
Abstract
The 2537-m-deep North Greenland Eemian Ice Drilling (NEEM) core provided a first-time opportunity to perform extensive microbiological analyses on selected, recently drilled ice core samples representing different depths, ages, ice structures, deposition climates and ionic compositions. Here, we applied cultivation, small subunit (SSU) rRNA gene clone library construction and Illumina next-generation sequencing (NGS) targeting the V4–V5 region, to examine the microbial abundance, viability and diversity in five decontaminated NEEM samples from selected depths (101.2, 633.05, 643.5, 1729.75 and 2051.5 m) deposited 300–80 000 years ago. These comparisons of the indigenous glacial microbial populations in the ice samples detected significant spatial and temporal variations. Major findings include: (a) different phylogenetic diversity of isolates, dominated by Actinobacteria and fungi, compared to the culture-independent diversity, in which Proteobacteria and Firmicutes were more frequent; (b) cultivation of a novel alphaproteobacterium; (c) dominance of Cyanobacteria among the SSU rRNA gene clones from the 1729.75-m ice; (d) identification of Archaea by NGS that are rarely detected in glacial ice; (e) detection of one or two dominant but different genera among the NGS sequences from each sample; (f) finding dominance of Planococcaceae over Bacillaceae among Firmicutes in the brittle and the 2051.5-m ice. The overall beta diversity between the studied ice core samples examined at the phylum/class level for each approach showed that the population structure of the brittle ice was significantly different from the two deep clathrated ice samples and the shallow ice core.
To access the supplementary material for this article, please see supplementary files under Article Tools Online.
Covering 1.7 million km2, the Greenland ice sheet is the second largest on Earth and provides the longest ice core palaeoclimate records in the Northern Hemisphere. Deep glacial ice also preserves a parallel record of microbial life for hundreds of thousands of years. However, microbiological studies of the few available deep Greenland ice cores are still limited in comparison to those from Antarctica and non-polar glaciers. In 1999, Willerslev et al. reported a diverse clone library of eukaryotic 18S rDNA in a North Greenland glacier and Castello et al. (Citation1999) detected plant and bacterial viruses in Dye3 and GISP2 ice core samples. Fungi and algae were also cultivated from these ice cores (Catranis & Starmer Citation1991; Christner et al. Citation2000; Starmer et al. Citation2005). Over 10% of viable bacterial spores were enumerated in GISP2 ice (Yung et al. Citation2007). Systematic studies of 3043-m-deep silty GISP2 ice detected an abundant and diverse microbial population and recovered isolates including ultrasmall and novel microorganisms (Sheridan et al. Citation2003; Miteva et al. Citation2004; Miteva & Brenchley Citation2005; Loveland-Curtze et al. Citation2008, Citation2010). Similar viable microbial populations were found in the basal ice of other Greenland glaciers (Yde et al. Citation2010; Stibal et al. Citation2012). In addition, the microbial diversity and abundance in selected GISP2 samples differed depending on deposition climate (Miteva et al. Citation2009). In the light of possible metabolic activity of glacial microbial populations, Price (2007) suggested that the excessively high CO2, N2O and CH4 values measured at some GISP2 depths were due to in situ production by microorganisms (Rohde et al. Citation2008). Systematic measurements of chlorophyll autofluorescence along the GISP2 and D4 Greenland ice cores suggested an opportunity to study the evolution of archived marine cyanobacteria back in time (Price & Bay 2012). Christner et al. (Citation2012) found that microbiological processes occurring in basal water from the NGRIP borehole can have a significant role in global biogeochemical cycles. These microbiological studies of the Greenland ice sheet along with other research on polar and temperate glaciers have provided valuable information and advanced our understanding of the factors shaping microbial diversity in continental ice sheets, the mechanisms of survival in glacial ice and how microbial metabolism might affect the trace gas records. However, most of the deep ice core studies were performed on cores that have been stored for decades.
The new North Greenland Eemian Ice Drilling (NEEM) deep ice core provided a unique opportunity to perform detailed microbiological studies during (and immediately after) the drilling process. Here, we applied cultivation and culture-independent approaches, including Illumina next-generation sequencing (NGS), to examine the microbial abundance, viability and diversity in five freshly retrieved ice samples from selected depths (100–2051 m) that have been deposited during different time periods, from 300 to 80 000 years ago. Our diversity studies of the NEEM ice core followed the careful assay of potential exogenous contamination during the drilling process and exclusion of identified contaminants from the current study of the indigenous microbial population composition (Miteva, Burlingame et al. Citation2014; Miteva, Sowers et al. Citation2014). We found significant spatial and temporal variations of microbial abundance, viability and diversity among the studied ice samples that we believe to be related to specific in situ characteristics and climate during deposition.
Materials and methods
Site characterization
The new 2537-m-deep NEEM ice core was drilled in north-western Greenland in 2009–2011 with the participation of an international consortium of scientists led by the University of Copenhagen, Denmark. The main goal of the project was to obtain new climate records reaching back to the Eemian, the last interglacial period 115 000–130 000 years ago (NEEM Community Members Citation2013). The present research was part of the biology consortium of the NEEM project.
Ice core samples
The studied ice core samples () represent different depths, ages and ice structures (bubbly, brittle and clathrated ice). They were retrieved in 2009 and 2010. A full 1-m-long ice core from 101.2 m obtained from a shallow drill (S2) and sub-sections of two deep clathrated ice samples from 1729.75 and 2051.5 m from the main NEEM borehole were transported from Greenland soon after processing in the field for continuous chemistry, gas, dust and water isotopes analyses. Our project was allocated 55-cm-long pieces (bags) of the “gas” section from 633.05, 643.5, 1729.75 and 2051.5 m. The two brittle ice samples from the main core were left in the camp for one year for relaxation before being processed in the field and transported to the Pennsylvania State University, where all samples were stored at −32°C.
Table 1 Characteristics of decontaminated North Greenland Eemian Ice Drilling core samples subjected to microbiological analyses.
Ice core decontamination
The ice core samples were aseptically prepared for microbiological analyses according to the procedure of Rogers et al. (Citation2004). Each core length and characteristics are listed in . First, the ice cores that had been stored at −32°C were transferred to −20°C for 18 h. Decontamination was performed under a laminar flow hood at room temperature using sterile tools and glassware. Each core was immersed in pre-chilled 5.25% sodium hypochlorite for 10 s followed by three consecutive washes in 2–3 L each of pre-chilled autoclaved MilliQ water and gradual melting in sterile funnels. Fractions of 50–60 ml were collected in individual sterile bottles and used immediately for flow cytometry analyses, plating and DNA extraction. Opened Reasoner's 2A agar (R2A) and trypticase soy agar (TSA) plates served as hood controls during the decontamination procedure. Remaining samples were kept frozen at −80°C.
Flow cytometry
Direct flow cytometric analyses of the microbial populations in all melted ice fractions was performed on a triple laser, five-colour FC500 Beckman flow cytometer (Miami Lakes, FL, USA) utilizing previously developed and extensively used protocols (Miteva et al. Citation2009). Briefly, 1-ml samples were stained with either 2-µl 0.5-mM green fluorescent SYTO13 (Molecular Probes, Eugene, OR, USA) or with the LIVE/DEAD kit (Invitrogen, Waltham, MA, USA) for 4–5 h in the dark and analysed using red fluorescent pre-counted 1-µm beads as a standard. We used autoclaved 0.1-µm filtered MilliQ H2O as a sheath solution and extended times (20 000–100 000 events) for each analysis.
Isolate cultivation
Samples (0.25 ml/plate) of the innermost melted ice fraction were plated onto the following agar media (5–8 plates each): R2A, 1/10 strength R2A, TSA without carbohydrate, 1/10 strength TSA and incubated aerobically at 25°C, 18°C and 5°C for up to two months. Control non-inoculated plates kept open during plating were also incubated at the same temperatures. Individual colonies were re-cultivated on TSA and R2A at 18°C. Pure cultures were grouped by colony morphology (colour, size, texture) and microscopy (cell shape, size, Gram-staining, presence of spores). In most cases, there was no growth on the control plates. The few colonies that appeared on the control plates were also purified and analysed. Glacial isolates, which were phylogenetically and morphologically similar to those from the blank controls, were excluded from further analyses.
Isolate genomic DNA extraction and small subunit rRNA gene sequence analyses
Genomic DNA was extracted from pure cultures of bacterial and fungal isolates using the Ultra Clean Microbial DNA kit (MoBio Laboratories, Solana Beach, CA, USA) applying 30 s of bead beating in a MiniBeadbeater-8 Cell Disrupter (Biospec Products, Bartlesville, OK, USA). The small subunit (SSU) rRNA genes were polymerase chain reaction (PCR) amplified with 515F-1492R universal primers (Reysenbach & Pace Citation1995; Baker et al. Citation2003) using Ready-to-go PCR Beads (GE Healthcare Biosciences, NJ, USA). Amplified rDNA restriction analysis (ARDRA) with RsaI (Promega, Madison, WI, USA) was used to group the phylotypes. The PCR products representing each distinct restriction pattern were purified and sequenced with 515F or 1492R primer on an ABI Hitachi 3730XL DNA Analyzer at the Pennsylvania State University Genomics Core Facility of the Huck Institute for Life Sciences. Sequences were compared with those from the GenBank database using the basic local alignment search tool BLAST. Sequences of isolates and closest relatives selected from the database were aligned with ClustalX 2.1 (Larkin et al. Citation2007). Phylogenetic trees were generated based on distance analysis using the neighbour-joining algorithm that is part of the Juke-Cantor model. Species richness (Chao1) and diversity indices (Shannon and Inverse Simpson) were determined using Mothur, a microbial community analysis platform (Schloss et al. Citation2009).
Ice core genomic DNA extraction and whole genome amplification
Cells from several ice core melt fractions (500 ml each from the brittle ice and ca. 200 ml each from the other three ice cores) were concentrated by centrifugation at 30 996 g for 40 min and additional collection of cells by double filtration of the supernatants through 0.2-µm filters. Pellets and cells collected on the filters were combined in a single tube and centrifuged again. Total DNA was extracted from the cell pellets with the Ultra Clean Microbial DNA kit (MoBio Laboratories) using two consecutive bead-beating steps, first, for 30 s, followed by centrifugation and removal of the lysate; then, adding a new portion of the bead solution and MD1 solution, a second bead beating was performed for 90 s to disrupt additional, previously un-lysed cells. In order to increase the low DNA yield, whole genome amplification was carried out using the REPLI-g™ kit (Qiagen, Germantown, MD, USA). Enterobacterial repetitive intergenic consensus (ERIC) PCR fingerprinting of genomic DNA samples before and after REPLI-g amplification showed identical profiles (Supplementary Fig. S1).
Construction of SSU rRNA genes clone libraries, ARDRA and sequence analyses
Whole genome amplification amplified genomic DNA from the melted ice samples was used to obtain SSU rRNA genes by PCR with universal primers 515F-1492R and Ready-to-go PCR Beads (GE Healthcare Biosciences). The PCR profile was: initial denaturation at 95°C for 5 min, 35 cycles, consisting of 95°C for 1 min, 55°C for 1 min and 72°C for 1.5 min with a final elongation step for 7 min at 72°C. Purified products were cloned using the pDrive vector and EZ competent cells from the Qiagen PCR cloning kit (Valencia, CA, USA). Transformants containing plasmids with inserts were grown in Luria broth with ampicillin (100 µg ml−1) at 37°C and plasmid DNA was extracted by the boiling procedure and analysed by electrophoresis. Candidate plasmids with inserts were then used for re-amplification with T7-SP6 primers. ARDRA of the cloned PCR products with RsaI or HaeIII was used to group the clones in operational taxonomic units (OTUs). Individual PCR amplified SSU rDNA products were purified and sequenced. Chimeric sequences were identified using the Mallard program (Ashelford et al. Citation2006) and DECIPHER (Wright et al. Citation2012). Sequences were compared with those from the GenBank database using BLAST. Sequences of clones and closest relatives selected from the database were aligned with ClustalX 2.1 (Larkin et al. Citation2007). Phylogenetic trees were generated based on distance analysis using the neighbour-joining algorithm with the Juke-Cantor model. Species richness (Chao1) and diversity indices (Shannon and Inverse Simpson) were determined using Mothur (Schloss et al. Citation2009).
Illumina MiSeq SSU rRNA gene sequencing
REPLI-g amplified products from samples 101.2, 633.05, 1729.75 and 2051.5 m were purified from random primers using the recommended Qiagen supplementary protocol RG21 (Qiagen Citation2012). DNA concentrations were measured on Nanodrop 1000 (Thermo Scientific, Waltham, MA, USA) and samples were diluted in H2O to 200 ng total DNA in 20 µl. Amplicon libraries and Illumina MiSeq paired-end sequencing was performed at the Pennsylvania State University Genomics Core Facility of the Huck Institute for Life Sciences.
Libraries were prepared using the universal primers 515F (GTGCCAGCMGCCGCGGTAA) and 926R (CCGTCAATTCMTTTRAGTTT) targeting the V4-V5 SSU rRNA gene hypervariable region, providing an amplicon of 412 bp. These primers were chosen because of their high coverage of almost all phyla in conventional and metagenomic studies (Baker et al. Citation2003; Liu et al. Citation2008; Wang & Qianh Citation2009). Preliminary PCR tests on extracted genomic DNA from the studied ice core samples, and a set of reference genomic DNA from bacterial, archaeal and fungal cultures showed positive results.
Amplicons were generated using the SSU rRNA gene specific primers with Illumina adaptors and FastStart high-fidelity polymerase (Roche, Basel, Switzerland) under the following protocol: activation at 94°C for 3 min, amplification for 28 cycles at 94°C for 30 s, 57°C for 30 s, 72°C 30 s and a final extension step at 72°C for 8 min. A second round of amplification added the barcodes and specific primers for Illumina sequencing using the Nextera Index Kit (Illumina, San Diego, CA, USA). The PCR products were purified using the Agencourt AMPure technology (Beckman Coulter, Brea, CA, USA). After clean-up, the products were first quantified by Qubit (Life Technologies, Carlsbad, CA, USA) and sized-checked using the DNA 7500 LabChip on the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Quantitative PCR was performed to quantify the libraries using the Kapa Biosystems Library Quantification Kit (Kapa Biosystems, Woburn, MA, USA) prior to pooling in equimolar amounts. Paired-end sequencing, 2×250, was performed using the Illumina MiSeq platform and standard sequencing conditions according to the MiSeq System users guide.
Sequence data analysis
The paired-end MiSeq Illumina reads from the four studied samples were aligned and converted to contigs (2×250 bp) yielding 5 640 930 reads. Sequence analysis of each combined single fasta file was performed using the implemented features of Mothur version 1.31.2 (Schloss et al. Citation2009). Mothur MiSeq standard operating procedure was followed in data screening, alignment to the Mothur version of SILVA bacterial reference sequences (Pruesse et al. Citation2007), pre-clustering and chimera removal using the UCHIME algorithm (Edgar et al. Citation2011). Classification of the high quality 3 152 080 sequences with average length of 412 nucleotides was done by the Mothur version of Bayesian classifier with the RDP training set version 9 (9665 bacterial +384 archaeal 16S rRNA sequences). Taxonomic classification was used to assign sequences to phylotypes. Rarefaction analysis in Mothur was used to examine the adequacy of the sampling depth for OTUs formed at the level of genus, family and order. Alpha diversity was assessed by calculating the richness estimator (Chao1) and the diversity indices (Shannon and Inverse Simpson). To normalize everything, we randomly selected 341 215 sequences (number of sequences in the smallest sample) from each sample 1000 times and calculated the average.
For comparisons of beta diversity between samples, dendrograms were created based on phylotyping at different taxonomic levels using both the membership-based Jaccard coefficient and the structure based thetayc calculators. Principal coordinates plots were generated using the pairwise weighted UniFrac distances.
Nucleotide sequences accession numbers
The SSU rRNA gene sequences for the NEEM ice isolates and clones used in the phylogenetic trees have been submitted to the GenBank database under accession numbers KF768970–KF768990, KF768993, KF768994, KF768996–KF769006, KF769008–KF769014 and KJ735650–KJ735667. The Illumina generated sequences were deposited in the Sequence Read Archive of the National Center for Biotechnology Information under accession number SRP041348 (PRJNA243903).
Results
The five studied samples originated from different depths (100–2051 m) of the NEEM Greenland core, represented different deposition time periods (300–80 000 years BP) and had different ice structure (bubbly, brittle and clathrated). Summary details of ice core properties and chemistry are presented in . Specifically, samples varied in calcium and dust concentrations. The present comparative microbiological analyses of the decontaminated NEEM ice samples showed variable microbial abundance, viability and diversity based on cultivation and culture-independent approaches, including Illumina NGS.
Microbial abundance
Flow cytometry counting data showed variable cell numbers in melted sub-samples from the interior of the decontaminated ice core samples. For all cores, the outer surface layer before decontamination contained one to two orders of magnitude higher cell numbers (not presented). As shown in , ice from 1729.75 m depth had the highest cell numbers, which corresponded to high Ca2 + and dust concentrations and as expected was deposited during a colder climate (Guillevic et al. Citation2012). The deepest ice core from 2051.5 m had lower cell abundance that correlated with lower Ca2 + and dust concentrations and a warmer deposition climate. These two samples represented clathrated ice deposited 36 000 and 80 000 years ago, respectively.
The two closely located samples from the brittle ice originated from 633.05 and 643.5 m, ca. 3000-year-old ice, which corresponds to the present interglacial period with generally warmer climate. The cell numbers in these samples were at least one order of magnitude lower than those in the other ice cores analysed.
Finally, the most shallow bubbly ice sample from the 101.2 m depth was deposited 355 years ago and had relatively high cell abundance.
Viability and cultivation of aerobic heterotrophs
The proportion of viable cells, determined by flow cytometry after differential staining with the LIVE/DEAD kit varied in the studied samples (). It was very high (>80%) in the 101.2-m shallow ice and showed a trend of declining with depth to 56.7% in the brittle ice samples and down to 39% in the 1729.75-m sample. However, the deepest ice core (2051.5 m) had a surprisingly high proportion of viable cells reaching 84.5%.
The enumeration of culturable aerobic heterotrophs showed variable results that did not directly correlate with the percentage of viable cells (). The highest number of cultivated colonies was obtained from the two brittle ice samples (10.25 colony-forming units [CFU]/ml and 15.75 CFU/ml) and the 1729.75-m ice (12.25 CFU/ml). A total of nine CFU (2.25 CFU/ml) was recovered from the shallow 101.2-m core plating and only four isolates (1 CFU/ml) were obtained from the deepest 2051.5-m ice despite the high percentage of viable cells detected in these ice cores. Culturability estimates based on the number of cultivated colonies versus total number of cells were generally low. Even the highest culturability, calculated for the two brittle ice samples, reached only 0.008 and 0.016% of the total number of cells, whereas for all other samples it was significantly lower, between 0.00009 and 0.00028%.
Morphological and phylogenetic diversity of intrinsic glacial ice isolates
More than 100 distinct pure cultures were obtained which showed a wide range of colony and cell morphologies. The few colonies that grew on the control plates were also re-cultivated and analysed so that several fungal isolates similar to these have been eliminated. Our strategy for identifying the truly indigenous glacial microorganisms included initial systematic comparative evaluation of each isolate against our database of NEEM drilling fluid contaminants based on growth characteristics, phylogenetic SSU rRNA gene relationships and ERIC PCR genomic fingerprinting (Miteva, Burlingame et al. Citation2014). Here, we present the diversity of the final set of 41 glacial isolates after strain-by-strain comparisons and contaminant exclusion.
The distribution of morphologically distinct CFU representing major phylogenetic groups differed among samples (). Significantly more isolates were cultivated from the brittle ice (633.05 and 643.5 m) and the 1729.75-m-deep clathrated ice than from the shallow and the deepest ice core samples. Still, all samples yielded phylogenetically diverse isolates though in low numbers, which differed between samples.
Fig. 1 Distribution of colony-forming units (CFU) representing different phyla/classes cultivated from each North Greenland Eemian Ice Drilling core sample.
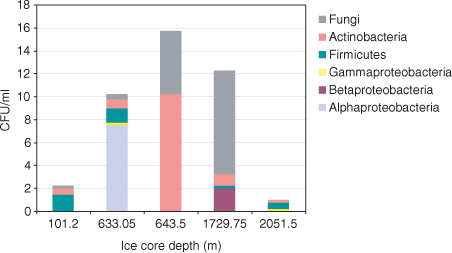
The phylogenetic analysis of the SSU rDNA sequences from isolates obtained from the five studied ice core samples showed considerable diversity represented by major phylogenetic groups: Alpha-, Beta- and Gammaproteobacteria (11 isolates), Firmicutes (six isolates), Actinobacteria (10 isolates) and fungi (14 isolates) (). Gram-positive bacteria were recovered from all samples with isolates related to genera Micrococcus, Kytococcus, Humicoccus, Arthrobacter, Oerskovia, Frigoribacterium, Streptomyces, Bacillus and Staphylococcus. Most of these bacteria were cultivated from the brittle ice samples and the 1729.75-m clathrated ice. Furthermore, as indicated in many closest relatives originated from cold and frozen environments, including snow, permafrost and glacial ice.
Fig. 2 Phylogenetic relationships of small subunit rRNA genes of isolates from decontaminated North Greenland Eemian Ice Drilling core samples from different depths. The tree is based on distance analysis (neighbour-joining algorithm with the Juke-Cantor model). Bootstrap values greater than 50% generated from 100 replicates are shown above the nodes. Accession numbers for these isolates and for closely related environmental cultured representatives retrieved from databases are included. Area in grey highlights the subcluster of the novel alphaproteobacterium and its closest relatives.
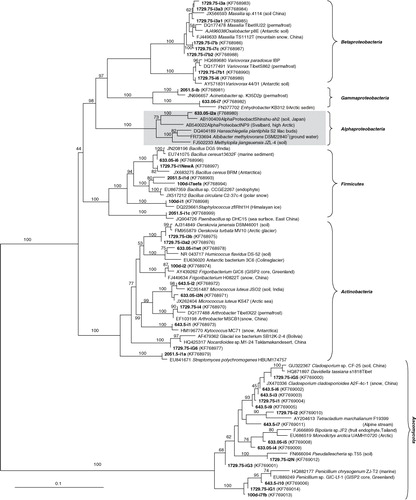
Fungal isolates were cultivated from four ice core samples mostly originating again from the brittle ice and the 1729.75-m-deep ice. They were identified as Ascomycota fungi related to genera Cladosporidium, Davidiella, Tetracladium, Bipolaris, Pseudallescheria and Penicillium (). It should be noted that no fungi were cultivated from the deepest NEEM ice sample.
Interestingly, all Betaproteobacteria isolates were obtained from the 1729.75-m ice only. Their phylogenetic affiliation with species belonging to Variovorax, Massilia and Oxalobacter suggests a capability for utilizing various substrates in either heterotrophic or chemolithotrophic metabolism. One gammaproteobacterium that was isolated from the 633.05-m ice was related to the gas-vacuole forming catalase-positive genus Enhydrobacter. Another isolate from the deepest ice core sample 2051.5 m was identified as Acinetobacter.
Despite the very low culturability, the calculated richness and diversity indices for the bacterial and fungal isolates for each ice core sample at genus level () indicated that sample 1729.75 m and the brittle ice had relatively high diversity.
Table 2 Number of isolate small subunit rRNA gene sequences observed operational taxonomic units (Sobs), richness and diversity indices for North Greenland Eemian Ice Drilling core samples from different depths.
Characterization of a novel alphaproteobacterium
The most intriguing finding was a novel isolate (633.05-i2a) obtained as a dominant culture from the 633.05-m brittle ice sample. This isolate belonged to the class Alphaproteobacteria, order Rhizobiales (, ). Sequence similarity calculations between 16S rRNA genes showed that isolate 633.05-i2a was most closely related to the validly described methylotrophic species Hansshlegelia plantiphila (91.3%; Ivanova et al. Citation2007), Agaricicola taiwanensis (88.6%; Chu et al. Citation2010), Methylopila jiangsuensis (87.3%; Li et al. Citation2011) and Albibacter methylovorans (86.5%; Doronina et al. Citation2001). These significant distances suggest that our isolate represents a novel genus or family. Initial characterization showed slow growth, transparent to light yellow small colonies (1 mm) on TSA and colonies with exopolymeric material on R2A (Supplementary Fig. S2a, b). Cells were visualized as very small Gram-negative rods. Further characterization of the novel alphaproteobacterium 633.05-i2a from the 633.05-m-deep NEEM core included growth and soft agar motility tests and electron microscopy which showed that the cells were non-motile, very short, small sized rods (0.6/0.4 µm) with at least one long flagellum-like structure (Supplementary Fig. S2c–f). Growth temperatures ranged from 2 to 30°C, with an optimum at 25°C. However, even at optimal incubation temperature, colonies became visible on TSA or R2A agar plates only after 10 days. Liquid aerobic cultures in Reasoner's 2B broth (R2B), trypticase soy broth and Luria broth did not reach high density and growth curves plateaued at 150–200 Klett units. The estimated doubling time was approximately 40 h, indicating a slow growth rate. No growth was found for our isolate in agar or liquid K medium for methylotrophs supplemented with either methanol or trimethylamine, as described by (Ivanova et al. Citation2007). The isolate also failed to grow anaerobically.
Similar characteristics were published by Hashizume et al. (Citation2004) for a rare soil bacterium Shinshu-ah2 of a possibly new genus isolated from prolonged soil enrichment cultures, which appeared to be the closest relative of isolate 633.05-i2a at 92.3%. Additionally, our isolate was distantly related phylogenetically to a filterable ultramicrobacterium alphaproteobactNP9 (88.5%), isolated from Svalbard (Nakai et al. Citation2013) and had similarly small cell sizes.
Diversity of SSU rRNA gene clone libraries
The second approach used in this study was the construction of five SSU rRNA gene clone libraries from total DNA extracted from the decontaminated ice core samples. A total of 351 clones were grouped by ARDRA restriction profiles and 148 were sequenced. The phylogenetic relationships of rRNA gene sequences and their quantitative distribution are presented in . One common observation for all libraries was that despite their differences from one another, they all had one dominant group of highly similar sequences. This may explain the relatively low Shannon diversity index (). The Inverse Simpson index was highest for sample 1729.75 m indicating greater diversity, followed by that of the shallow 101.2-m ice.
Fig. 3 Neighbour-joining tree of SSU rRNA gene clone sequences from different depths of North Greenland Eemian Ice Drilling glacial ice. For groups with highly similar clone sequences from the same ice core sample, a single representative was chosen. Bootstrap values greater than 50% generated from 1000 replicates are shown above the nodes.
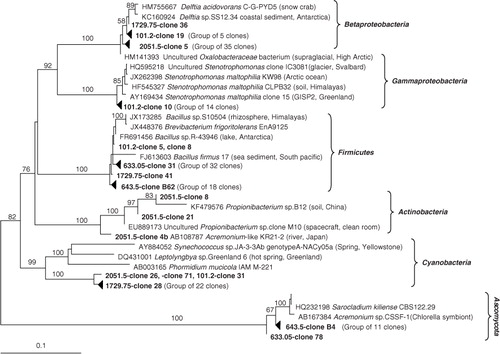
Table 3 Number of small subunit rRNA gene clone sequences, observed operational taxonomic units (Sobs), richness and diversity indices at genus level for North Greenland Eemian Ice Drilling core samples from different depths.
A dominant group of 14 sequences in the 101.2-m library belonged to Gammaproteobacteria, with closest species Stenotrophomonas maltophilia, originating from different cold and frozen environments (). Two 101.2-m clones were related to Brevibacterium frigoritolerans and one clone matched Cyanobacteria related to the genus Phormidium. Betaproteobacteria clones related to Delftia were found in this and two other libraries from the deeper ice cores including a dominant group of 35 clones in the 2051.5-m sample. Actinobacteria were detected only in the deepest (2051.5 m) ice core sample ().
The libraries from the two brittle ice samples separated in depth/age by 10 m/50 years showed very similar diversity dominated by Brevibacterium and Bacillus related Firmicutes (50 clones) followed by 12 clones clustering with fungi Acremonium. In a separate study, we identified certain bacterial and fungal species as potential contaminants originating mostly from the drilling fluid (Miteva, Burlingame et al. Citation2014). It should be noted that a few Bacillus related sequences (likely contaminants) were found in the 101.2- and 1729.75-m ice clone libraries but the majority was detected in the brittle ice samples. However, Bacillus sequences were not unanimously excluded from the clone libraries phylogenetic tree solely on the basis of sequence similarity to cloned SSU rRNA genes from the potentially contaminating drilling fluid. Unlike bacilli related isolates, which could be compared by a polyphasic approach (morphology, 16S rRNA gene sequencing and genomic fingerprinting), identifying a Bacillus sequence in a clone library could be either a result of contamination or an intrinsic member of the glacial population due to their ubiquitous distribution in natural environment.
Interestingly, the dominant group of clones from the 1729.75-m sample was related to filamentous Cyanobacteria commonly found in Antarctica and the Arctic (Quesada & Vincent 2012; Strunecky et al. Citation2012), including in Greenland glacial ice (Knowlton et al. Citation2013). Several cyanobacteria were also found in the 101.2- and 2051.5-m-deep NEEM ice samples. Because of the relative low similarity, we constructed another clone library from this ice core sample using the cyanobacteria specific 16S rRNA gene primers CYA106F-CYA781R (Nubel et al. Citation1997). Analysis of 30 sequences showed highest similarity to the genus Calothrix, order Nostocales, also common in glacial environments (Sihvonen et al. Citation2007; Komárek et al. Citation2012).
Illumina MiSeq phylogenetic diversity
The third approach used in this study explored the phylogenetic diversity of four NEEM samples by Illumina paired-end sequencing targeting the V4-V5 hypervariable region of the SSU rRNA gene. Importantly, this region overlapped with the one used for the construction of our conventional clone libraries. A total of 3 152 080 reads of average length 412 nucleotides were obtained after quality filtering the original 5 640 930 paired-end reads. They were relatively equally distributed among the three deeper ice cores in the range of 800 000–900 000 each, whereas the most shallow sample from 101.2 m had one-third the number of reads ().
Table 4 Number of Illumina small subunit rRNA gene sequences, observed operational taxonomic units (Sobs), richness and diversity indices for North Greenland Eemian Ice Drilling core samples from different depths. The average values (±SD) were calculated for genus level. A normalized equal number of sequences (341 215) from each sample were randomly selected for analysis.
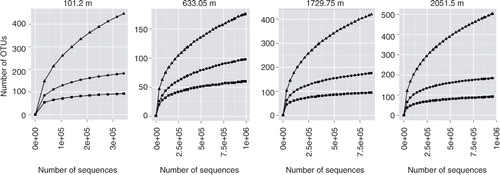
To address how well the Illumina sequencing data represent the actual NEEM microbial populations, rarefaction curves of alpha diversity were built for OTUs formed at the level of genus, family and order using Mothur. As shown in , rarefaction curves did not reach saturation at a genus level but were flat at the level of family or order. These results demonstrate that the sampling was sufficient at the higher phyla level but did not capture the total diversity at the genus level. Species richness (Chao1) was highest for the 101.2-m ice, followed by the deepest ice core sample. The average Shannon and Inverse Simpson diversity estimates were highest for the 1729.75-m ice (similarly to the isolates and clone libraries) followed by the other clathrated ice sample from 2051.5 m ().
Fig. 4 Rarefaction curves of phylotype based operational taxonomic units (OTUs) of the small subunit rRNA gene Illumina sequence reads for North Greenland Eemian Ice Drilling core samples from different depths. Phylotype levels include genus (triangles), family (dots) and order (rectangles).
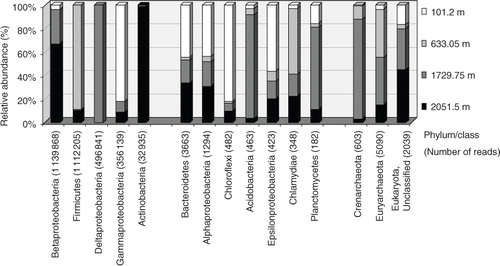
Analysis of the classified sequences showed variable representation and quantitative distribution of different taxa of Bacteria, Archaea and Eukaryota among samples (). Overall, Betaproteobacteria and Firmicutes comprised the most abundant phylogenetic groups with over 1 million reads each, followed by Deltaproteobacteria and Gammaproteobacteria with 496 845 and 356 139 reads, respectively. Actinobacteria were identified only in the deepest ice core at a lower number (32 935). As in the case with Sanger clone libraries, each ice core sample had one numerically dominant group: Gammaproteobacteria in the 101.2-m-deep ice; Firmicutes in the brittle ice (633.05 m); Deltaproteobacteria in the 1729.75-m ice; and Betaproteobacteria and Actinobacteria in the 2051.5-m deepest ice core. Other taxa, including Bacteriodetes, Alphaproteobacteria, Chloroflexi, Acidobacteria, Epsilonproteobacteria, Chlamydia, Planctomyces, were detected in lower numbers in all samples. Over 5000 Archaeal sequences were represented mostly by Euryarchaeota. Archaea reads were highest (2555) in the 1729.75-m-deep ice followed by the brittle ice (2161), the 2051.5-m sample (800) and fewest (177) in the shallow 101.2-m ice. Some Eukaryota and unclassified sequences (2039) were found as well. As shown in , most of these groups were detected in different proportions in all or some of the studied ice core samples.
Fig. 5 Distribution of major phyla/classes of small subunit rRNA gene sequences generated by Illumina MiSeq sequencing. The number of reads for each column is given in parentheses. Groups are shown in numerically descending order of sequence reads.
Further comparisons within each sample and between samples were done at genus level, also distinguishing dominant genera (above 1%) and low represented genera (0.02–1%) (). The category of dominant genera included seven bacterial genera from different phyla/classes. Two genera dominated in the 101.2-m sample: Delftia (Betaproteobacteria) and Stenotrophomonas (Gammaproteobacteria). They were also found in the two clathrated ice samples from 1729.75 and 2051.5 m but in different proportions. Specifically, Delftia related sequences comprised nearly 40 and 80%, respectively. One intriguing observation was the very high number of reads related to the genus Vampirovibrio (Deltaproteobacteria), a known Chlorella parasite, which dominated in the 1729.75 m. The two dominant Firmicutes genera in the 633.05-m ice core were Bacillus (87.7%) and Planococcus (8.8%). It should be noted that the deepest ice core showed highest diversity with six of the seven dominant genera present, from phyla Actinobacteria, Betaproteobacteria, Gammaproteobacteria and Firmicutes.
Fig. 6 Relative abundance of genera in North Greenland Eemian Ice Drilling core samples from different depths based on Illumina MiSeq sequencing. Left panels show the most abundant genera (>1%). Right panels show less frequently detected genera (0.02–1%).
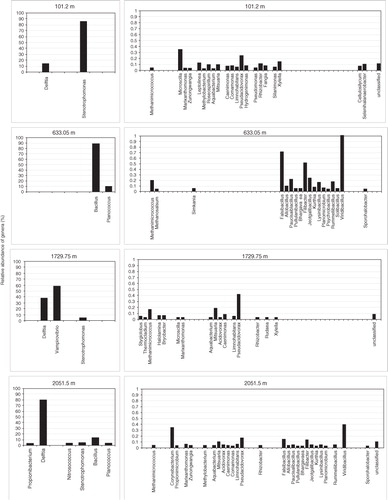
Forty-seven genera comprised the category of low representation (). Numerically they ranged from 100 to 200 reads (0.02%) to 9970 reads (1%). Methanomicrobia related Archaea genera were found in all samples. Furthermore, only the 1729.75-m-deep ice also contained two Crenarchaeota genera Stygiolobus and Thermocladium and one genus Halolamina represented Halobacteria. Diverse bacterial genera were detected in all samples but their distribution varied significantly. Again, the highest number of genera (26), representing all major bacterial phylogenetic groups (Actinobacteria, Bacteroidetes, Alpha-, Beta- and Gammaproteobacteria, Firmicutes, Bacillaceae, Planococcaceae and Clostridia) were identified in the deepest ice core from 2051.5 m. Conversely, the bacterial genera found in the 633.05-m-deep ice were limited to Firmicutes (15 genera) and one Chlamydiae genus Simkania. Notably different was the 1729.75-m sample, where nine Beta- and Gammaproteobacteria genera were most prominent but no Firmicutes were identified.
One notable finding was the identification of several similar Betaproteobacteria genera in three of the studied samples. These genera (Aquabacterium, Mitsuaria, Acidovorax, Caenimonas, Comamonas, Limnohabitans, Pseudacidovorax), along with the dominant in the same sample Delftia and the whole class Betaproteobacteria, are known for their versatile morphological and metabolic properties and rapid response to changing environmental characteristics (Hell et al. Citation2013; Kasalicky et al. Citation2013). Another similarity between these three samples was the detection of several Bacteroidetes genera of marine origin: Microscilla, Marixantomonas and Zunongwangia.
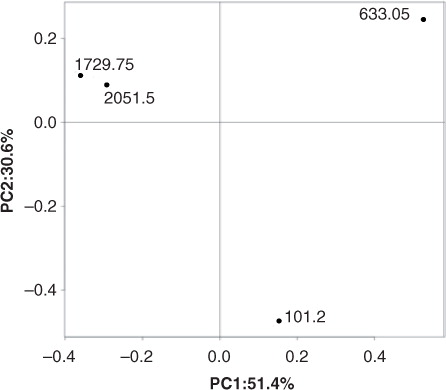
The community structure of individual samples was compared using principal coordinate analysis of beta diversity with weighted UniFrac metrics after even sampling. The plot in revealed a clear separation between samples with high similarity between the two clathrated ice samples, whereas the 101.2-m and the 633.05-m samples were significantly different.
Fig. 7 Principle coordinate analysis of operational taxonomic unit abundance in all samples. Plots of beta diversity were calculated based on weighted Unifrac metrics. The percent variations explained by each coordinate are shown on each axis.
Comparisons of the phylogenetic composition of NEEM ice core samples as revealed by different methods
The application of cultivation plus two culture-independent approaches resulted in different pictures of the phylogenetic diversity of the studied NEEM ice core samples. summarizes the distribution of dominant phyla in different samples revealed by each method and illustrates the overall population structure similarity between the samples based on the thetayc coefficients.
Fig. 8 Distribution of dominant phylogenetic groups in populations from different North Greenland Eemian Ice Drilling ice depths revealed by: (a) cultivation/direct plating; (b) Sanger clone libraries; (c) Illumina next-generation sequencing. Column graphs show percentage of small subunit rRNA gene sequences assigned to different phyla/classes. Dendrograms represent the population structure similarity between the samples at class level based on the thetayc coefficient.
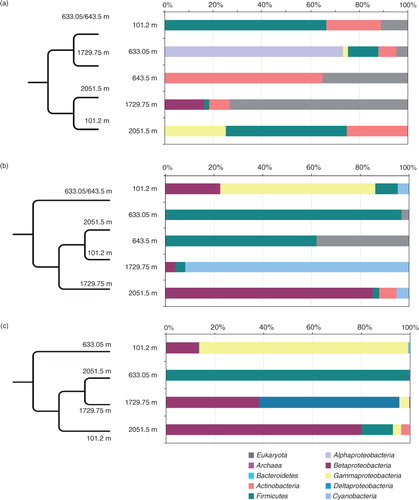
Not surprisingly, the phylogenetic diversity of cultivated isolates differed from the diversity revealed by non-cultivation methods, in which case a significantly similar distribution of major phyla was observed. Specifically, Actinobacteria and fungi (with the exception of the 2051.5-m ice) were more abundant among isolates than among clones and Illumina generated sequences while Proteobacteria were more frequent among non-cultivation based sequences. The relatively higher proportion of Proteobacteria among the 633.05-m ice isolates was due to the larger number of identical CFUs from the novel alphaproteobacterium (a).
The phylogenetic composition similarity between the clone libraries and Illumina sequences (b, c) observed for all studied samples indicates that these approaches reveal similar diversity patterns though at different resolutions. One unusual finding was the identification of a numerically dominant group of Illumina reads related to Vampirovibrio (Deltaproteobacteria), whereas the clone library from the same sample was dominated by Cyanobacteria (mostly Phormidium) related sequences. In this relation, several authors have pointed out that this taxon (Vampirovibrio), initially described as a Chlorella symbiont and member of Proteobacteria, requires a taxonomical/gene sequence revision (Goeke et al. Citation2013). Recent genome sequencing and database comparisons have indicated that Vampirovibrio is a deep branching cyanobacterium (Hemp Citation2012; Soo et al. Citation2014).
The overall beta diversity between the ice core microbial populations from increasing depths was examined at different taxonomic levels for each of the approaches used. Comparisons of the phylotype dendrograms at the class level in showed nearly similar clustering between samples based on clone libraries and Illumina NGS with the brittle ice significantly different from the other three ice core samples. In contrast, the isolate phylotype clustering differed showing closer similarity between the brittle ice and the 1729.75-m sample possibly due to the large number of fungal isolate sequences compared to the other subcluster of the shallow 101.2-m ice and the deepest 2051.5-m ice.
Discussion
Interdisciplinary studies of high-resolution deep ice cores are important because they can yield insights into palaeoclimate records and dynamics while examining the relationships between the parallel chronological archive of microbial life and the environment (both in situ and palaeo-deposition). The new NEEM Greenland ice core provided a first-time opportunity to perform extensive microbiological analyses on selected, recently drilled ice core samples representing different depths, ages, deposition climates, in situ temperatures, and ionic compositions soon after drilling. The results from our comprehensive approach including cultivation, SSU rRNA gene clone library construction and NGS allowed different comparisons: (a) between NEEM samples (i.e., depths, ages, in situ temperature, ice structures); (b) between analytical methods for each NEEM sample; and (c) diversity of the NEEM ice core compared to GISP2 and other Greenland cores. These comparisons helped provide insights on the existence of spatial patterns of microbial distribution and diversity within the ice and their dependence on environmental factors.
Ensuring the authenticity of microbiological results was a major goal in these studies. First, we followed a stringent decontamination procedure and routinely used a series of controls at each step. More importantly, these analyses were preceded by detailed assessment of any potential forward contamination, originating from the drilling fluid and other sources during the drilling process (Miteva, Burlingame et al. Citation2014). This allowed us to obtain specific information about the quantity and nature of the microbial contaminants that could have been introduced during drilling and use the created database of contaminants to screen our isolates and clones in attempts to distinguish possible contaminants from the indigenous glacial populations.
The conclusive finding of the present study was the existence of abundant, viable and diverse indigenous microbial populations in the NEEM Greenland ice core, at depths ranging from 100 to 2051 m. Our study also provided new evidence confirming the previously found direct correlation between microbial cell abundance in polar and non-polar ice cores and deposition climate, with higher numbers of cells found in ice layers deposited during colder climate periods versus lower cell abundance in ice core layers deposited during warmer climates (Abyzov Citation1993; Xiang et al. Citation2005; Yao et al. Citation2006; Miteva et al. Citation2009; Xiang et al. Citation2009). Furthermore, detecting viable cells in ice deposited 300–80 000 years ago and recovering diverse isolates (albeit at a low culturability levels) showed once again that microbial cells are capable of long-term survival trapped in one of the harshest environments on Earth. In this relation, glacial ice isolates provide excellent models for genomic and physiological studies of the mechanisms of survival.
Importantly, the detection of variable phylogenetic diversity of the intrinsic microbial populations in the studied NEEM ice core samples was achieved by different methodological approaches: analysis of cultivated isolates, SSU rRNA gene clone libraries and Illumina next-generation sequences. The application of these methods was oriented towards obtaining a comparable multivariate view of the microbial diversity in the studied samples rather than at evaluating these approaches and their well-known biases and discrepancies. The observed low levels of culturability on the media used provided yet another example that the majority of environmental microorganisms are not easily culturable on standard laboratory media. We were also aware that the combination of the DNA extraction technique (MoBio kit), the whole genome amplification and SSU rRNA gene cloning with the selected PCR primers (515F-1492R and 515F-926R) could recover only a subset of the real microbial diversity, which would differ from other subsets obtained by other methods/primer sets (Hong et al. Citation2009). However, REPLI-g has been shown to be useful and sometimes essential for obtaining sufficient DNA from environmental samples of low biomass or limited volume and to generate the least bias in NGS (Pinard et al. Citation2006). Furthermore, the selection of universal PCR primers targeting highly conserved regions and shown to amplify the SSU rRNA genes from bacteria, archaea and eukarya (Isenbarger et al. Citation2008) was important for our comparisons.
It should be noted that the Illumina sequencing targeting the V4-V5 SSU rRNA gene region provided an unprecedentedly high number of sequences and improved coverage. To our knowledge, this is the first study to provide a considerable amount of sequence information comparing the microbial population diversity at several depths of a 2537-m-deep polar ice core using NGS. A recent report of Knowlton et al. (Citation2013) performed a 454 metagenomic/metatransciptomic analysis of two sections of the GISP2 ice core from 1601- and 3014-m depths yielding a limited number of 33 quality sequences unique to the ice. Several other metagenomic studies of glacial ice were performed on near surface glacial ice (Simon et al. Citation2009; Choudhari et al. Citation2013) or on subglacial Arctic ice and accretion Vostok ice (Hamilton et al. Citation2013; Shtarkman et al. Citation2013).
As stated earlier, a number of studies, including this one, support the notion that deposition climate is a prominent factor impacting the abundance and diversity of the indigenous glacial microbial populations. Palaeoclimate fluctuations between colder and warmer periods representing glacial millennial scale variability have been reported for the NEEM and other Greenland cores (Guillevic et al. Citation2012; NEEM Community Members Citation2013). Here, the two deep clathrated ice samples (1729.75 and 2051.5 m) provide a good example of contrasting deposition climates and correspondingly a one order of magnitude microbial cell abundance difference. The in situ physico-chemical parameters established so far along the NEEM core may also influence the survival of the microorganisms initially deposited on the glacial surface and gradually embedded deeper into the ice. The habitat where microbial cells reside and survive in glacial ice are the liquid veins at the triple junctions between ice grains and the grain surfaces which are covered with a thin liquid layer (Price Citation2000, Citation2007; Dani et al. Citation2012). Notable differences between the studied ice core samples from different depths include their age and ice structure (bubbly, brittle and clathrated ice). The shallow NEEM bubbly ice, enclosing numerous air bubbles (sample 101.2 m), was shown to have relatively large ice crystals (2–3 mm) and consequently larger veins, which can grow further in size during the dynamic transition to bubble-free ice accompanied by the formation of multiple micro-fractures during relaxation in the brittle ice zone (Faria et al. Citation2010; Binder, Garber et al. Citation2013; Binder, Weikusat et al. Citation2013). The deeper clathrated ice (samples 1729.75 and 2051.5 m) typically exhibits fine-grain structure and thinner veins, which may also vary depending on climate changes (Durand et al. Citation2006; Montagnat et al. Citation2014). These remarkable ice structure differences can be a major factor shaping the composition and diversity of the microbial populations.
Overall, the composition of the NEEM microbial populations reported here resemble the microbial composition found in previous studies of deep glacial ice with commonly detected Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes and fungi (Miteva Citation2008; Junge et al. Citation2011; Buzzini et al. Citation2012). This is best illustrated by the large number of species and genera closely related to our NEEM isolates and clones that have origins from glacial ice or other frozen environments (, ), strongly suggesting adaptive capabilities for long-term survival under extreme conditions. Specifically, comparisons between the NEEM ice core diversity and the previously studied GISP2 deep clathrated ice (57 000–68 000 years old) showed similar population composition among isolates and in clone libraries, constructed with the same universal SSU rRNA gene primers (Miteva et al. Citation2009). However, the proportional distribution of major phyla among ice samples differed in both studies, which could be attributed to the different depths/ages and geochemical characteristics of the samples. It also indicates the existence of enormous variability in the microbial diversity of deep ice cores on a narrow scale, corresponding to annual snow deposition.
Some specific characteristics of the indigenous microbial population structure in the studied NEEM ice core samples and major findings revealed by our three independent approaches are outlined below.
The phylogenetic diversity of isolates recovered from all studied NEEM ice core samples was dominated by Actinobacteria and fungi that differed from the culture-independent diversity with more frequent Proteobacteria and Firmicutes. It should be noted that because of the extremely low culturability the isolates represented a very small portion of the real indigenous microbial diversity.
One intriguing discovery was the isolation and characterization of a novel organism belonging to Alphaproteobacteria that may represent a new genus or family. Sequence similarities between 16S rRNA genes showed that this isolate was most closely related to the methylotrophic species Hansshlegelia plantiphila (91.3%; Ivanova et al. Citation2007), Agaricicola taiwanensis (88.6%; Chu et al. Citation2010), Methylopila jiangsuensis (87.3%; Li et al. Citation2011) and Albibacter methylovorans (86.5%; Doronina et al. Citation2001).
Finding prevailing cyanobacterial sequences in one clone library and detecting a dominant group of a provisional deep branching clade of Cyanobacteria (Vampirovibrio) among the Illumina sequences from the same 1729.75-m ice core sample is of special interest. Cyanobacteria are common in the cryosphere because they are known to have extremely high cold tolerance and a remarkable range of mechanisms ensuring long-term survival (Vincent Citation2007; Vishnivetskaya Citation2009; Quesada & Vincent 2012). However, cyanobacteria are most frequently found in surface ice layers, specifically dominating the active assemblages of cryoconite holes. They are rarely detected in such glacial depths as the 1729.75-m NEEM ice sample.
We identified over 5000 Archaeal sequences by Illumina NGS that were distributed among all studied samples and related to the methanogenic genus Methanimicrococcus. This is one of the first studies to detect Archaea within glacial ice after the report of Choudhari et al. (Citation2013) for the Byron Glacier, Alaska. Previous metagenomic studies of a glacier in Germany did not find Archaea (Simon et al. Citation2009).
Another interesting finding was the dominance of Planococcaceae (10 genera) as compared to Bacillaceaea (five genera) among Firmicutes that were detected only in the brittle ice and the 2051.5-m ice. Among Planococcaceae were many non-spore-forming genera, including the dominant Planococcus, as well as Bhargavaea, Filibacter, Kurthia, Jeotgalibacillus and Planomicrobium. Some of these Firmicutes were related to known psychrophiles such as Planococcus antarcticus and Psychrobacillus.
Sequences related to Delftia betaproteobacteria were found both among clones and by NGS in three of the studied samples (101.2, 1729.75 and 2051.5 m) with significant dominance in the deepest ice. Members of this genus are aerobic, capable of degrading organic matter and denitrification (Leibeling et al. Citation2010; De Sousa & Bhosle Citation2012). The latter possibility is supported by ion composition data showing relatively high content of nitrate in the two clathrated ice samples (109.17 and 78.15 ng/g, respectively) (Bigler, pers. comm.). Representatives of Delftia have been previously found in glacial ice (Skidmore et al. Citation2005; Zhang et al. 2010; Zeng et al. 2013).
Although taxonomic composition based on SSU rRNA gene sequences cannot be directly linked to functional activity, some correlations can be observed. Furthermore, microbial communities in deep glacial ice are not considered dynamic as cells rarely divide and any metabolic activity mostly maintains survival for extended time periods. We can speculate about some potential functional traits, highly specific to certain organisms. For example, the presence of Nitrosococcus in the deepest ice suggests possible nitrification and it has been established that this ice sample contains 5.98 ng/g NH4 (Bigler, pers. comm.). Similarly, finding the genus Methanimicrococcus in all studied samples indicates a possibility for methane production.
In conclusion, these first polyphasic studies of the indigenous microbial diversity in the NEEM Greenland ice core showed spatial and temporal variations in microbial abundance, viability and population structure along different core depths. The application of cultivation and culture-independent methods showed complementary results that helped identifying numerous members of the glacial microbial populations. The overall beta diversity examined at the phylum/class level for each approach showed that the population structure of the brittle ice was significantly different from the two deep clathrated ice samples and the shallow ice core. These results extended previous findings from GISP2 and other Greenland ice cores and contributed to building a more comprehensive picture of the vertical microbial distribution within the Greenland ice sheet. Microbiological research of the NEEM core will be particularly valuable because extensive, high-resolution geochemical information is becoming available to correlate our microbiological findings to the ecological settings and past climate records within ice.
Supplementary Material
Download PDF (508.8 KB)Acknowledgements
The ice core samples used in this study were retrieved during the NEEM project in Greenland. NEEM is directed and organized by the Centre for Ice and Climate at the Niels Bohr Institute and the Office of Polar Programs of the US National Science Foundation (NSF). It is supported by funding agencies and institutions in Belgium (FNRS-CFB and FWO), Canada (NRCan/GSC), China (CAS), Denmark (FIST), France (IPEV, CNRS/INSU, CEA and ANR), Germany (AWI), Iceland (RannIs), Japan (NIPR), Korea (KOPRI), The Netherlands (NWO/ALW), Sweden (VR), Switzerland (SNF), UK (NERC) and the USA (US NSF, Office of Polar Programs). This study was supported by NSF grants ARC0909323 and 0806407 and the National Aeronautics and Space Administration—National Astrobiology Institute NASA-NAI agreement NNA09DA76A. We thank Greg Grove, Deb Grove, and Craig Praul from the Pennsylvania State University Genomics Facility for the libraries preparation and next-generation sequencing. We acknowledge the valuable help of Istvan Albert with the Illumina sequence analyses. We thank M. Bigler (University of Bern, Switzerland) for sharing data on NEEM continuous flow analysis dust and ion concentration. We also thank Caroline Burlingame and Amanda Gifford for their input in this research.
Notes
To access the supplementary material for this article, please see supplementary files under Article Tools Online.
Related Research Data
References
- Abyzov S.S. Friedman E.I. Microorganisms in the Antarctic ice. Antarctic microbiology. 1993; New York: Wiley. 265–295.
- Ashelford K.E., Chuzhanova N.A., Fry J.C., Jones A.J., Weightman A.J. New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Applied and Environmental Microbiology. 2006; 72: 5734–5741. [PubMed Abstract] [PubMed CentralFull Text].
- Baker G.C., Smith J.J., Cowan D.A. Review and re-analysis of domain-specific 16S primers. Journal of Microbiological Methods. 2003; 55: 541–555. [PubMed Abstract].
- Binder T., Garbe C.S., Wagenbach D., Freitag J., Kipfstuhl S. Extraction and parametrization of grain boundary networks in glacier ice using a dedicated method of automatic image analysis. Journal of Microscopy. 2013; 2502: 130–141.
- Binder T., Weikusat I., Freitag J., Garbe C.S., Wagenbach D., Kipfstuhl S. Microstructure through an ice sheet. Materials Science Forum. 2013; 753: 481–484.
- Buzzini P., Branda E., Goretti M., Turchetti B. Psychrophilic yeasts from worldwide glacial habitats: diversity, adaptation strategies and biotechnological potential. FEMS Microbiology Ecology. 2012; 82: 217–241. [PubMed Abstract].
- Castello J., Rogers S., Starmer W., Catranis C., Ma L., Bachand W.G., Zhao Y., Smith J. Detection of tomato mosaic tobamovirus RNA in ancient glacial ice. Polar Biology. 1999; 22: 207–212.
- Catranis C., Starmer W. Microorganisms entrapped in glacier ice. Antarctic Journal of the United States. 1991; 26: 234–236.
- Choudhari S., Smith S., Owens S., Gilbert J.A., Shain D.H., Dial R.J., Grigoriev A. Metagenome sequencing of prokaryotic microbiota collected from Byron Glacier, Alaska. Genome Announcements. 2013; 1(2): 00099–13.
- Christner B.C., Montross G.G., Priscu J.C. Dissolved gases in frozen basal water from the NGRIP borehole, implications for biogeochemical processes beneath the Greenland ice sheet. Polar Biology. 2012; 35: 1735–1741.
- Christner B.C., Mosley-Thompson E., Thompson L.G., Zagorodnov V., Sandman K., Reeve J.N. Recovery and identification of viable bacteria immured in glacial ice. Icarus. 2000; 144: 479–485.
- Chu J.-N., Arun A.B., Chen W.-M., Chou J.-H., Shen F.-T., Rekha P.D., Kämpfer P., Young L.-S., Lin S.-Y., Young C.-C. Agaricicola taiwanensis gen. nov., sp. nov., an alphaproteobacterium isolated from the edible mushroom Agaricus blazei. International Journal of Systematic and Evolutionary Microbiology. 2010; 60: 2032–2035. [PubMed Abstract].
- Dani K.G.S., Mader H.M., Wolff E.W., Wadham J.L. Modeling the liquid-water veins system within polar ice sheets as a potential microbial habitat. Earth and Planetary Science Letters. 2012; 333–334: 238–249.
- De Sousa T., Bhosle S. Satyanarayana T. Microbial denitrification and its ecological implications in the marine system. Microorganisms in environmental management, microbes and environment. 2012; New York: Springer. 683–700.
- Doronina N., Trotsenko Y.A., Tourova T.P., Boris B., Kuznetsov B.B., Leisinger T. Albibacter methylovorans gen. nov., sp. nov., a novel aerobic, facultatively autotrophic and methylotrophic bacterium that utilizes dichloromethane. International Journal of Systematic and Evolutionary Microbiology. 2001; 51: 1051–1058. [PubMed Abstract].
- Durand G., Weiss J., Lipenkov V., Barnola J., Krinner G., Parrenin F., Delmonte B., Ritz C., Duval P., Röthlisberger R., Bigler M. Effect of impurities on grain growth in cold ice sheets. Journal of Geophysical Research—Earth Surface. 2006; 111: 01015.
- Edgar R.C., Haas B.J., Clemente J.C., Quince C., Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011; 27: 2194–2200. [PubMed Abstract] [PubMed CentralFull Text].
- Faria S.H., Freitag J., Kipfstuhl S. Polar ice structure and the integrity of ice-core paleoclimate records. Quaternary Science Reviews. 2010; 29: 338–351.
- Goeke F., Thiel V., Wiese J., Labes A., Imhoff J.F. Algae as an important environment for bacteria—phylogenetic relationships among new bacterial species isolated from algae. Phycologia. 2013; 52: 14–24.
- Guillevic M., Bazin L., Landais A., Kindler P., Orsi A., Masson-Delmotte V., Blunier T., Buchardt S.L., Capron E., Leuenberger M., Martinerie P., Prie F., Vinther B.M. Spatial gradient of temperature, accumulation and δ18O-ice in Greenland over a series of Dansgaard-Oeschger events. Climate of the Past. 2012; 8: 5209–5261.
- Hamilton T.L., Peters J.W., Skidmore M.L., Boyd E.S. Molecular evidence for an active endogenous microbiome beneath glacial ice. The ISME Journal. 2013; 7: 1402–1412. [PubMed Abstract] [PubMed CentralFull Text].
- Hashizume A., Fudou R., Jojima Y., Nakai R., Hiraishi A., Tabuchi A., Sen K., Shibai H. Rare bacterium of a new genus isolated with prolonged enrichment culture. Bioscience, Biotechnology and Biochemistry. 2004; 681: 28–35.
- Hell K., Edwards A., Zarsky J., Podmirseg S.M., Girdwood S., Pachebat J.A., Insam H., Sattler B. The dynamic bacterial communities of a melting High Arctic glacier snowpack. The ISME Journal. 2013; 7: 1814–1826. [PubMed Abstract] [PubMed CentralFull Text].
- Hemp J. The search for ancestrally anoxygenic or non-phototrophic Cyanobacteria. 2012. Accessed on the internet at www.mbl.edu/microbialdiversity/files/2012/08/Jim-Hemp.pdf on 28 April 2014.
- Hong S.H., Bunge J., Leslin C., Jeon S., Epstein S.S. Polymerase chain reaction primers miss half of rRNA microbial diversity. The ISME Journal. 2009; 3: 1365–1373. [PubMed Abstract].
- Isenbarger T.A., Carr C.E., Johnson S.S., Finney M., Church G.M., Gilbert W., Zuber M.T., Ruvkun G. The most conserved genome segments for life detection on Earth and other planets. Origins of Life and Evolution of Biospheres. 2008; 38: 517–533.
- Ivanova E., Doronina N., Trotsenko Y. Hansshlegelia plantiphila gen. nov. sp. nov., a new aerobic restricted facultative methylotrophic bacterium associated with plants. Systematic and Applied Microbiology. 2007; 30: 444–452. [PubMed Abstract].
- Junge K., Christner B., Staley J.T. Horikoshi K. Diversity of psychrophilic bacteria from sea ice and glacial ice communities. Extremophiles handbook. 2011; New York: Springer. 794–815.
- Kasalicky V., Jezbera J., Hahn M.W., Simek K. The diversity of the Limnohabitans genus, an important group of freshwater bacterioplankton, by characterization of 35 isolated strains. PLoS One. 2013; 83: 58209.
- Komárek J., Nedbalová L., Hauer T. Phylogenetic position and taxonomy of three heterocytous cyanobacteria dominating the littoral of deglaciated lakes, James Ross Island, Antarctica. Polar Biology. 2012; 35: 759–774.
- Knowlton C., Veerapaneni R., D'Elia T., Rogers S.O. Microbial analyses of ancient ice core sections from Greenland and Antarctica. Biology. 2013; 2: 206–232. [PubMed Abstract] [PubMed CentralFull Text].
- Larkin M.A., Blackshields N.P., Brown R.C., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez A.R., Thompson J.D., Gibson T.J., Higgins D.G. Clustal W and Clustal X version 2.0. Bioinformatics. 2007; 2321: 2947–2948.
- Leibeling S., Schmidt F., Jehmlich N., von Bergen M., Muller R.H., Harms H. Declining capacity of starving Delftia acidovorans MC1 to degrade phenoxypropionate herbicides correlates with oxidative modification of the initial enzyme. Environmental Science & Technology. 2010; 44: 3793–3799. [PubMed Abstract] [PubMed CentralFull Text].
- Li L., Zheng J.-W., Hang B.-J., Doronina N.V., Trotsenko Y.A., He J., Li S.-P. Methylopila jiangsuensis sp. nov., an aerobic, facultatively methylotrophic bacterium. International Journal of Systematic and Evolutionary Microbiology. 2011; 61: 1561–1566. [PubMed Abstract].
- Liu Z., DeSantis T.Z., Andersen G.L., Knight R. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Research. 2008; 36: 120.
- Loveland-Curtze J., Miteva V., Brenchley J. Herminiimonas glaciei sp. nov., a novel ultramicrobacterium from 3042 m deep Greenland glacial ice. International Journal of Systematic and Evolutionary Microbiology. 2008; 59: 1272–1277.
- Loveland-Curtze J., Miteva V., Brenchley J. Novel ultramicrobacterial isolates from a deep Greenland ice core represent a proposed new species, Chryseobacterium greenlandense sp. nov. Extremophiles. 2010; 14: 61–69. [PubMed Abstract].
- Miteva V. Margesin R. Bacteria in snow and glacier ice. Psychrophiles, from biodiversity to biotechnology. 2008; Berlin: Springer. 31–50.
- Miteva V., Burlingame C., Sowers T., Brenchley J. Comparative evaluation of the indigenous microbial diversity versus drilling fluid contaminants in the NEEM Greenland ice core. FEMS Microbiology Ecology. 2014; 89: 238–256. [PubMed Abstract].
- Miteva V., Sowers T., Brenchley J. Penetration of fluorescent microspheres into the NEEM North Eemian Greenland ice core to assess the probability of microbial contamination. Polar Biology. 2014; 37: 47–59.
- Miteva V., Teacher C., Sowers T., Brenchley J. Comparison of microbial diversity at different depths of the GISP2 Greenland ice core in relation to deposition climate. Environmental Microbiology. 2009; 11: 640–656. [PubMed Abstract].
- Miteva V.I., Brenchley J.B. Detection and isolation of ultrasmall microorganisms from a 120,000 years old Greenland glacier ice core. Applied and Environmental Microbiology. 2005; 71: 7806–7818. [PubMed Abstract] [PubMed CentralFull Text].
- Miteva V.I., Sheridan P.P., Brenchley J.B. Phylogenetic and physiological diversity of microorganisms isolated from a deep Greenland ice core. Applied and Environmental Microbiology. 2004; 70: 202–213. [PubMed Abstract] [PubMed CentralFull Text].
- Montagnat M., Azuma N., Dahl-Jensen D., Eichler J., Fujita S., Gillet-Chaulet F., Kipfstuhl S., Samyn D., Svensson A., Weikusat I. Fabric measurement along the NEEM ice core, Greenland, and comparison with GRIP and NGRIP ice cores. The Cryosphere Discussions. 2014; 8: 307–335.
- Nakai R., Shibuya E., Justel A., Rico E., Quesada A., Kobayashi F., Iwasaka Y., Shi G.-Y., Amano Y., Iwatsuki T., Naganuma T. Phylogeographic analysis of filterable bacteria with special reference to Rhizobiales strains that occur in cryospheric habitats. Antarctic Science. 2013; 25: 219–228.
- NEEM Community Members. Eemian interglacial reconstructed from a Greenland folded ice core. Nature. 2013; 493(7433): 489–494.
- Nubel U., Garcia-Pichel F., Muyzer G. PCR primers to amplify 16S rRNA genes from cyanobacteria. Applied and Environmental Microbiology. 1997; 63: 3327–3332. [PubMed Abstract] [PubMed CentralFull Text].
- Quesada A., Vincent W.F. Whitton B.A. Cyanobacteria in the cryosphere, snow, ice and extreme cold. Ecology of cyanobacteria. II. Their diversity in space and time. 2012; New York: Springer. 387–399.
- Pinard R., de Winter A., Sarkis G.J., Gerstein M.B., Tartaro K.R., Plant R.N., Egholm M., Rothberg J.M., Leamon J.H. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 2006; 7: 216–237. [PubMed Abstract] [PubMed CentralFull Text].
- Price P.B. A habitat for psychrophiles in deep Antarctic ice. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97: 1247–1251. [PubMed Abstract] [PubMed CentralFull Text].
- Price P.B. Microbial life in glacier ice and implications for a cold origin of life. FEMS Microbiology Ecology. 2007; 59: 217–231. [PubMed Abstract].
- Price P.B., Bay R.C. Marine bacteria in deep Arctic and Antarctic ice cores, a proxy for evolution in oceans over 300 million generations. Biogeosciences. 2012; 9: 3799–3815.
- Pruesse E., Quast C., Knittel K., Fuchs B.M., Ludwig W., Peplies J., Glöckner F.O. SILVA, a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research. 2007; 35: 7188–7196. [PubMed Abstract] [PubMed CentralFull Text].
- Qiagen. Qiagen supplementary protocol. Purification of DNA amplified with REPLI-g kits (RG21 Nov-12). 2012; Hilden, Germany: Qiagen.
- Rasmussen S.O., Abbott P., Blunier T., Bourne A., Brook E., Buchardt S.L., Buizert C., Chappellaz J., Clausen H.B., Cook E., Davies S., Guillevic M., Kipfstuhl S., Laepple T., Dahl-Jensen D., Seierstad I.K., Severinghaus J.P., Steffensen J.P., Stowasser C., Svensson A., Vallelonga P., Vinther B.M., Wilhelms F., Winstrup M. A first chronology for the NEEM ice core. Climate Past Discussions. 2013; 9: 2967–3013.
- Reysenbach A.-L., Pace N.R. Robb F.T. Reliable amplification of hyperthermophilic Archaeal 16S rRNA genes by the polymerase chain reaction. Archaea: a laboratory manual. 1995; Cold Spring Harbor: Cold Spring Harbor Laboratory Press. 101–105.
- Rogers S.O., Theraisnathan V., Ma L.J., Zhao Y., Zhang G., Shin S., Castelo J.D., Starmer W.T. Comparisons of protocols for decontamination of environmental ice samples for biological and molecular examinations. Applied and Environmental Microbiology. 2004; 70: 2540–2544. [PubMed Abstract] [PubMed CentralFull Text].
- Rohde R.A., Price P.B., Bay R.C., Bramall N.E. In-situ microbial metabolism as a cause of gas artifacts in ice. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105: 8667–8672. [PubMed Abstract] [PubMed CentralFull Text].
- Schloss P.D., Westcott S.L., Ryabin T., Hall J.R., Hartmann M., Hollister E.B., Lesniewski R.A., Oakley B.B., Parks D.H., Robinson C.J., Sahl J.W., Stres B., Thallinger G.G., Van Horn D.J., Weber C.F. Introducing mothur, open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology. 2009; 75: 7537–7541. [PubMed Abstract] [PubMed CentralFull Text].
- Sheridan P.P., Miteva V.I., Brenchley J.B. Phylogenetic analysis of anaerobic psychrophilic enrichment cultures obtained from a Greenland ice core. Applied and Environmental Microbiology. 2003; 69: 2153–2160. [PubMed Abstract] [PubMed CentralFull Text].
- Shtarkman Y.M., Kocer Z.A., Edgar R., Veerapaneni R.S., D'Elia T., Morris P.F., Rogers S.O. Subglacial Lake Vostok Antarctica. Accretion ice contains a diverse set of sequences from aquatic, marine and sediment-inhabiting Bacteria and Eukarya. PLoS One. 2013; 8: 67221.
- Sihvonen L.M., Lyra C., Fewer D.P., Rajaniemi-Wacklin P., Lehtimaki J.M., Wahlsten M., Sivonen K. Strains of the cyanobacterial genera Calothrix and Rivularia isolated from the Baltic Sea display cryptic diversity and are distantly related to Gloeotrichia and Tolypothrix. FEMS Microbiology Ecology. 2007; 61: 74–84. [PubMed Abstract].
- Simon C., Strittmatter A.W., Daniel R. Phylogenetic diversity and metabolic potential revealed in a glacier ice metagenome. Applied and Environmental Microbiology. 2009; 75: 7519–7526. [PubMed Abstract] [PubMed CentralFull Text].
- Skidmore M., Anderson S.P., Sharp M., Foght J., Lanoil B.D. Comparison of microbial community compositions of two subglacial environments reveals a possible role for microbes in chemical weathering processes. Applied and Environmental Microbiology. 2005; 71: 6986–6997. [PubMed Abstract] [PubMed CentralFull Text].
- Soo R.M., Skennerton C.T., Sekiguchi Y., Imelfort M., Paech S.J., Dennis P.G., Steen J.A., Parks D.H., Tyson G.W., Hugenholtz P. Photosynthesis is not a universal feature of the phylum Cyanobacteria. Peer Journal PrePrints. 2014; 2: 204v2.
- Starmer W., Fell J., Catranis C., Aberdeen V., Ma L.-J., Zhou S., Rogers S. Castello J.D., Rogers S.O. Yeasts in the genus Rhodotorula recovered from the Greenland ice sheet. Life in ancient ice. 2005; Princeton: Princeton University Press. 181–196.
- Stibal M., Hasan F., Wadham J.L., Sharp M.J., Anesio A.M. Prokaryotic diversity in sediments beneath two polar glaciers with contrasting organic carbon substrates. Extremophiles. 2012; 16: 255–265. [PubMed Abstract].
- Strunecky O., Elster J., Komarek J. Molecular clock evidence for survival of Antarctic cyanobacteria Oscillatoriales, Phormidium autumnale. from Paleozoic times. FEMS Microbiology Ecology. 2012; 82: 482–490. [PubMed Abstract].
- Vincent W.F. Seckbach J. Cold tolerance in cyanobacteria and life in the cryosphere. Algae and cyanobacteria in extreme environments. 2007; Berlin: Springer. 287–301.
- Vishnivetskaya T.A. Margesin R. Viable cyanobacteria and green algae from the permafrost darkness. Permafrost soils. 2009; Berlin: Springer. 73–84.
- Wang Y., Qianh P.Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One. 2009; 4: 7401.
- Willerslev E., Hansen A., Christensen B., Steffensen J.P., Arctander P. Diversity of Holocene life forms in fossil glacier ice. Proceedings of the National Academy of Sciences of the United States of America. 1999; 96: 8017–8021. [PubMed Abstract] [PubMed CentralFull Text].
- Wright E.S., Yilmaz L.S., Noguera D.R. DECIPHER, a search-based approach to chimera identification for 16S rRNA Sequences. Applied and Environmental Microbiology. 2012; 78: 717–725. [PubMed Abstract] [PubMed CentralFull Text].
- Xiang S., Yao T., An L., Xu B., Wang J. 16S rRNA sequences and difference in bacteria isolated Muztag Ata Glacier at increasing depths. Applied and Environmental Microbiology. 2005; 71: 4619–4627. [PubMed Abstract] [PubMed CentralFull Text].
- Xiang S.R., Shang T.C., Chen Y., Yao T.D. Deposition and post-deposition mechanisms as possible drivers of microbial population variability in glacier ice. FEMS Microbiology Ecology. 2009; 70: 165–176.
- Yao T., Xiang S., Zhang X., Wang N. Microorganisms in the Malan ice core and their relation to climatic and environmental changes. Global Biogeochemical Cycles. 2006; 20: 1004.
- Yde J.C., Finster K.W., Raiswell R., Steffensen J.P., Heinemeier J., Olsen J., Gunnlaugsson H.P., Nielsen O.B. Basal ice microbiology at the margin of the Greenland ice sheet. Annals of Glaciology. 2010; 51: 71–79.
- Yung P.T., Shafaat H.S., Connon S.A., Ponce A. Quantification of viable endospores from a Greenland ice core. FEMS Microbiology Ecology. 2007; 59: 300–306. [PubMed Abstract].
- Zeng Y., Yan M., Yu Y., Li H., He J., Sun K., Zhang F. Diversity of bacteria in surface ice of Austre Lovenbreen glacier, Svalbard. Archives of Microbiology. 2013; 195: 313–322. [PubMed Abstract].
- Zhang S., Yang G., Wang Y., Hou S. Abundance and community of snow bacteria from three glaciers in the Tibetan Plateau. Journal of Environmental Science China. 2010; 22: 1418–1424.