
Abstract
Age-related cardiomyopathy accounts for a significant part of heart failure cases. Imbalance of the energetic equilibrium of the heart along with mitochondrial dysfunction and impaired β-adrenergic receptor signaling contributes in the aggravation of cardiac function in the elderly. In this review article, studies that correlate cardiac aging with lipotoxicity are summarized. The involvement of inhibition of peroxisome proliferator-activated receptor-α, β-adrenergic receptor desensitization, and mitochondrial dysfunction as underlying mechanisms for the lipid-driven age-related cardiomyopathy are presented with the aim to indicate potential therapeutic targets for cardiac aging.
Age-related cardiac dysfunction is a major factor in heart failure. The elderly accounts for at least 80% of patients with ischemic heart disease, 75% of patients with congestive heart failure, and 70% of patients with atrial fibrillation (Citation1). Heart failure with either lower or preserved ejection fraction is common for hospitalized patients with cardiac abnormalities. Cardiac aging, which is evident in both humans and mice, plays an important role for both types of heart failure (Citation2). Mouse cardiac aging is characterized by a reduction in fractional shortening, diastolic dysfunction, left ventricular hypertrophy, increased left ventricular end-diastolic pressure, fibrosis, cardiomyocyte hypertrophy, and increased apoptosis. Several components of cardiac function, including energetic homeostasis, adrenergic signaling, and mitochondrial dysfunction, can be compromised during aging (Citation3). Balanced cardiac lipid metabolism is critical for normal function of the heart. Any deviation toward either increased or reduced fatty acid metabolism may be detrimental for cardiac function, primarily depending on the type of pathophysiological challenge. Aging-related cardiomyopathy has been associated with downregulation of peroxisome proliferator-activated receptor (PPAR)-α (Citation4), which is a central regulator of cardiac fatty acid metabolism (Citation5) and cardiac lipid accumulation (Citation6, Citation7). Thus, impairment of fatty acid metabolism may at least partially account for the aggravation of cardiac function that occurs with aging.
Aging-related cardiomyopathy
The cardiovascular system's functions are altered with aging, and a significant portion of the observed changes affects myocardial biology. Decreased elasticity and increased stiffness of the arterial system increases afterload on the left ventricle, as well as systolic blood pressure and leads to left ventricular hypertrophy. Although systolic function is preserved in the older humans, diastolic dysfunction and lower exercise capacity have been reported (Citation2). Diastolic dysfunction accounts for the lower exercise capacity and increased mortality (Citation8). Mice with cardiac aging demonstrate increased left ventricular mass with slightly lower systolic function, diastolic dysfunction, reduced exercise capacity, and worse myocardial performance (Citation9, Citation10). Aged mouse hearts obtained from either 129/SvJ-C57BL/6 or C57BL/6J mice have increased fibrosis, hypertrophic cardiomyocytes, increased apoptosis, and amyloid deposition (Citation3, Citation11). C57BL/6J mice develop contractile dysfunction approximately at 18 months of age (Citation12). This is associated with increased cardiac fibrosis, apoptosis, and inflammation-related gene expression (Citation12). Cardiac fibrosis is generally associated with increased Transforming growth factor (TGF)-β levels, which is also thought to be a critical mediator of age-related cardiac fibrosis (Citation13). In addition, other proteins such as matrix metalloproteinases (MMPs), osteopontin, and periostin have a significant contribution in cardiac fibrosis (Citation14). Induction of inflammatory markers has also been associated with aging (Citation15), although it seems that there is a significant mouse-strain-dependent variation on the extent of inflammation (Citation16). Similarly, the existence of apoptotic cardiomyocytes seems to vary with studies showing either increased (Citation17) or unchanged (Citation12) apoptosis during cardiac aging. Also, oxidative stress has been associated with cardiac aging and particularly with aging-related cardiac hypertrophy (Citation18). Finally, reduced responsiveness to adrenergic stimulus and increased plasma catecholamine levels have been reported with aging (Citation3). Thus, cardiac aging is accompanied by dysregulation of various pathways that have been attributed causative role in several types of cardiomyopathy.
Therefore, the aged myocardium is more susceptible to hemodynamic and ischemic stress compared to young myocardium (Citation19, Citation20). The vulnerability of aged hearts to stress has been correlated with increased reactive oxygen species (ROS) levels (Citation21) and higher susceptibility to mitochondrial permeability transition pore (mPTP) opening (Citation22). Nevertheless, mPTP opening may eventually cause mitochondrial swelling, ATP depletion, apoptosis, and cell death (Citation23, Citation24). Thus, cardiac aging is associated with myocardial dysfunction and increased sensitivity to cardiac stress.
Cardiac fatty acid metabolism and lipotoxicity
The heart normally consumes a large amount of ATP in order to pump more than 7,000 liters of blood on a daily basis (Citation25). For the production of ATP that is needed for this massive amount of work, the heart oxidizes fatty acids, glucose, lactate, ketone bodies, and amino acids as energy-providing substrates. Fatty acid oxidation (FAO) is a major component of the energy production process as it accounts for the generation of approximately 70% of cardiac ATP (Citation25, Citation26).
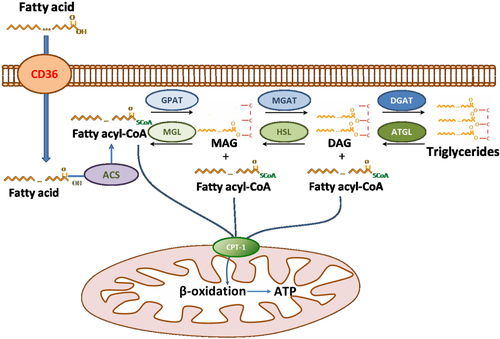
FA utilization in healthy hearts is a complex process that includes several steps: FA uptake, conversion of free FA to FA-CoA, storage of FAs in triglycerides (TG), TG lipolysis, transfer of fatty acids into the mitochondria, β-oxidation, and oxidative phosphorylation for ATP production (). The flawless transfer of fatty acids from cellular uptake to mitochondrial oxidation prevents accumulation of excess lipids. A study using positron emission tomography in humans showed that aging decreases myocardial FA utilization and FAO without any difference in myocardial glucose utilization (Citation27). Several types of cardiac dysfunction, such as ischemia, obesity, diabetes, sepsis, and heart failure, are associated with impaired FAO, which frequently leads to lipid accumulation characterized as cardiac lipotoxicity (Citation28, Citation29). Although cardiac lipotoxicity is accompanied by increased accumulation of neutral lipids, several studies have dissociated myocardial TG accumulation from lipid-driven cardiac dysfunction (Citation30–Citation33). In fact, cardiac lipotoxicity has been attributed to other lipids, such as saturated free fatty acids, ceramides, diacylglycerols (DAGs), and acylcarnitines (Citation29, Citation32) (Citation34, Citation35), which change in parallel with cardiac TG.
Fig. 1 Summary of cardiomyocyte fatty acid metabolism in healthy hearts.
Although cardiac toxic lipids have been associated with cardiac dysfunction (Citation29), it has not been studied thoroughly whether they mediate aging-related cardiomyopathy, as well as what lipid-driven signaling mechanisms may be involved. Various studies have established a correlation between cardiac lipid accumulation and aging in humans and animal models (Citation6, Citation7). Aged hearts have increased expression of the cardiac fatty acid transporter, cluster of differentiation 36 (CD36) (Citation36). Genetic ablation of CD36 prevented age-related cardiomyopathy, which indicates the involvement of increased cardiomyocyte lipid uptake in this process (Citation36). Several lipids have been related to lipotoxic cardiomyopathy, such as palmitic acid, acylcarnitine, unesterified cholesterol, lysolecithin, ceramide, and DAGs. These lipids can trigger apoptosis, inflammation, and mitochondrial dysfunction (Citation29). Interestingly, ob/ob mice, which are models of hyperphagia and type 2 diabetes, develop symptoms of cardiac aging much earlier, compared to wild-type mice fed with regular chow diet (Citation37). This phenotype is associated with increased cardiac TG and DAG accumulation. Cardiac ceramide levels have been shown to be increased in a senescence-accelerated mouse model that develops cardiac hypertrophy (Citation38). Thus, toxic lipids, such as DAGs and ceramides, are increased with cardiac aging.
Either ceramides or DAGs can activate Protein Kinase C (PKC) signaling (Citation39) that has been associated with myocardial aging (Citation40) PKCs constitute a lipid-sensitive Ser/Thr kinase family, which is involved in a broad range of G protein–coupled receptor (GPCR)-mediated responses and have been linked with several types of cardiomyopathy (Citation24, Citation41–Citation46). All PKC isoforms share a common structural motif consisting of a conserved kinase domain linked at the N-terminal domain, via a hinge region, with a more variable regulatory domain that in most cases contains lipid/phorbol ester binding and calcium-dependent phospholipid binding subdomains. PKCs are classified into three subcategories: conventional PKCs (cPKCs – PKCα, PKCβI, PKCβII, and PKCγ), novel PKCs (nPKCs – PKCδ, PKCɛ, PKCθ, and PKCη), and atypical PKCs (aPKCs – PKCζ and PKCι/λ) (Citation39). All PKC isoforms, except PKCθ and PKCι/λ, have been detected in cardiomyocytes from humans or other animal models (Citation41, Citation44) (Citation46–Citation48). A number of studies have demonstrated that PKC activation relies on binding of DAG or ceramide and translocation to the membrane. Briefly, PKC movement to the membrane involves activation by Ca2+ binding to the C2 domain and DAG or ceramide binding to the C1 domain for membrane penetration. DAG binding results in formation of a hydrophobic cap facilitating membrane insertion and stabilizing the protein-membrane interface without causing a conformational change.
Role of PPARα in aging-related cardiomyopathy
Several proteins of the energy production machinery that mediates processing of FAs and ATP production are regulated at the transcriptional level by PPARα (Citation49). PPARα is activated by FAs that are released via lipolysis from the intracellular TG (Citation50, Citation51). Inhibition of PPARα, as shown in PPARα−/− mice, decreases myocardial fatty acid metabolism (Citation52–Citation54) and reduces cardiac ATP levels (Citation53, Citation55). Although some studies have reported normal cardiac function at baseline for the PPARα−/− mice (Citation54–Citation56), others showed that these mice have reduced cardiac function at baseline associated with fibrosis (Citation53, Citation57) and oxidative stress (Citation58, Citation59). Antioxidant therapy alleviated left ventricular dysfunction, indicating that oxidative damage accounts for the cardiac dysfunction seen in mice with PPARα ablation (Citation59).
Cardiac abnormalities that occur in PPARα−/− mice progress further during aging (Citation53) and decrease longevity (Citation60). Nevertheless, aging is associated with decreased cardiac PPARα levels (Citation4, Citation61). Metabolomic analysis indicated an age-dependent decrease in cardiac glucose content, signs of decreased ketone supply, and altered FA synthesis (Citation62). The importance of PPARα inhibition in accelerating cardiac aging was demonstrated in 20-month-old rats that were treated with the lipid lowering drug atorvastatin, which increases PPARα expression (Citation63). The treatment with atorvastatin reduced cardiac hypertrophy, collagen deposition, oxidative stress, expression of inflammatory cytokines, and the aging marker β-galactosidase. The conclusion about the association of PPARα inhibition and cardiac aging was further emphasized when pretreatment with PPAR inhibitors attenuated the effect of atorvastatin on the inhibition of inflammatory cytokines (Citation63). This indicated that the beneficial effects of atorvastatin on cardiac aging may be attributed to either lipid lowering effect or the anti-inflammatory role of PPAR signaling (Citation64, Citation65).
Furthermore, cardiac aging that is observed in mice with cardiomyocyte-specific overexpression of human aldose reductase (αMHC-hAR) is associated with reduced cardiac expression of PPARα gene targets, such as pyruvate dehydrogenase kinase 4 and acetyl-CoA oxidase (Citation66), and increased PPARγ signaling that promotes cardiac lipid accumulation characterized by increased TG, ceramides, and acyl-carnitine levels (Citation67). Moreover, genetic ablation of Pparα in αMHC-hAR mice resulted in earlier onset of cardiac dysfunction (Citation66). Increased cardiac toxic lipid levels, such as ceramides, have also been reported in senescence-accelerated mice that have lower expression of PPARα (Citation38). Another study also showed that PPARα activation reduces inflammation in aged mice (Citation61). Thus, although reduced cardiac PPARα expression has been associated with aging-related cardiomyopathy, the underlying mechanisms that mediate the beneficial effect of PPARα have not been fully elucidated.
β-Adrenergic signaling and cardiac aging
A component of cardiac aging pathophysiology is the impairment of β-AR signaling (Citation3). Normally, stress increases the release of adrenal norepinephrine and epinephrine that target cardiac β-ARs, which belong to the GPCR family. Increased release of catecholamines by the sympathetic nervous system stimulates β-ARs and increases contractile force and heart rate. Activated β-ARs induce adenylyl cyclase activation and cAMP formation. β-ARs are subsequently phosphorylated and deactivated by kinases designated G protein–coupled receptor kinases (GRKs) (Citation68). GRKs can be activated by PKCs (Citation69). However, GRKs do not seem to be involved in cardiac aging in humans (Citation70). PKCs can also deactivate β-ARs directly via a ligand-independent cascade (heterologous desensitization) (Citation71). Desensitization of β-AR is followed by internalization of the receptor. This is a key step required either for restoration (Citation72) or for its proteasomal degradation (Citation73). Failing hearts demonstrate reduced cardiac β-AR-mediated responsiveness to catecholamines and abnormal myocardial β-AR signaling (Citation74), which coincide with increased catecholamine production.
Age-related inhibition of β-AR responsiveness occurs in both animals and humans and is characterized by reduced β-AR density and internalization (Citation75). Isolated left ventricular cardiomyocytes from hearts of animals at different age showed that the age-related contractility impairment during β-adrenergic stimulation was associated with reduced cAMP levels.
It has been shown (Citation34, Citation76) that excessive cardiac lipid accumulation is associated with dilated cardiomyopathy in several animal models of cardiac lipotoxicity, which is consistent with observations in humans. Cardiac lipotoxicity is accounted for by accumulation of toxic lipids, such as DAGs and ceramides, which activate PKCα and PKCδ and impair catecholamine-stimulated cardiac contractility and relaxation (Citation34, Citation35). Various studies have identified palmitic acid as the FA species that primarily induces formation of DAGs and ceramides, activates PKC signaling, and promotes β-AR desensitization and cardiac dysfunction (Citation34, Citation51) (Citation76, Citation77). Interestingly, a metabolomics study showed that aged rat hearts have increased utilization of palmitic acid (Citation78) indicating a potential role for this toxic FA in aging-related cardiac dysfunction. Thus, lipid-driven mechanisms that may involve PKC signaling may account for the impairment of β-AR signaling that occurs in aged hearts.
Mitochondria and cardiac aging
Impaired mitochondrial oxidative capacity is another component of cardiac lipotoxicity that seems to have a causative role in aging (Citation6). Aging-related cardiac mitochondrial defects have been primarily attributed to interfibrillar rather than subsarcolemmal mitochondria (Citation79, Citation80), which have lower abundance in aged hearts (Citation80). Increased mitochondrial ROS generation has been proposed to be a central event in cellular aging as it was described as a major determinant of lifespan several decades ago (Citation81). Formation of ROS accompanies dysregulation of oxidative phosphorylation and mitochondrial dysfunction. Excess electrons from complex I and III can be transferred directly to O2 to generate superoxide anion (O−), which is then converted to H2O2 that diffuses into cytosol and nucleus and activates redox signaling. H2O2 can be converted to a hydroxyl radical, which is the most reactive ROS species that targets mitochondrial DNA, lipids, and proteins and contributes in mitochondrial dysfunction and aging (Citation82).
Changes in mitochondrial biology have a significant influence in cardiomyocyte function. Nevertheless, cardiomyocytes are rich in mitochondria that serve as ATP generators. Mitochondrial function deteriorates with aging due to electron transport chain defects (Citation83). Particularly, reduced activity of complexes I and IV has been reported and leads to mitochondrial superoxide formation, mitochondrial ROS production, and protein damage (Citation84, Citation85). Aged hearts have increased frequency of mitochondrial DNA mutations, oxidative damage of mitochondrial proteins, and mitochondrial structural defects (Citation86), which have been associated with aging (Citation87). Accordingly, mice that overexpress catalase in the mitochondria, which attenuates ROS formation and protects mitochondria from protein damage and structural defects, have increased lifespan (Citation21); and they are protected from cardiac dysfunction (Citation86).
Cardiolipin is a critical lipid component of the inner mitochondrial protein, where the electron transport chain exists; thus, it has a central role in oxidative phosphorylation and cellular energy production (Citation88). It remains controversial whether the content or composition of cardiolipin is altered in mitochondria of aged hearts as there are studies that show reduced content (Citation89–Citation91), as well as other studies that do not (Citation92). While it is not clear whether aging itself alters cardiolipin it seems that when stress, such as ischemia, is applied to aged hearts, cardiolipin oxidation is augmented compared to non-aged ischemic hearts (Citation93, Citation94). As cardiolipin oxidation, which occurs during oxidative stress, may account for disruption of the electron transport chain (Citation95) and mitochondrial damage (Citation96), this mechanism may underlie cardiac lipotoxicity-driven mitochondrial dysfunction during aging.
Increased frequency of mitochondrial DNA defects, particularly deletions (Citation97), has also been associated with cardiac aging. This was demonstrated in mice with homozygous mutation of mitochondrial polymerase γ. These mice have increased mtDNA mutations and deletions, and they develop age-dependent cardiomyopathy and have shorter lifespan (Citation98). The importance of mitochondrial DNA mutations for cardiac aging has also been demonstrated in humans as shown by age-associated accumulation of mtDNA deletions that have been reported in various tissues, including the heart (Citation99, Citation100). Thus, mitochondrial dysfunction that can be triggered by either oxidative stress or mutations of mitochondrial DNA is a contributing factor in cardiac aging.
Epilogue
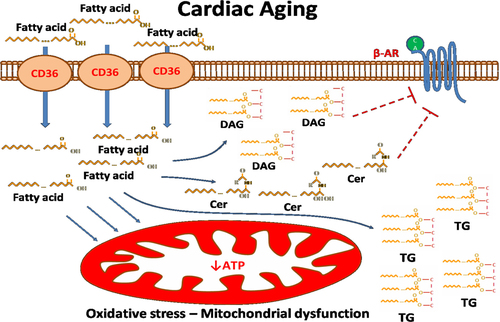
In summary, cardiac FAO is important for lipid metabolism homeostasis and normal cardiac function. Inhibition of FAO leads to increased cardiac lipid content, which is often accompanied by increased levels of toxic lipids, such as Cer and DAGs. These lipids compromise cardiac function via β-AR desensitization, which is driven by activation of the PKC signaling pathway. Aging-related cardiomyopathy is associated with reduced cardiac levels of PPARα, a master regulator of cardiac FAO, as well as with inhibition of β-AR signaling and mitochondrial dysfunction. These components of cardiac lipotoxicity that are also involved in cardiac aging indicate therapeutic targets that may alleviate age-related cardiomyopathy ().
Fig. 2 Cardiac lipotoxicity contributes to cardiac aging by promoting formation of toxic lipids, ceramides (Cer), and diacylglycerols (DAGs) that compromise β-AR function and by causing mitochondrial dysfunction.
Conflict of interest and funding
This work was supported by a NHLBI ‘Pathway to Independence’ R00 award (HL112853) and HL130218. The author has no conflict of interest to disclose.
References
- Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, etal. Heart disease and stroke statistics – 2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007; 115: e69–171.
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation. 2003; 107: 346–54.
- Yan L, Vatner DE, O'Connor JP, Ivessa A, Ge H, Chen W, etal. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007; 130: 247–58.
- Iemitsu M, Miyauchi T, Maeda S, Tanabe T, Takanashi M, Irukayama-Tomobe Y, etal. Aging-induced decrease in the PPAR-alpha level in hearts is improved by exercise training. Am J Physiol Heart Circ Physiol. 2002; 283: H1750–60.
- Pol CJ, Lieu M, Drosatos K. PPARS: protectors or opponents of myocardial function?. PPAR Res. 2015; 2015: 19.
- Zhao L, Zou X, Feng Z, Luo C, Liu J, Li H, etal. Evidence for association of mitochondrial metabolism alteration with lipid accumulation in aging rats. Exp Gerontol. 2014; 56: 3–12.
- van der Meer RW, Rijzewijk LJ, Diamant M, Hammer S, Schar M, Bax JJ, etal. The ageing male heart: myocardial triglyceride content as independent predictor of diastolic function. Eur Heart J. 2008; 29: 1516–22.
- Bursi F, Weston SA, Redfield MM, Jacobsen SJ, Pakhomov S, Nkomo VT, etal. Systolic and diastolic heart failure in the community. JAMA. 2006; 296: 2209–16.
- Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, etal. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009; 119: 2789–97.
- Schefer V, Talan MI. Oxygen consumption in adult and AGED C57BL/6J mice during acute treadmill exercise of different intensity. Exp Gerontol. 1996; 31: 387–92.
- Treuting PM, Linford NJ, Knoblaugh SE, Emond MJ, Morton JF, Martin GM, etal. Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J Gerontol A Biol Sci Med Sci. 2008; 63: 813–22.
- Boyle AJ, Shih H, Hwang J, Ye J, Lee B, Zhang Y, etal. Cardiomyopathy of aging in the mammalian heart is characterized by myocardial hypertrophy, fibrosis and a predisposition towards cardiomyocyte apoptosis and autophagy. Exp Gerontol. 2011; 46: 549–59.
- Chen W, Frangogiannis NG. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev. 2010; 15: 415–22.
- Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012; 92: 635–88.
- Swindell WR. Genes and gene expression modules associated with caloric restriction and aging in the laboratory mouse. BMC Genomics. 2009; 10: 585.
- Park SK, Kim K, Page GP, Allison DB, Weindruch R, Prolla TA. Gene expression profiling of aging in multiple mouse strains: identification of aging biomarkers and impact of dietary antioxidants. Aging Cell. 2009; 8: 484–95.
- Anversa P, Palackal T, Sonnenblick EH, Olivetti G, Meggs LG, Capasso JM. Myocyte cell loss and myocyte cellular hyperplasia in the hypertrophied aging rat heart. Circ Res. 1990; 67: 871–85.
- Venkataraman K, Khurana S, Tai TC. Oxidative stress in aging – matters of the heart and mind. Int J Mol Sci. 2013; 14: 17897–925.
- Isoyama S, Nitta-Komatsubara Y. Acute and chronic adaptation to hemodynamic overload and ischemia in the aged heart. Heart Fail Rev. 2002; 7: 63–9.
- Mariani J, Ou R, Bailey M, Rowland M, Nagley P, Rosenfeldt F, etal. Tolerance to ischemia and hypoxia is reduced in aged human myocardium. J Thorac Cardiovasc Surg. 2000; 120: 660–7.
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, etal. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005; 308: 1909–11.
- Juhaszova M, Rabuel C, Zorov DB, Lakatta EG, Sollott SJ. Protection in the aged heart: preventing the heart-break of old age?. Cardiovasc Res. 2005; 66: 233–44.
- Di Lisa F, Bernardi P. Mitochondria and ischemia-reperfusion injury of the heart: fixing a hole. Cardiovasc Res. 2006; 70: 191–9.
- Vagnozzi RJ, Hoffman NE, Elrod JW, Madesh M, Force T. Protein kinase signaling at the crossroads of myocyte life and death in ischemic heart disease. Drug Discov Today Ther Strateg. 2012; 9: e173–82.
- Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010; 90: 207–58.
- Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005; 85: 1093–129.
- Kates AM, Herrero P, Dence C, Soto P, Srinivasan M, Delano DG, etal. Impact of aging on substrate metabolism by the human heart. J Am Coll Cardiol. 2003; 41: 293–9.
- Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012; 15: 805–12.
- Drosatos K, Schulze PC. Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep. 2013; 10: 109–21.
- Bosma M, Dapito DH, Drosatos-Tampakaki Z, Huiping-Son N, Huang LS, Kersten S, etal. Sequestration of fatty acids in triglycerides prevents endoplasmic reticulum stress in an in vitro model of cardiomyocyte lipotoxicity. Biochim Biophys Acta. 2014; 1841: 1648–55.
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr., Ory DS, etal. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. 2003; 100: 3077–82.
- Son NH, Yu S, Tuinei J, Arai K, Hamai H, Homma S, etal. PPARgamma-induced cardiolipotoxicity in mice is ameliorated by PPARalpha deficiency despite increases in fatty acid oxidation. J Clin Invest. 2010; 120: 3443–54.
- Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, etal. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. 2009; 284: 36312–23.
- Drosatos K, Bharadwaj KG, Lymperopoulos A, Ikeda S, Khan R, Hu Y, etal. Cardiomyocyte lipids impair beta-adrenergic receptor function via PKC activation. Am J Physiol Endocrinol Metab. 2011; 300: E489–99.
- Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, etal. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation. 2012; 125: 2844–53.
- Koonen DP, Febbraio M, Bonnet S, Nagendran J, Young ME, Michelakis ED, etal. CD36 expression contributes to age-induced cardiomyopathy in mice. Circulation. 2007; 116: 2139–47.
- Wang X, West JA, Murray AJ, Griffin JL. Comprehensive metabolic profiling of age-related mitochondrial dysfunction in the high-fat-fed ob/ob mouse heart. J Proteome Res. 2015; 14: 2849–62.
- Rodriguez-Calvo R, Serrano L, Barroso E, Coll T, Palomer X, Camins A, etal. Peroxisome proliferator-activated receptor alpha down-regulation is associated with enhanced ceramide levels in age-associated cardiac hypertrophy. J Gerontol A Biol Sci Med Sci. 2007; 62: 1326–36.
- Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008; 88: 1341–78.
- Hunter JC, Korzick DH. Age- and sex-dependent alterations in protein kinase C (PKC) and extracellular regulated kinase 1/2 (ERK1/2) in rat myocardium. Mech Ageing Dev. 2005; 126: 535–50.
- Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, etal. Increased protein kinase C activity and expression of Ca2+− sensitive isoforms in the failing human heart. Circulation. 1999; 99: 384–91.
- Boyle AJ, Kelly DJ, Zhang Y, Cox AJ, Gow RM, Way K, etal. Inhibition of protein kinase C reduces left ventricular fibrosis and dysfunction following myocardial infarction. J Mol Cell Cardiol. 2005; 39: 213–21.
- Braz JC, Bueno OF, De Windt LJ, Molkentin JD. PKC alpha regulates the hypertrophic growth of cardiomyocytes through extracellular signal-regulated kinase1/2 (ERK1/2). J Cell Biol. 2002; 156: 905–19.
- Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, etal. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004; 10: 248–54.
- Connelly KA, Kelly DJ, Zhang Y, Prior DL, Advani A, Cox AJ, etal. Inhibition of protein kinase C-beta by ruboxistaurin preserves cardiac function and reduces extracellular matrix production in diabetic cardiomyopathy. Circ Heart Fail. 2009; 2: 129–37.
- Rouet-Benzineb P, Mohammadi K, Perennec J, Poyard M, Bouanani Nel H, Crozatier B. Protein kinase C isoform expression in normal and failing rabbit hearts. Circ Res. 1996; 79: 153–61.
- Kohout TA, Rogers TB. Use of a PCR-based method to characterize protein kinase C isoform expression in cardiac cells. Am J Physiol. 1993; 264: C1350–9.
- Shin HG, Barnett JV, Chang P, Reddy S, Drinkwater DC, Pierson RN, etal. Molecular heterogeneity of protein kinase C expression in human ventricle. Cardiovasc Res. 2000; 48: 285–99.
- Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol. 2008; 44: 968–75.
- Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, etal. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med. 2011; 17: 1076–85.
- Lahey R, Wang X, Carley AN, Lewandowski ED. Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation. 2014; 130(20): 1790–9.
- Djouadi F, Weinheimer CJ, Saffitz JE, Pitchford C, Bastin J, Gonzalez FJ, etal. A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator-activated receptor alpha-deficient mice. J Clin Invest. 1998; 102: 1083–91.
- Watanabe K, Fujii H, Takahashi T, Kodama M, Aizawa Y, Ohta Y, etal. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age- dependent cardiac toxicity. J Biol Chem. 2000; 275: 22293–9.
- Campbell FM, Kozak R, Wagner A, Altarejos JY, Dyck JR, Belke DD, etal. A role for peroxisome proliferator-activated receptor alpha (PPARalpha) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem. 2002; 277: 4098–103.
- Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation. 2005; 112: 2339–46.
- Liu J, Wang P, He L, Li Y, Luo J, Cheng L, etal. Cardiomyocyte-restricted deletion of PPARbeta/delta in PPARalpha-null mice causes impaired mitochondrial biogenesis and defense, but no further depression of myocardial fatty acid oxidation. PPAR Res. 2011; 2011: 372854.
- Loichot C, Jesel L, Tesse A, Tabernero A, Schoonjans K, Roul G, etal. Deletion of peroxisome proliferator-activated receptor-alpha induces an alteration of cardiac functions. Am J Physiol Heart Circ Physiol. 2006; 291: H161–6.
- Guellich A, Damy T, Lecarpentier Y, Conti M, Claes V, Samuel JL, etal. Role of oxidative stress in cardiac dysfunction of PPARalpha−/− mice. Am J Physiol Heart Circ Physiol. 2007; 293: H93–102.
- Guellich A, Damy T, Conti M, Claes V, Samuel JL, Pineau T, etal. Tempol prevents cardiac oxidative damage and left ventricular dysfunction in the PPAR-alpha KO mouse. Am J Physiol Heart Circ Physiol. 2013; 304: H1505–12.
- Howroyd P, Swanson C, Dunn C, Cattley RC, Corton JC. Decreased longevity and enhancement of age-dependent lesions in mice lacking the nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha). Toxicol Pathol. 2004; 32: 591–9.
- Poynter ME, Daynes RA. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J Biol Chem. 1998; 273: 32833–41.
- Atherton HJ, Gulston MK, Bailey NJ, Cheng KK, Zhang W, Clarke K, etal. Metabolomics of the interaction between PPAR-alpha and age in the PPAR-alpha-null mouse. Mol Syst Biol. 2009; 5: 259.
- Han L, Li M, Liu Y, Han C, Ye P. Atorvastatin may delay cardiac aging by upregulating peroxisome proliferator-activated receptors in rats. Pharmacology. 2012; 89: 74–82.
- Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002; 2: 748–59.
- Youssef J, Badr M. Role of peroxisome proliferator-activated receptors in inflammation control. J Biomed Biotechnol. 2004; 2004: 156–66.
- Son NH, Ananthakrishnan R, Yu S, Khan RS, Jiang H, Ji R, etal. Cardiomyocyte aldose reductase causes heart failure and impairs recovery from ischemia. PLoS One. 2012; 7: e46549.
- Thiagarajan D, Ananthakrishnan R, Zhang J, O'Shea KM, Quadri N, Li Q, etal. Aldose reductase acts as a selective derepressor of PPARgamma and the retinoic acid receptor. Cell Rep. 2016; 15: 181–96.
- Inglese J, Freedman NJ, Koch WJ, Lefkowitz RJ. Structure and mechanism of the G protein-coupled receptor kinases. J Biol Chem. 1993; 268: 23735–8.
- Chuang TT, LeVine H 3rd , De Blasi A. Phosphorylation and activation of beta-adrenergic receptor kinase by protein kinase C. J Biol Chem. 1995; 270: 18660–5.
- Leineweber K, Klapproth S, Beilfuss A, Silber RE, Heusch G, Philipp T, etal. Unchanged G-protein-coupled receptor kinase activity in the aging human heart. J Am Coll Cardiol. 2003; 42: 1487–92.
- Sibley DR, Lefkowitz RJ. Beta-adrenergic receptor-coupled adenylate cyclase. Biochemical mechanisms of regulation. Mol Neurobiol. 1987; 1: 121–54.
- Krueger KM, Daaka Y, Pitcher JA, Lefkowitz RJ. The role of sequestration in G protein-coupled receptor resensitization. Regulation of beta2-adrenergic receptor dephosphorylation by vesicular acidification. J Biol Chem. 1997; 272: 5–8.
- Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science. 2001; 294: 1307–13.
- Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, etal. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982; 307: 205–11.
- Christou DD, Seals DR. Decreased maximal heart rate with aging is related to reduced {beta}-adrenergic responsiveness but is largely explained by a reduction in intrinsic heart rate. J Appl Physiol. 2008; 105: 24–9.
- Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, etal. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res. 2008; 49: 2101–12.
- Okere IC, Chandler MP, McElfresh TA, Rennison JH, Sharov V, Sabbah HN, etal. Differential effects of saturated and unsaturated fatty acid diets on cardiomyocyte apoptosis, adipose distribution, and serum leptin. Am J Physiol Heart Circ Physiol. 2006; 291: H38–44.
- Sample J, Cleland JG, Seymour AM. Metabolic remodeling in the aging heart. J Mol Cell Cardiol. 2006; 40: 56–63.
- Lemieux H, Vazquez EJ, Fujioka H, Hoppel CL. Decrease in mitochondrial function in rat cardiac permeabilized fibers correlates with the aging phenotype. J Gerontol A Biol Sci Med Sci. 2010; 65: 1157–64.
- Fannin SW, Lesnefsky EJ, Slabe TJ, Hassan MO, Hoppel CL. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 1999; 372: 399–407.
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956; 11: 298–300.
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005; 120: 483–95.
- Tatarkova Z, Kuka S, Racay P, Lehotsky J, Dobrota D, Mistuna D, etal. Effects of aging on activities of mitochondrial electron transport chain complexes and oxidative damage in rat heart. Physiol Res. 2011; 60: 281–9.
- Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007; 292: C670–86.
- Kuka S, Tatarkova Z, Racay P, Lehotsky J, Dobrota D, Kaplan P. Effect of aging on formation of reactive oxygen species by mitochondria of rat heart. Gen Physiol Biophys. 2013; 32: 415–20.
- Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med. 2009; 19: 213–20.
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, etal. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005; 309: 481–4.
- Houtkooper RH, Vaz FM. Cardiolipin, the heart of mitochondrial metabolism. Cell Mol Life Sci. 2008; 65: 2493–506.
- Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Age-dependent decline in the cytochrome C oxidase activity in rat heart mitochondria: role of cardiolipin. FEBS Lett. 1997; 406: 136–8.
- Paradies G, Petrosillo G, Ruggiero FM. Cardiolipin-dependent decrease of cytochrome C oxidase activity in heart mitochondria from hypothyroid rats. Biochim Biophys Acta. 1997; 1319: 5–8.
- Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Age-dependent decrease in the cytochrome C oxidase activity and changes in phospholipids in rat-heart mitochondria. Arch Gerontol Geriatr. 1993; 16: 263–72.
- Moghaddas S, Stoll MS, Minkler PE, Salomon RG, Hoppel CL, Lesnefsky EJ. Preservation of cardiolipin content during aging in rat heart interfibrillar mitochondria. J Gerontol A Biol Sci Med Sci. 2002; 57: B22–8.
- Lesnefsky EJ, Hoppel CL. Cardiolipin as an oxidative target in cardiac mitochondria in the aged rat. Biochim Biophys Acta. 2008; 1777: 1020–7.
- Lesnefsky EJ, Minkler P, Hoppel CL. Enhanced modification of cardiolipin during ischemia in the aged heart. J Mol Cell Cardiol. 2009; 46: 1008–15.
- Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Peroxidative damage to cardiac mitochondria: cytochrome oxidase and cardiolipin alterations. FEBS Lett. 1998; 424: 155–8.
- Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. 2007; 292: C33–44.
- Mohamed SA, Hanke T, Erasmi AW, Bechtel MJ, Scharfschwerdt M, Meissner C, etal. Mitochondrial DNA deletions and the aging heart. Exp Gerontol. 2006; 41: 508–17.
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, etal. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004; 429: 417–23.
- Corral-Debrinski M, Stepien G, Shoffner JM, Lott MT, Kanter K, Wallace DC. Hypoxemia is associated with mitochondrial DNA damage and gene induction. Implications for cardiac disease. JAMA. 1991; 266: 1812–16.
- Zhang C, Bills M, Quigley A, Maxwell RJ, Linnane AW, Nagley P. Varied prevalence of age-associated mitochondrial DNA deletions in different species and tissues: a comparison between human and rat. Biochem Biophys Res Commun. 1997; 230: 630–5.