2i0.17470.
- Katz JD, Agrawal S, Velasquez M. Getting to the heart of the matter: osteoarthritis takes its place as part of the metabolic syndrome. 2010; 225512-9. 10.3402/pba.v2i0.17470.
- Lago F, Gomez R, Conde J, Scotece M, Gomez-Reino JJ, Gualillo O. Cardiometabolic comorbidities and rheumatic diseases: focus on the role of fat mass and adipokines. 2011; 6381083-90. 10.3402/pba.v2i0.17470.
- Davis MA, Ettinger WH, Neuhaus JM. The role of metabolic factors and blood pressure in the association of obesity with osteoarthritis of the knee. 1988; 15121827-32.
- Griffin TM, Fermor B, Huebner JL, Kraus VB, Rodriguiz RM, Wetsel WC et al. Diet-induced obesity differentially regulates behavioral, biomechanical, and molecular risk factors for osteoarthritis in mice. 2010; 124R13010.3402/pba.v2i0.17470.
- Griffin TM, Huebner JL, Kraus VB, Guilak F. Extreme obesity due to impaired leptin signaling in mice does not cause knee osteoarthritis. . 2009;60(10):2935–44 .10.3402/pba.v2i0.17470.
- Griffin TM, Huebner JL, Kraus VB, Yan Z, Guilak F. Induction of osteoarthritis and metabolic inflammation by a very high-fat diet in mice: effects of short-term exercise. 2012; 642443-53. 10.3402/pba.v2i0.17470.
- Hui W, Litherland GJ, Elias MS, Kitson GI, Cawston TE, Rowan AD et al. Leptin produced by joint white adipose tissue induces cartilage degradation via upregulation and activation of matrix metalloproteinases. 2012; 713455-62. 10.3402/pba.v2i0.17470.
- Mooney RA, Sampson ER, Lerea J, Rosier RN, Zuscik MJ. High-fat diet accelerates progression of osteoarthritis after meniscal/ligamentous injury. 2011; 136R19810.3402/pba.v2i0.17470.
- Pallu S, Francin PJ, Guillaume C, Gegout-Pottie P, Netter P, Mainard D et al. Obesity affects the chondrocyte responsiveness to leptin in patients with osteoarthritis. 2010; 123R11210.3402/pba.v2i0.17470.
- Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. 1998; 3956704763-70. 10.3402/pba.v2i0.17470.
- Otero M, Gomez Reino JJ, Gualillo O. Synergistic induction of nitric oxide synthase type II: in vitro effect of leptin and interferon-gamma in human chondrocytes and ATDC5 chondrogenic cells. 2003; 482404-9. 10.3402/pba.v2i0.17470.
- Simopoulou T, Malizos KN, Iliopoulos D, Stefanou N, Papatheodorou L, Ioannou M et al. Differential expression of leptin and leptin's receptor isoform (Ob-Rb) mRNA between advanced and minimally affected osteoarthritic cartilage; effect on cartilage metabolism. 2007; 158872-83. 10.3402/pba.v2i0.17470.
- Dumond H, Presle N, Terlain B, Mainard D, Loeuille D, Netter P et al. Evidence for a key role of leptin in osteoarthritis. 2003; 48113118-29. 10.3402/pba.v2i0.17470.
- Bastiaansen-Jenniskens YM, Clockaerts S, Feijt C, Zuurmond AM, Stojanovic-Susulic V, Bridts C et al. Infrapatellar fat pad of patients with end-stage osteoarthritis inhibits catabolic mediators in cartilage. 2012; 712288-94. 10.3402/pba.v2i0.17470.
- Clockaerts S, Bastiaansen-Jenniskens YM, Runhaar J, Van Osch GJ, Van Offel JF, Verhaar JA et al. The infrapatellar fat pad should be considered as an active osteoarthritic joint tissue: a narrative review. 2010; 187876-82. 10.3402/pba.v2i0.17470.
- Distel E, Cadoudal T, Durant S, Poignard A, Chevalier X, Benelli C. The infrapatellar fat pad in knee osteoarthritis: an important source of interleukin-6 and its soluble receptor. 2009; 60113374-7. 10.3402/pba.v2i0.17470.
- Klein-Wieringa IR, Kloppenburg M, Bastiaansen-Jenniskens YM, Yusuf E, Kwekkeboom JC, El-Bannoudi H et al. The infrapatellar fat pad of patients with osteoarthritis has an inflammatory phenotype. 2011; 705851-7. 10.3402/pba.v2i0.17470.
- Presle N, Pottie P, Dumond H, Guillaume C, Lapicque F, Pallu S et al. Differential distribution of adipokines between serum and synovial fluid in patients with osteoarthritis. Contribution of joint tissues to their articular production. 2006; 147690-5. 10.3402/pba.v2i0.17470.
- Urban JP. The chondrocyte: a cell under pressure. 1994; 3310901-8. 10.3402/pba.v2i0.17470.
- Darling EM, Athanasiou KA. Biomechanical strategies for articular cartilage regeneration. 2003; 3191114-24. 10.3402/pba.v2i0.17470.
- Loeser RF, Carlson CS, Del Carlo M, Cole A. Detection of nitrotyrosine in aging and osteoarthritic cartilage: correlation of oxidative damage with the presence of interleukin-1beta and with chondrocyte resistance to insulin-like growth factor 1. 2002; 4692349-57. 10.3402/pba.v2i0.17470.
- Buckwalter JA, Martin JA, Brown TD. Perspectives on chondrocyte mechanobiology and osteoarthritis. 2006; 433–4603-9.
- Leong DJ, Hardin JA, Cobelli NJ, Sun HB. Mechanotransduction and cartilage integrity. 2011; 1240132-7. 10.3402/pba.v2i0.17470.
- Loeser RF. Molecular mechanisms of cartilage destruction in osteoarthritis. 2008; 84303-6.
- Martin JA, McCabe D, Walter M, Buckwalter JA, McKinley TO. N-acetylcysteine inhibits post-impact chondrocyte death in osteochondral explants. 2009; 9181890-7. 10.3402/pba.v2i0.17470.
- Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA. The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. 2011; 119607-15. 10.3402/pba.v2i0.17470.
- Fogelholm M. Physical activity, fitness and fatness: relations to mortality, morbidity and disease risk factors. A systematic review. 2010; 113202-21. 10.3402/pba.v2i0.17470.
- Lubberts E, Joosten LA, Van Den Bersselaar L, Helsen MM, Bakker AC, Xing Z et al. Intra-articular IL-10 gene transfer regulates the expression of collagen-induced arthritis (CIA) in the knee and ipsilateral paw. 2000; 1202375-83. 10.3402/pba.v2i0.17470.
- Ferretti M, Gassner R, Wang Z, Perera P, Deschner J, Sowa G et al. Biomechanical signals suppress proinflammatory responses in cartilage: early events in experimental antigen-induced arthritis. 2006; 177128757-66.
- Nam J, Aguda BD, Rath B, Agarwal S. Biomechanical thresholds regulate inflammation through the NF-kappaB pathway: experiments and modeling. 2009; 44e526210.3402/pba.v2i0.17470.
- Agarwal S, Deschner J, Long P, Verma A, Hofman C, Evans CH et al. Role of NF-kappaB transcription factors in antiinflammatory and proinflammatory actions of mechanical signals. 2004; 50113541-8. 10.3402/pba.v2i0.17470.
- Akanji OO, Sakthithasan P, Salter DM, Chowdhury TT. Dynamic compression alters NFkappaB activation and IkappaB-alpha expression in IL-1beta-stimulated chondrocyte/agarose constructs. 2010; 59141-52. 10.3402/pba.v2i0.17470.
- Nam J, Perera P, Liu J, Wu LC, Rath B, Butterfield TA et al. Transcriptome-wide gene regulation by gentle treadmill walking during the progression of monoiodoacetate-induced arthritis. 2011; 6361613-25. 10.3402/pba.v2i0.17470.
- Torzilli PA, Bhargava M, Park S, Chen CT. Mechanical load inhibits IL-1 induced matrix degradation in articular cartilage. 2010; 18197-105. 10.3402/pba.v2i0.17470.
- Jin G, Sah RL, Li YS, Lotz M, Shyy JY, Chien S. Biomechanical regulation of matrix metalloproteinase-9 in cultured chondrocytes. 2000; 186899-908. 10.3402/pba.v2i0.17470.
- Deschner J, Hofman CR, Piesco NP, Agarwal S. Signal transduction by mechanical strain in chondrocytes. 2003; 63289-93.
- Johnson K, Jung A, Murphy A, Andreyev A, Dykens J, Terkeltaub R. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. 2000; 4371560-70. 10.3402/pba.v2i0.17470.
- Stefanovic-Racic M, Stadler J, Georgescu HI, Evans CH. Nitric oxide and energy production in articular chondrocytes. 1994; 1592274-80. 10.3402/pba.v2i0.17470.
- Blanco FJ, Ochs RL, Schwarz H, Lotz M. Chondrocyte apoptosis induced by nitric oxide. 1995; 146175-85.
- Regan EA, Bowler RP, Crapo JD. Joint fluid antioxidants are decreased in osteoarthritic joints compared to joints with macroscopically intact cartilage and subacute injury. 2008; 164515-21. 10.3402/pba.v2i0.17470.
- Vaillancourt F, Fahmi H, Shi Q, Lavigne P, Ranger P, Fernandes JC et al. 4-Hydroxynonenal induces apoptosis in human osteoarthritic chondrocytes: the protective role of glutathione-S-transferase. 2008; 105R10710.3402/pba.v2i0.17470.
- Morquette B, Shi Q, Lavigne P, Ranger P, Fernandes JC, Benderdour M. Production of lipid peroxidation products in osteoarthritic tissues: new evidence linking 4-hydroxynonenal to cartilage degradation. 2006; 541271-81. 10.3402/pba.v2i0.17470.
- Del Carlo MJr, Loeser RF. Increased oxidative stress with aging reduces chondrocyte survival: correlation with intracellular glutathione levels. 2003; 48123419-30. 10.3402/pba.v2i0.17470.
- Loeser RF, Jr. Aging cartilage and osteoarthritis – what's the link?. 2004; 2004(29): pe31.10.3402/pba.v2i0.17470.
- Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. 2005; 75–6619-26. 10.3402/pba.v2i0.17470.
- Miki Y, Teramura T, Tomiyama T, Onodera Y, Matsuoka T, Fukuda K, et al. Hyaluronan reversed proteoglycan synthesis inhibited by mechanical stress: possible involvement of antioxidant effect. . 2010; 59:471–7.10.3402/pba.v2i0.17470.
- Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. 2011; 33360481440-5. 10.3402/pba.v2i0.17470.
- Goodwin W, McCabe D, Sauter E, Reese E, Walter M, Buckwalter JA et al. Rotenone prevents impact-induced chondrocyte death. 2010; 2881057-63.
- Baker MS, Feigan J, Lowther DA. The mechanism of chondrocyte hydrogen peroxide damage. Depletion of intracellular ATP due to suppression of glycolysis caused by oxidation of glyceraldehyde-3-phosphate dehydrogenase. 1989; 1617-14.
- Kurz B, Lemke A, Kehn M, Domm C, Patwari P, Frank EH et al. Influence of tissue maturation and antioxidants on the apoptotic response of articular cartilage after injurious compression. 2004; 501123-30. 10.3402/pba.v2i0.17470.
- Nakagawa S, Arai Y, Mazda O, Kishida T, Takahashi KA, Sakao K et al. N-acetylcysteine prevents nitric oxide-induced chondrocyte apoptosis and cartilage degeneration in an experimental model of osteoarthritis. 2009; 282156-63.
- Pratta MA, Ackerman NR, Arner EC. Effect of ebselen on IL-1-induced alterations in cartilage metabolism. 1998; 473115-21. 10.3402/pba.v2i0.17470.
- Ramakrishnan P, Hecht BA, Pedersen DR, Lavery MR, Maynard J, Buckwalter JA et al. Oxidant conditioning protects cartilage from mechanically induced damage. 2010; 287914-20. 10.3402/pba.v2i0.17470.
- Kamata H, Hirata H. Redox regulation of cellular signalling. 1999; 1111-14. 10.3402/pba.v2i0.17470.
- Lo YY, Conquer JA, Grinstein S, Cruz TF. Interleukin-1 beta induction of c-fos and collagenase expression in articular chondrocytes: involvement of reactive oxygen species. 1998; 69119-29. 10.3402/pba.v2i0.17470.
- Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. . 2012; 24:981–90.10.3402/pba.v2i0.17470.
- Osburn WO, Wakabayashi N, Misra V, Nilles T, Biswal S, Trush MA et al. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. 2006; 45417-15. 10.3402/pba.v2i0.17470.
- Gomez-Cabrera MC, Domenech E, Vina J. Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. 2008; 442126-31. 10.3402/pba.v2i0.17470.
- Moyes CD, Hood DA. Origins and consequences of mitochondrial variation in vertebrate muscle. 2003; 65: 177-201. 10.3402/pba.v2i0.17470.
- Hotamisligil GS. Inflammation and metabolic disorders. 2006; 4447121860-7. 10.3402/pba.v2i0.17470.
- Dunlop DD, Song J, Semanik PA, Chang RW, Sharma L, Bathon JM et al. Objective physical activity measurement in the osteoarthritis initiative: are guidelines being met?. 2011; 63113372-82. 10.3402/pba.v2i0.17470.
- Hootman JM, Helmick CG. Projections of US prevalence of arthritis and associated activity limitations. 2006; 541226-9. 10.3402/pba.v2i0.17470.
- Handschin C, Spiegelman BM. The role of exercise and PGC1alpha in inflammation and chronic disease. 2008; 4547203463-9. 10.3402/pba.v2i0.17470.