
Abstract
This study focuses on sulphurous and carbonaceous aerosol, the major constituents of particulate matter in the lowermost stratosphere (LMS), based on in situ measurements from 1999 to 2008. Aerosol particles in the size range of 0.08–2 µm were collected monthly during intercontinental flights with the CARIBIC passenger aircraft, presenting the first long-term study on carbonaceous aerosol in the LMS. Elemental concentrations were derived via subsequent laboratory-based ion beam analysis. The stoichiometry indicates that the sulphurous fraction is sulphate, while an O/C ratio of 0.2 indicates that the carbonaceous aerosol is organic. The concentration of the carbonaceous component corresponded on average to approximately 25% of that of the sulphurous, and could not be explained by forest fires or biomass burning, since the average mass ratio of Fe to K was 16 times higher than typical ratios in effluents from biomass burning. The data reveal increasing concentrations of particulate sulphur and carbon with a doubling of particulate sulphur from 1999 to 2008 in the northern hemisphere LMS. Periods of elevated concentrations of particulate sulphur in the LMS are linked to downward transport of aerosol from higher altitudes, using ozone as a tracer for stratospheric air. Tropical volcanic eruptions penetrating the tropical tropopause are identified as the likely cause of the particulate sulphur and carbon increase in the LMS, where entrainment of lower tropospheric air into volcanic jets and plumes could be the cause of the carbon increase.
1. Introduction
Aerosol particles play an important role in the radiation balance of the earth, with cooling and warming effects depending on particle composition. The combined aerosol effects are complex and estimated to provide a net negative radiative forcing (IPCC, Citation2007), which implies cooling of the earth surface. Because of the complexity and the scarcity of measurement data, the associated uncertainties are still large. Hence, it is important to learn more about particle chemical and elemental composition, not only for assessing the direct radiative effects, but also with regard to aerosol microphysical properties and, ultimately, cloud formation.
The major part of the particulate mass in the stratosphere is carried by submicrometer diameter particles (Deshler, Citation2008). Sedimentation velocities for such particles at these altitudes are low compared to the residence times of the air masses (Martinsson et al., Citation2005). Also for the upper troposphere (UT) and lowermost stratosphere (LMS), transport of air masses is of particular relevance for particles and precursor gases. We first briefly summarise some of the main aspects of the dynamics affecting this region of the atmosphere. The lower bound of the LMS is the tropopause and the upper one the 380 K isentropic surface (Hoskins, Citation1991; Holton et al., Citation1995). Large-scale transport in the stratosphere occurs via the Brewer–Dobson (BD) circulation (Brewer, Citation1949; Dobson, Citation1956), in which tropical tropospheric air is first transported upwards to the tropical stratosphere and subsequently polewards, to descend finally through the LMS into the extratropical troposphere. Air reaching the tropical stratosphere can undergo fast transport (month) to mid-latitudes in a transitional branch or be moved diabatically upwards in deeper branches of the BD circulation (years) (Gettelman et al., Citation2011). The downward mass flux across the 380 K isentrope varies with season. In the northern hemisphere, a maximum is found in January and a minimum in July (Appenzeller et al., Citation1996). The subsidence through the LMS takes months, resulting in a seasonal maximum of stratospheric influence at the tropopause in May (Tang et al., Citation2011). In addition, direct two-way mass exchange between the UT and LMS, through the extratropical tropopause, occurs along isentropic surfaces (Dessler et al., Citation1995; Sprenger and Wernli, Citation2003). The LMS can thus be regarded as a mixture of air from the troposphere and the stratosphere. Mixing of air in the LMS creates concentration gradients of trace gases (Lelieveld et al., Citation1997; Zahn and Brenninkmeijer, Citation2003) and particulate matter (Martinsson et al., Citation2005).
Studies on stratospheric aerosol composition started with Junge et al. (Citation1961). They found a water-soluble sulphurous component, subsequently identified as sulphuric acid and water (Rosen, Citation1971), formed via gas to particle conversion of mainly OCS and SO2 (Crutzen, Citation1976; Weisenstein et al., Citation1997). OCS photo-oxidises at high altitudes, within the deep BD branch, while SO2 oxidises at all altitudes in the stratosphere. A recent study by Brühl et al. (Citation2012) argues that OCS is the major source of non-volcanic particulate sulphur in the stratosphere, while Chin and Davis (Citation1995) advocated that the emission rate of OCS alone is insufficient to explain the large amounts of particulate sulphur in the stratosphere. It remains unclear to what degree SO2 from fossil fuel combustion penetrates the tropical troposphere because of difficulties to distinguish its contribution to the stratospheric aerosol from that of OCS and volcanism (Solomon et al., Citation2011).
Large volcanic eruptions represent the strongest source of variability in stratospheric aerosol as they can inject substantial amounts of particulate matter and SO2 into the stratosphere (Robock, Citation2000). The eruption of Mount Pinatubo in 1991 was observed in the tropics (Dutton and Bodhaine, Citation2001) and delayed by more than half a year in the stratospheric column at mid-latitudes (Trickl et al., Citation2013). It perturbed the stratosphere for several years, with an estimated global surface cooling in the order of 0.5°C (McCormick et al., Citation1995) the year after eruption. A large decline in the aerosol load of the stratosphere was then observed in the following years (Deshler et al., Citation2006), until reaching a period of near-background conditions in the late 1990s.
LIDAR measurements (Hofmann et al., Citation2009) indicated increasing amounts of particulate matter in the stratosphere at mid-latitudes in the northern hemisphere after the year 2000. Vernier et al. (Citation2011b) used satellite observations to address this increase and found that the trend increased after year 2005 in connection with volcanic eruptions, reaching the tropical stratosphere. The volcanic aerosol was eventually transported to mid-latitudes (Vernier et al., Citation2009), increasing the aerosol abundance in both the northern (Hofmann et al., Citation2009; Bazhenov et al., Citation2012) and southern hemisphere (Nagai et al., Citation2010). Solomon et al. (Citation2011) estimated the effect of the stratospheric volcanic aerosol for the time period 2000–2010, to have had a mean radiative forcing of −0.1 W m−2. In addition to the direct injection via explosive volcanic eruptions, Bourassa et al. (Citation2012) found evidence that the eruption of the tropical volcano Nabro in 2011 reached the UT and was lifted to the lower stratosphere, by the South Asian monsoon, where the effluents were transported to the extratropical stratosphere within 4 weeks of the eruption. Neely et al. (Citation2013) later found that the contribution to the stratospheric aerosol load in this period from anthropogenic emissions and that of transport within the South Asian monsoon to be small, further validating volcanic eruptions in the tropics as the main cause of perturbation.
In addition to sulphate, a carbonaceous component in stratospheric particles was observed more recently (Murphy et al., Citation1998) and was found to be a large fraction of the UT/LMS aerosol (Nguyen and Martinsson, Citation2007; Schmale et al., Citation2010). Using electron microscopy, PIXE (particle-induced X-ray emission) and PESA (particle elastic scattering analysis), Nguyen et al. (Citation2008) found a mixture of sulphurous and carbonaceous components in UT and LMS particles, with LMS particles containing a framework of carbonaceous material. Martinsson et al. (Citation2009) and Schmale et al. (Citation2010) found a large carbonaceous component in the aerosol from the Kasatochi eruption. In a recent study, based on model results and satellite measurements of extinction ratios from SAGE, Brühl et al. (Citation2012) suggest that organic particulate matter contributes a significant fraction to the light extinction in the LMS, as particulate sulphur concentrations from the model are insufficient to fully explain the observed light extinction in the stratosphere.
In situ measurements for elemental characterisation of particulate matter in the tropopause region are scarce. CARIBIC (Civil Aircraft for the Regular Investigation of the atmosphere Based on an Instrument Container, www.caribic-atmospheric.com) is the first project providing in situ elemental characterisation of particulate matter in the UT and LMS on a regular basis.
In this study, the LMS background or close-to-background aerosol is investigated using the CARIBIC platform. For this purpose, datasets from the period 1999–2002 and 2005–2008 are used, spanning almost a decade (data after 2008 are omitted from this study as the LMS became perturbed by a direct injection of volcanic aerosol in Aug 2008). These data for sulphurous and carbonaceous fractions of LMS particles provide, as far as we know, the longest time series on particulate sulphur and the first long-term quantitative measurements of the particulate carbon content in the tropopause region. Aerosol concentrations are presented relative to the position of the tropopause, as temporal trends, seasonal differences and inter-annual variations. In addition, the chemical composition of the carbonaceous component is presented as stoichiometric ratios of oxygen to carbon, and aerosol sources are discussed and identified.
2. Methods
The first generation CARIBIC measurements (CARIBIC phase #1) (Brenninkmeijer et al., Citation1999) using intercontinental flights were performed in 1997–2002 on board a Boeing 767–300 ER from LTU International Airways providing the first long-term in situ observations of the chemical composition of aerosol particles, combined with particle number and trace gas concentrations. The present CARIBIC system (CARIBIC phase #2) is the second generation consisting of a 1.6-ton container with an extended scientific payload, sampling at cruise altitudes of 9–12 km (Brenninkmeijer et al., Citation2007). This new CARIBIC container is installed in a Lufthansa Airbus 340–600 on a monthly basis for four consecutive flights, since May 2005 (no aerosol samples were collected in the period May 2002–April 2005). This aircraft is equipped with a sophisticated inlet system for gases and particles. In situ measurements of aerosol particles and of trace gases (e.g. O3, CO, NO/NOy, VOCs, gaseous and condensed water) are carried out. Collected air samples are analysed for greenhouse gases, hydro- and halo carbons (Brenninkmeijer et al., Citation2007; Schuck et al., Citation2009; Baker et al., Citation2010; Oram et al., Citation2012). Monthly measurement flights depart from Frankfurt for destinations in South East Asia, East Asia, Southern Africa and North and South America, thus covering a large geographical area, mainly in the northern hemisphere.
2.1. Aerosol sampling
The aerosol data presented in this work from CARIBIC phase #1 and #2 have been obtained using two different inlet systems. Based on experimental studies and modelling, the CARIBIC phase #1 inlet efficiency for particle sizes of 0.1–1 µm was estimated to be 90% (Hermann et al., Citation2001). The present CARIBIC inlet system is described by Brenninkmeijer et al. (Citation2007). Its sampling efficiency is estimated to be 60% at 5 µm particle diameter (Rauthe-Schöch et al., Citation2012), and based on modelling and experience with other aerosol inlets, its efficiency is estimated be at least 90% for particles of 0.01–1 µm diameter.
Collection of aerosol particles was undertaken by automated impactors (Nguyen et al., Citation2006), implemented in the CARIBIC instrument containers, collecting particles of 0.08–2 µm aerodynamic diameters. The CARIBIC phase #1 (phase #2) impactors have 14 (16) channels for sequential and integral samples. Each sequential sample is collected during typically 150 (100) minutes corresponding to flight distances of approximately 2200 (1500) km at cruise speed. The integral samples [1 (2) out of 14 (16) samples] are collected for the purpose of checking for sample contamination. The air sampling volume for a sequential sample is approximately 0.09 (0.25) m3 STP [standard temperature (273.15 K) and pressure (1013.25 hPa)]. Sampling is suspended during take-off and landing, whenever pressure exceeds 350 hPa, to prevent mixed samples of lower tropospheric (or even boundary layer) and UT/LMS aerosol particles. Particles accelerated in 0.5 mm diameter impactor orifices are deposited in four spots (phase #2), forming a square pattern with centre distances between spots of 1.3 mm, whereas the CARIBIC phase #1 sampler collected aerosol from a single orifice (Martinsson et al., Citation2001). The sampling substrate consists of a 0.2 µm thin AP1™ polyimide film, a material well suited for ion beam analysis (IBA) (Papaspiropoulos et al., Citation1999; Nguyen and Martinsson, Citation2007).
2.2. Analyses of aerosol particles
Analyses were undertaken with a 2.55 MeV proton beam at the Lund IBA accelerator. The IBA measurement techniques used are PIXE and PESA. PIXE (Johansson and Campbell, Citation1988) was used for elements with an atomic number larger than 13 and PESA for measurements of lighter elements (hydrogen, carbon, nitrogen and oxygen). Analyses were run in two steps, using the technique described by Nguyen and Martinsson (Citation2007). Typical elemental minimum detection limits (MDL) in units of ng m−3 STP are 1 (hydrogen), 15 (carbon), 7 (oxygen), 2 (sulphur) and 0.1 (iron). The accuracy of PIXE and PESA is estimated to be 10% (Nguyen and Martinsson, Citation2007). Concentrations below MDL were set to MDL/2. The detection frequencies, that is, the fraction of the samples with elemental concentrations above the MDL are: 100% (hydrogen), 83% (carbon), 100% (oxygen), 100% (sulphur) and 50% (iron).
Films with low deposited aerosol mass occasionally lead to significantly higher MDLs compared to the majority of the samples in the PESA analysis as a result of problems in identifying the aerosol deposit spot, which prevents analysis. This MDL problem is primarily associated with short sampling times. MDLs, in ng m−3, were derived from sampled mass density, in µg cm−2, on the exposed films and are thus highly dependent on sampling time. Samples with sampling times shorter than 60 minutes (caused for instance by aircraft landings) were therefore excluded to prevent these effects of high MDLs on the data set.
The analytical system used for detection of carbon was developed after 2002. Hence, no analysis of carbon was possible for samples collected with the first generation collection system. The carbon data in the present study is thus obtained from measurements with the second-generation collection system running since 2005.
2.3. Ozone measurements
We use the concurrent CARIBIC in situ ozone measurements to assist the interpretation of the variations in particulate carbon and sulphur concentrations in the UT/LMS. Two ozone analysers are currently installed in the CARIBIC container; an accurate, dual-beam UV-photometer serving as a calibrated standard instrument and a fast solid state chemiluminescence detector. The accuracy was estimated to be 2% for 10 Hz measurements (Brenninkmeijer et al., Citation2007; Zahn et al., Citation2012), while the uncertainty for the first generation CARIBIC system was 4% or 4 ppbv, whichever was larger (Zahn et al., Citation2002).
2.4. The tropopause definition
The dynamical tropopause, based on potential vorticity (PV) with a typical threshold value of 1.5–3.5 PVU (Potential Vorticity Units; 1 PVU = 10−6 K m2 kg−1 s−1) (Hoerling et al., Citation1991; Hoinka, Citation1997), is used for identifying samples collected in the LMS. PV values were derived from archived ECMWF (European Centre for Medium-range Weather Forecast) analyses with a resolution of 1×1 degree in the horizontal at 91 vertical hybrid sigma-pressure model levels and calculated by the Royal Netherlands Meteorological Institute, de Bilt, the Netherlands (KNMI). PV values were interpolated linearly in latitude, longitude, log pressure and time to the location of the aircraft and for each sample averaged over the duration of sampling. Samples at an average PV of over 2 PVU are accordingly regarded as stratospheric.
2.5. Exclusion of periods affected by direct injection into the LMS by extratropical, strong volcanic eruptions
Previous studies have shown a large impact on the LMS aerosol mass concentrations from volcanoes injecting directly into the LMS (Martinsson et al., Citation2009; Schmale et al., Citation2010; Andersson et al., Citation2013). The largest such eruptions in the period studied here (1999–2008) were Okmok (July 12, 2008) and Kasatochi (August 7–8, 2008), of which the latter strongly affected the LMS aerosol load (Martinsson et al., Citation2009). The first samples containing volcanic aerosol from Okmok and Kasatochi were collected by CARIBIC on August 15, 2008. Samples containing aerosol from these eruptions are excluded in our study. This refers to all LMS samples collected after August 15, 2008. After excluding samples affected by fresh volcanic clouds, this study on the background aerosol is based on 181 LMS samples, whereof 38 were from the period 1999–2002.
3. Results
3.1. Particulate sulphur and carbon concentrations in the LMS
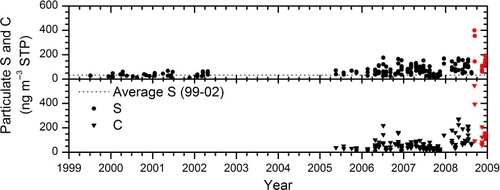
shows the temporal evolution of particulate sulphur and carbon concentrations in the LMS for the period considered. The strong effect on the aerosol concentrations via a direct injection of volcanic aerosol into the extratropical LMS by the Kasatochi eruptions in August 2008, as reported by Martinsson et al. (Citation2009) is evident, while the rest of the period represents an episode of low volcanic activity at mid-latitudes. Comparing the early sulphur data (1999–2002) with later data (2005–2008), a clear increase in concentration and variation is observed. Moreover, the concentrations increase substantially in the period 2005–2008, for both sulphur and carbon, with on average higher concentrations in 2008 than in any previous year investigated.
Fig. 1 Temporal variations of concentrations of particulate sulphur and carbon (ng m−3 STP) in the northern hemisphere LMS. Red dots illustrate observations after the eruption of Kasatochi. The dashed line shows the geometric average particulate sulphur concentrations during 1999–2002.
3.2. Vertical gradients and seasonal variations in the LMS
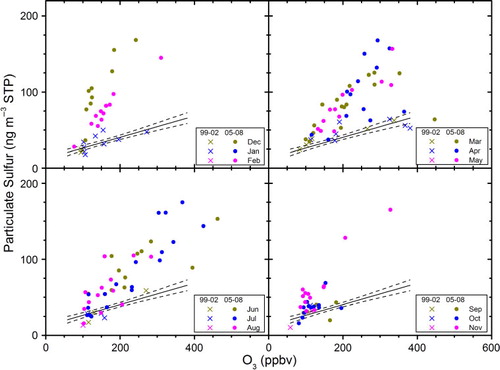
Ozone is formed deep in the stratosphere and is transported down to the LMS within the large-scale DB circulation. In the LMS, ozone has a chemical lifetime of >1 yr (Solomon et al., Citation1997; Smith et al., Citation2001); that is, it is basically an inert transport tracer and is solely diluted with inflowing ozone-poor tropospheric air, locally or via the lower BD branch. Thus, in the LMS ozone constitutes a reliable tracer for the degree of mixing with tropospheric air and an accurate measure for the distance above the tropopause (Sprung and Zahn, Citation2010). In , we use LMS ozone concentrations, thus expressing distance to the tropopause, for the four different seasons. Increase in aerosol concentrations is investigated via a scatter plot of particulate sulphur vs. ozone to illustrate the vertical gradient of sulphur from the tropopause into the LMS. A linear fit is applied to the 1999–2002 data for comparison to the 2005–2008 data. This fit is based on data for all four seasons to obtain a significant number of results. shows the difference between the two periods much more clearly. For all seasons, over 90% of the observed concentrations in the period 05–08 is found to be higher than during 99–02, whereas measurements at lowest ozone concentrations, that is, close to the tropopause, show similar concentrations in the two periods. The difference between the periods is less pronounced during fall. In the period 05–08, the observed gradient is strongest during winter and decreases in strength during spring and summer, to reach significantly lower levels in fall.
Fig. 2 Concentration of particulate sulphur vs. ozone for stratospheric samples (potential vorticity > 2 PVU) collected in the northern hemisphere. Upper left: winter (December, January, February). Upper right: spring (March, April, May). Lower left: summer (June, July, August). Lower right: fall (September, October, November). A regression model (full line), with 95% confidence interval (dashed lines) based on the 1999–2002 data is used to facilitate the comparison to the 2005–2008 data.
3.3. Inter-annual variations
To further explore seasonality of particulate sulphur in the LMS, the distance from the tropopause needs to be considered. We consider this using PV, a meteorological parameter with a strong gradient from the tropopause into the LMS. Hence, the sulphur to PV ratio is used in b and is subsequently compared to the corresponding ozone and particulate carbon ratios. The O3/PV ratio in the LMS is primarily determined by the seasonally varying subsidence of stratospheric air in the BD circulation and thus maximises in mid-latitudes in late spring (Tang et al., Citation2011), at the seasonal maximum influence from the stratosphere. We observe no difference in the average O3/PV for the periods 99–02 and 05–08, indicating that no major changes occurred in the transport from the stratosphere during this time. A sine curve fitting is applied with the relative amplitude of 0.31, as shown in a, which holds throughout the period 1999–2008, corroborating the absence of major changes in transport.
Fig. 3 Temporal trends of ratios to potential vorticity (PV) for a) ozone (ppbv PVU−1), b) particulate sulphur (ng m−3 STP PVU−1) and c) particulate carbon (ng m−3 STP PVU−1). Sine functions, based on ozone to PV ratios in a) are used in b) and c) to guide the eye.
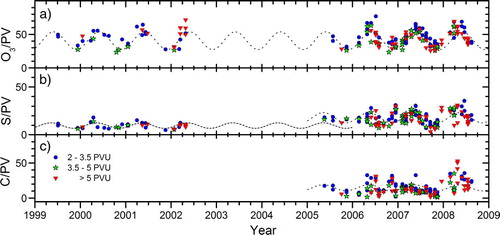
The contribution by downward transport of sulphurous aerosol formed in the stratosphere is evaluated in b, using S/PV. To account for the increase in sulphur concentrations from 99–02 to 05–08, two sine functions are used with the same relative amplitude and phase as for the O3/PV model. In the period 05–08, the S/PV shows considerable agreement with the seasonal variation of O3/PV with two large deviations (end of 2006 and 2007), and a tendency of higher values during 2008 compared to the two preceding years. Whereas the contribution from the deep branch is expected to follow the phase of the model, transport of SO2 and particles in the shallow BD branch can cause deviation from this seasonal pattern. The data show that this may have been the case. Interestingly, for carbon the picture seems more complicated, as the C/PV ratio (c) lacks any clear resemblance to that of O3/PV (a). Some larger deviations are found at the end of 2006 and 2007 and during 2008. These deviations will be further investigated in the Discussion section.
3.4. Composition of carbonaceous aerosol
Besides the very presence of carbonaceous aerosol in conjunction with sulphur and its variability, any information on its actual chemical moiety is valuable. Optical measurements of black carbon in the LMS (Schwarz et al., Citation2010) showed concentrations in the range of 0.1–4 ng m−3 STP. We are however dealing with concentrations that are up to two orders of magnitudes higher (), indicating that soot is only a minor constituent of the carbonaceous fraction in the LMS. The major part of the carbonaceous fraction is therefore likely to be organic.
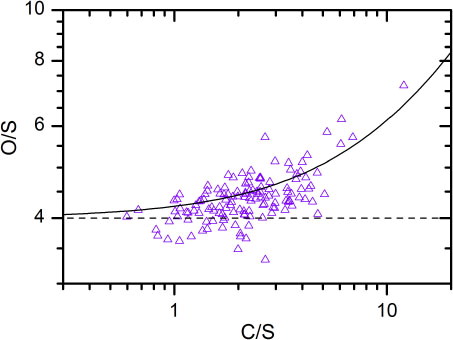
By means of stoichiometric calculations we can use the PIXE and PESA elemental analyses to infer main properties of the chemical compounds in the samples. Previous CARIBIC measurements have shown that LMS particles have two major components, namely a sulphurous and a carbonaceous one (Nguyen and Martinsson, Citation2007). In addition several minor constituents can be found, with mineral particles from the earth's crust being the most frequently observed. illustrates the stoichiometric ratios of oxygen to sulphur against carbon to sulphur, for all LMS samples with concentrations above the MDLs of these elements (C, O and S). Oxygen is present in the sulphurous fraction and also in the crustal fraction. A crustal component was identified in 19 samples based on elemental concentration ratios between potassium, calcium, titanium and iron. The crustal contribution to the oxygen content was subtracted from the total stoichiometric amounts of oxygen shown in . The correction was based on the average mineral composition of the earth crust estimated by Rudnick and Fountain (Citation1995). The contribution from the sulphurous fraction can be identified at an oxygen to sulphur ratio of 4 (O/S = 4), based on the assumption that all sulphur is present in the form of sulphate. indeed supports this, as most of the O/S ratios scatter at values exceeding 4. A positive trend from the line of O/S = 4 with increasing C/S ratio is present, which reflects the fraction of oxygen in the carbonaceous component. A relationship for the O/S ratios in relation to C/S ratios can be derived using a linear regression. Thus, the oxygen content in the carbonaceous fraction is obtained at an average O/C ratio of 0.2. Combined with the low concentrations of black carbon (Schwarz et al., Citation2010), this corroborates that the main part of carbonaceous LMS aerosol is organic.
Fig. 4 Oxygen to sulphur vs. carbon to sulphur molar ratios. The dashed line represents O/S = 4, and the full line shows the best fit, i.e. O/S = 4+0.2 C/S.
4. Discussion
An upward trend of 5–6% per year in the background aerosol at 20–25 km altitude for the period 2000–2009 was observed using LIDAR measurements. Increasing coal combustion associated with sulphur emissions, primarily in China, was hypothesised as an explanation (Hofmann et al., Citation2009). However, Vernier et al. (Citation2011b) used satellite observations to study the trend of increasing amounts of particulate matter in the stratosphere and found that several moderate volcanic eruptions with explosions reaching the tropical stratosphere had affected the aerosol load. In addition to volcanism, Vernier et al. (Citation2011a) identified the South Asian summer monsoon as a likely transport path for aerosol and precursor gases to the tropical tropopause layer (TTL). The aerosol load in the TTL in that study was found to be fairly constant in the period 2006–2008, with a minor increase in 2008, discussed to be an effect of volcanic aerosol.
The particulate sulphur concentration increases from the tropopause into the LMS and is significantly higher at the 380 K isentrope than at the tropopause or the UT (Martinsson et al., Citation2005). The same pattern but magnified appears in the period 2005–2008 (). The increase in aerosol concentration in the LMS in the period 2000–2009 thus indicates transport from the stratosphere, rather than from local mixing of air from the UT.
The difference in sulphur concentrations between the periods 99–02 and 05–08 is smallest in fall (), when ozone and particulate sulphur concentrations minimises, indicating the lowest contribution from the deep BD branch. Air mass budget analyses based on aircraft data demonstrated a strongly varying seasonal partitioning of stratospheric and tropospheric air in the LMS with a minimum influence from the deep BD branch in fall (Hoor et al., Citation2005). In summer and fall, the LMS is flushed with ozone-poor air from the TTL (Bönisch et al., Citation2009), additionally reducing the impact from the deep BD branch. This further emphasises our finding that the particulate sulphur is of stratospheric origin and varies in phase with the downward flux within the BD circulation. Bönisch et al. (Citation2009) found the LMS above the extratropical transition layer (ExTL) to be more connected to the stratosphere than to the extratropical UT. Hence, the composition of the LMS air is essentially dependent on transport that occurs above the 380 K isentrope, that is, mixing of the deep and shallow BD branches. The shallow BD branch transports air from the tropical stratosphere to northern mid-latitudes within about 1–2 months (Bourassa et al., Citation2012; Flury et al., Citation2013), whereas transport in the deep branch takes a year or more (Holton et al., Citation1995).
Air transported polewards from the tropical stratosphere, experiences different exposure to photo-chemistry depending on altitude and speed along the transport paths. Whereas sulphuric acid is produced from SO2 at all altitudes, ozone production and particulate sulphur production from OCS occur predominantly within the deep BD branch. Trace gas concentration distributions bare evidence of meridional mixing of air at high and low altitudes in the stratosphere (Gettelman et al., Citation1997). The ozone concentration at the 380 K isentrope varies by a factor of 2 (Fortuin and Kelder, Citation1998; Martinsson et al., Citation2005), with its maximum in March and minimum in September. That variation compares well to the corresponding O3/PV variation in a, with a phase shift of 2 months (relative to the 380 K isentrope) and a peak-to-peak ratio of 1.9, indicating a strong coupling between the LMS and the stratosphere. Hence, the low ozone and sulphur concentrations in fall are results of less ozone and particulate sulphur in the air that is down-welling to the LMS as an effect of less transport via the deep BD branch, and an increasing importance of the shallow branch, during summer (Lin and Fu, Citation2013).
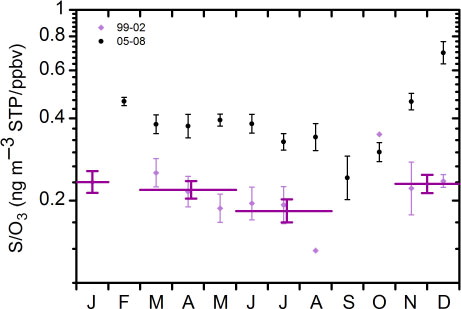
Regarding the sulphurous aerosol component we note that the approach used in b, based on the O3/PV variability, does capture the overall pattern of the S/PV values. Nonetheless, several deviations from the general pattern indicate that not only OCS contributed to the observed sulphur concentrations. Regarding the C/PV values in c, larger deviations are seen and only a weak seasonal cycle exists. This suggests that oxidation of precursor gases in the deep BD branch in relative terms is less important for formation of particulate carbon, compared to particulate sulphur. It also indicates that other processes cause the variability in the carbon concentration. The O3 concentration at the 380 K isentrope shows its seasonal minimum in September and maximum in March (Fortuin and Kelder, Citation1998; Martinsson et al., Citation2005). This seasonal variation is expected also in the concentration of sulphate aerosol formed from OCS, as it follows the deep BD branch, while transport of sulphate aerosol to the 380 K isentrope via the shallow BD branch can cause high S/O3 ratios also during fall. Low ozone concentration in the LMS in winter concurrent with elevated particulate sulphur concentration is thus an indication of formation of particulate matter in SO2-rich air that was transported in the stratosphere to mid-latitudes by the shallow BD branch. Hence to study the importance of particulate matter formation and transport in the shallow branch we use the seasonal variations of S/O3 (). For the period 05–08 we have enough data to produce monthly averages. The S/O3 is once again shown to be significantly higher in the period 05–08 than in the 99–02 ratios with a strong seasonal cycle. In the period 99–02, the S/O3 remained fairly constant with a small increase at the turn of the year. For 05–08, the S/O3 shows a clear saw-tooth pattern with a rapid rise from September to December. This increase in the S/O3 indicates production and transport of sulphurous aerosol within the shallow BD branch. Revisiting the strong particulate-sulphur-to-ozone gradient in during winter, we conclude that this is the consequence of down-welling of air that was transported in the shallow branch, from the overlying stratosphere, while the gradient in spring and in summer corresponds to mixing also via the deep branch.
Fig. 5 Ratios of particulate sulphur to ozone mixing ratio (Unit: ng m−3 STP ppbv−1) for the periods 1999–2002 and 2005–2008, expressed as monthly geometric averages (black and light purple). Purple lines denote averages in the period 1999–2002 for the months; January, March–May, June–August and November–December. Error bars represents standard errors.
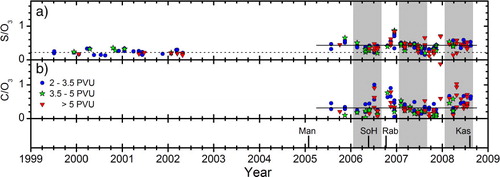
Using , we can inspect deviations of particulate sulphur and carbon from seasonality in down-welling of O3 from the stratosphere over a longer time span. During 05–08, there are periods (a) when S/O3 values are similar to those of the period 99–02, in particular during July–October 2007. There are also large positive deviations from the geometric average of May 2005–August 2008 in the end of 2006 and in December 2007, while 2008 shows higher S/O3 ratios compared to 2006 and 2007. A similar pattern is evident for the C/O3 ratios in b. Deviations from the geometric average of the C/O3 are essentially found in the end of 2006 and in December 2007, and during most of 2008. In fall 05–08 (), two of the observations have ozone and sulphur concentrations well above the average of the season. These were sampled on November 14–15, close to the end of the defined season and look more connected to the observations in the winter season.
Fig. 6 Ratios of particulate sulphur and carbon concentration to ozone mixing ratio (Unit: ng m−3 STP ppbv−1), for a) particulate sulphur and b) particulate carbon for stratospheric samples. Full and dashed horizontal lines represent geometric averages for the periods 2005–2008 and 1999–2002, respectively. The February–August period is indicated by a grey background. The four discussed volcanic eruptions (Manam, Soufriere Hills, Rabaul and Kasatochi) are indicated by vertical lines.
While sulphuric acid is generally considered the main component in stratospheric and LMS aerosol much less is known of the other large component; the carbonaceous aerosol. The following two sections will address the cause of high concentrations of particulate carbon, and of the temporal trends of particulate sulphur and carbon in the LMS aerosol.
4.1. Forest fires and biomass burning
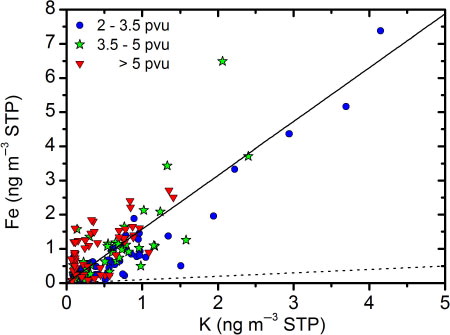
Combustion of biomass emits large amounts of soot and organic trace gases that lead to secondary aerosol formation (Andreae and Merlet, Citation2001). Episodes of increased scattering ratios coupled to pyroconvection from forest fires have been observed at altitudes above the tropopause, remaining for a month or more. A number of authors (Jost et al., Citation2004; Fromm et al., Citation2005, Citation2010; Damoah et al., Citation2006) discuss the possibility and magnitude of forest fire injection of smoke particles into the LMS via pyroconvective transport. Based on optical measurements Fromm et al. (Citation2008) estimate that a massive fire storm in Canada in the end of May 2001 injected a mass of smoke, corresponding to more than 5% of the background aerosol in the northern hemisphere lower stratosphere, that persisted until the end of summer. Such events are sporadic with uncertain occurrence frequency. Guan et al. (Citation2010) estimate that approximately 140 plumes reached altitudes above 5 km, in the northern hemisphere, in the period 1979–2009, that is, on average less than 5 per year. Potassium (K) is one of the elements, besides carbon that is generally found in aerosol from biomass burning (Andreae and Merlet, Citation2001). Importantly, there is no correlation between C and K in the stratospheric CARIBIC samples (R 2=0.004), indicating that biomass burning had a negligible contribution to the LMS aerosol during the period considered here. The other large potential source of K in the LMS is crustal material. Besides K crustal particles also contain silicon, iron, titanium and calcium. Out of these elements, iron (Fe) is the element with the highest detection frequency in our samples. In , a scatter plot of Fe vs. K in stratospheric samples is used to study the possible impact of biomass burning and crust particles on the LMS aerosol. Aerosol from biomass burning would result in ratios <0.1 ng Fe/ng K (Andreae et al., Citation1998), while crustal material would be found at ratios >1 (Rudnick and Fountain, Citation1995). Only a few observations lie close to the 0.1 line (dashed), indicating that biomass burning has a minor impact on the LMS aerosol. A linear regression model shows a strong correlation between Fe and K, with an average ratio of 1.58 ng Fe/ng K. These observations give evidence that biomass burning is insufficient to explain the high carbon concentrations observed.
Fig. 7 Mass concentrations of Fe vs. K (ng m−3 STP) for stratospheric samples. Regression model shown by full line illustrating a ratio of 1.58 ng Fe/ng K. Dotted line shows Fe/K mass ratio of 0.1, a typical ratio for effluents from biomass burning.
4.2. Impact of mid-latitude volcanism
Volcanic plumes bring high concentrations of particulate matter, SO2 and other gases to the stratosphere. Gas to particle conversion of SO2 in the stratosphere is fast compared to the residence times of the air mass and the entrained submicron particles. Volcanic injections of aerosol to the LMS from mid-latitude eruptions are expected to perturb the LMS for a few months or less depending on the altitude of the volcanic injection. Kasatochi's eruption in August 2008 increased the sulphurous aerosol concentrations by a factor of two in the following months. The SO2 emissions from Okmok were estimated to be 5% (0.1 Tg) of that of Kasatochi's (2 Tg) (Yang et al., Citation2010; Thomas et al., Citation2011). In addition some minor eruptions on mid-latitudes were reported to have reached the tropopause region (Massie, Citation2014) in the period January 2005–August 2008. Satellite images (from OMI, GOME-2 and AIRS) tell that the SO2 emissions from any of these eruptions were at least a factor of five lower than that from the eruption of Okmok, implying a maximum perturbation of the LMS aerosol of less than 2% in the months following one of these eruptions. Hence, it is most unlikely that the increases in aerosol concentrations from the periods 99–02 to 05–08 () are caused by mid-latitude eruptions.
4.3. Impact of tropical volcanism
Mixing in the extratropical stratosphere occurs as air from high altitudes descends and mixes with air at lower altitudes. As a result, aerosol and trace gases injected into the tropical stratosphere can reach mid-latitudes in a time scale of months by stratospheric low altitude meridional mixing, via the shallow branch of the BD circulation, and in 1–2 yr via the deep BD branch. The time of the year of a tropical volcano's eruption is expected to affect the period of time for the effects to be observable in the LMS. Main effects from volcanic aerosol transported via the high-altitude branch may appear 2 yr after the eruption. Three tropical volcanoes had eruptions that reached the stratosphere in the time period 2003–2008, namely Manam, Soufriere Hills and Rabaul (). Based on the transport characteristics of the stratosphere the Manam eruption in January 2005 could affect the mid-latitude stratosphere primarily in 2005–2006, whereas the effects of the two eruptions in 2006 () might be observable during late 2006, with the major effects in the downward transport in 2008. These patterns are essentially observed in measurements from the NASA satellite CALIPSO (Vernier et al., Citation2009, Citation2011b).
Table 1. Explosive volcanic eruptions most relevant for this study in the period 2005–August 2008
Volcanic aerosol is commonly considered to be composed of sulphate from the conversion of SO2, combined with a minor fraction of ash constituents (e.g. potassium, calcium, titanium and iron) even though large amounts of carbonaceous aerosol have been observed in volcanic clouds on several occasions. Martinsson et al. (Citation2009) found particulate carbon-to-sulphur-ratios (C/S, in terms of mass) of 2.6 in fresh plumes in the LMS, decreasing to 1 when most of the SO2, from the eruption of Kasatochi had been converted, corresponding to an organic content of 25–50% (assuming all particulate carbon to have been organic). Measurements with mass spectrometric methods indicated organic fractions of 20–40% (Carn et al., Citation2011) and 20% (Schmale et al., Citation2010) in fresh and aged volcanic clouds, respectively. Hence, these studies indicate that particulate carbon is a major constituent of the aerosol in volcanic clouds, with a substantial effect on the stratosphere given the observations of a volcanically perturbed stratosphere in 2006–2008, as reported by Vernier et al. (Citation2011b).
Despite a number of observations of elevated concentrations of particulate carbon in the LMS connected to volcanic clouds, little is known about the origin of this particulate carbon. In a recent study on volcanic aerosol, Andersson et al. (Citation2013) argue, based on the global distribution of organic aerosol in the lower troposphere, that volcanic jets and plumes could bring large amounts of particulate carbon and organic trace gases to the tropopause region via entrainment of air from low altitudes. Entrainment of low altitude air to the tropical stratosphere, via jets from the three tropical volcanoes could be the cause of the observations of high particulate carbon concentrations in the present study, whereas particulate sulphur is expected from transformation of SO2 emitted from the volcanoes. The term ‘volcanic aerosol’ will therefore be used for attributing, not only sulphurous, but also carbonaceous aerosol in the following sections.
It is not possible to attribute every deviation in to a specific event of volcanic activity, but the main patterns will be discussed. The elevated ratios of S/O3 and C/O3 during the end of 2006 and December 2007, suggest downward transport of volcanic aerosol from the low altitude branch of the BD circulation, carrying aerosol from the Soufriere Hills eruption. Towards the end of 2006, in the build-up to the maximum downward transport to the LMS in December, Rabaul could also have contributed via the shallow branch of the BD. When downward transport increased during the end of 2007, the volcanic aerosol was located at higher altitudes (Vernier et al., Citation2009) compared to the end of 2006, and it therefore took longer for the volcanic aerosol to be transported to the LMS, resulting in deviations occurring later in the annual cycle than in 2006. The February–August periods for 2008 S/O3 ratios are clearly elevated, and even more so for C/O3 ratios compared to 2006 and 2007, in connection with the down-welling of aerosol from the 2006 eruptions of Soufriere Hills and Rabaul.
4.4. Inter-annual trends
The trend of increasing concentrations of particulate matter above the LMS, as reported by Hofmann et al. (Citation2009) and Vernier et al. (Citation2011b), should be expected to be visible in the particulate sulphur and/or carbon concentrations also in the LMS. The strong influence on the LMS aerosol from the Kasatochi eruption effectively eliminated the possibility to study background stratospheric aerosol after August 2008 (Martinsson et al., Citation2009), shown in . Our data have 3 yr of good coverage for the months February to the beginning of August for both sulphur and carbon, in the period 05–08, and for sulphur 2 yr in the period 99–02. This part of the year, which is also the part of the year when the LMS is strongly influenced by air from higher stratospheric altitudes, is used to study the evolution with time of particulate sulphur and carbon concentrations in the LMS. The time dependence of the ‘background aerosol’ concentration is investigated using a and b, where the averaged concentration ratios of particulate sulphur and carbon to ozone are plotted, for the February–August periods. The years 1999, 2002 and 2005 are excluded as they lack observations for several months, while the data in the February to August periods of the years 2000 and 2001 are combined in one average, because of the small number of data for these years. The observed ratios of S/O3 and C/O3 for the February–August periods are increasing from 2006 to 2008. This coincides with the influence from volcanic eruptions on the LMS as discussed above, and also found by Vernier et al. (Citation2011b) in studies of the stratosphere.
Fig. 8 Geometric average of ratios (a) particulate sulphur (b) carbon and (c) carbon + 3 sulphur concentration to ozone mixing ratio (ng m−3 STP ppbv−1) for the February–August periods of 2000/2001, 2006, 2007 and 2008.
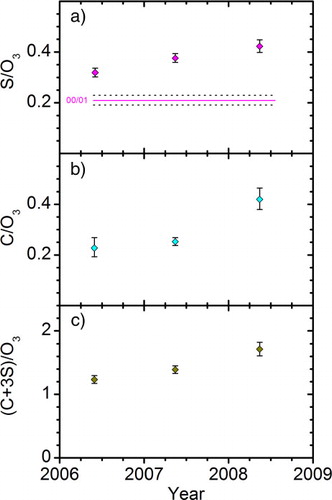
The optical measurements on aerosol in the stratosphere used by Hofmann et al. (Citation2009) and Vernier et al. (Citation2011b) do not reveal the chemical composition of particulate matter, but rather capture the average optical effects of the particulate matter in the studied region. The trend revealed from the optical measurements might therefore be more connected to the sum of the concentrations of the major constituents rather than with individual constituents. Hence, the trend for the sum of the two major components of the LMS aerosol, the carbonaceous and sulphurous fractions, is investigated in c. The sulphurous fraction is assumed to be composed of sulphate, with a mass three times that of sulphur, while the mass of the carbonaceous fraction is represented by the mass of carbon. The ratio of the sum of carbonaceous and sulphurous mass concentrations to ozone [(C + 3S)/O3] is shown in c for the February–August periods. A significant increase from 2006 to 2008 can be identified. This increase was 30%, with a stronger relative increase for carbon (70%; b) than for sulphur (23%; a).
Overall, a doubling is shown in the S/O3 from the 2000/2001 to the year 2008 (a). Hofmann et al. (Citation2009) found an increase at 20–25 km altitude of 6.3% year−1 in the mid-latitudes (Boulder, 40°N) over the years 2000–2009, corresponding to a total increase of 73%. The differences to our data can be caused by difference in altitude, time period studied or measurement method. The particulate mass concentrations presented here are particle volume oriented, whereas optical methods are basically particle surface oriented with a complicated response to particle size, shape and chemical composition. Another reason is that the data presented here pertain to the time of the year when the influence from the deep BD branch is strong.
The steady increase in S/O3 in the LMS (a) combined with the sudden rise in C/O3 from 2007 to 2008 (b) indicates different compositions for stratospheric aerosol in 2007 than in 2008. A comparison of particulate carbon-to-sulphur-ratios (C/S) for these years, shows a significant difference in the geometric average of 0.66–0.99, with geometric standard deviations of 1.06 and 1.08, respectively (i.e. a 1-sigma-range of 0.62–0.70 and 0.92–1.08). Could this increase in the C/S be an effect of different aerosol compositions from the different volcanic clouds?
The time span for the tropical volcanoes impact on the stratosphere is expected to depend on their penetration depth into the stratosphere. Volcanic clouds reaching high altitudes are expected to affect aerosol via the deep BD branch, while aerosol injected to lower altitudes would follow the shallow branch. Based on CALIPSO observations Vernier et al. (Citation2009) illustrated that aerosol from the Soufriere Hills eruption reached higher altitude (>20 km) compared to that from the Rabaul eruption (≤18 km), by the end of 2006. Hence, a larger fraction of the Soufriere Hills aerosol is expected to be transported via the deep branch, whilst the Rabaul aerosol would to a larger extent have been transported to mid-latitudes via the shallow BD branch. As the shallow branch transports air from the tropics to mid-latitudes within months, the deep branch affects the mid-latitude stratosphere after a year or more. The composition of the mid-latitude stratospheric aerosol, down-welling to the LMS in 2007, would thus be more connected to aerosol from the eruption of Rabaul, whereas the main effects of the Soufriere Hills aerosol would be visible during 2008. Studies based on satellite measurements (Stramska, Citation2009) and models (Spracklen et al., Citation2008) indicate higher regional organic aerosol concentrations in lower tropospheric air at the time of the Soufriere Hills eruption than for that of Rabaul. The eruption of Rabaul thus had a lower potential to entrain particulate carbon in the volcanic jet and plume. As these two eruptions emitted similar amounts of SO2 () a higher C/S concentration ratio could be expected for the Soufriere Hills eruption in agreement with our observations.
5. Conclusions
The present work is based on measurements of LMS aerosol particle composition sampled by CARIBIC (Civil Aircraft for the Regular Investigation of the atmosphere Based on an Instrument Container, www.caribic-atmospheric.com) in the periods 1999–2002 and 2005–2008. IBA was used to derive detailed elemental concentrations, finding a sulphurous and a carbonaceous component to be dominant. The concentration of the carbonaceous component corresponded on average to approximately 25% of that of the sulphurous, in terms of mass, and could not be explained by forest fires or other types of biomass burning. Stoichiometric O/C ratios of 0.2 and the fact that the measured particulate carbon concentrations greatly exceed literature data on black carbon concentrations indicate that the carbonaceous aerosol is organic in nature.
Particulate sulphur showed a distinct concentration gradient from the tropopause into the mid-latitude LMS. The gradient was strongest during winter and weakest during fall, indicating transport from the stratosphere as the cause of high concentration of particulate sulphur in the LMS. The gradient was also significantly stronger in the period 2005–2008 than in the earlier (1999–2002) period. Variations in the particulate sulphur and carbon concentrations were compared to that of O3 to track the effect of transport from high altitudes of the stratosphere via the deep Brewer-Dobson branch. That comparison indicated an increased importance for transport of sulphurous aerosol via the shallow BD branch in the period 2005–2008 compared to that of the earlier period, suggesting oxidation of SO2 as a likely cause for the strengthening of the concentration gradient.
The elevation and the variations in aerosol concentrations in the period 2005–2008 are associated with varying concentrations of particulate matter above the LMS, depending on mainly three volcanic eruptions in the tropics, with plumes reaching the tropical stratosphere. Subsequent transport by the BD circulation then carried the volcanic aerosol to the LMS, resulting in the elevated concentrations of particulate matter observed by CARIBIC in the following years. Entrainment of air from low altitudes, with high concentrations of particulate carbon and organic trace gases, in volcanic jets and plumes is proposed as the cause of high concentrations of particulate carbon in the stratosphere, while sulphur is expected from the volcanic effluents. The eruption in 2006 by Soufriere Hills reached deeper into the stratosphere than Rabaul later the same year, causing a longer duration of the influence from Soufriere Hills. The higher C/S ratios during 2008 could thus be connected with higher concentrations of carbonaceous aerosol at low altitude around the Soufriere Hills volcano.
Comparison of aerosol concentrations in the LMS during the February–August periods of 2006, 2007 and 2008 reveals an increase of 30% from 2006 to 2008. The rate of increase over that period was stronger for carbon (70%) than for the other major constituent sulphur (23%), hence indicating that volcanism had a stronger relative impact on the concentration of carbonaceous than on sulphurous aerosol in the stratosphere, although the increase in the absolute amount of sulphurous aerosol in the form of sulphate was larger than that of the carbonaceous aerosol.
Mass concentrations of sulphurous and carbonaceous aerosol, the two main components in the northern hemisphere mid-latitude LMS aerosol, increased in the period studied here. The former approximately doubled from the period 2000–2001 to the year 2008. This could to a large degree be attributed to intermediate volcanic eruptions in the tropics. Previous studies found an increasing stratospheric aerosol burden above 15 km altitude in the period 2000–2009, which potentially cooled the climate. This study extends these findings to the LMS.
6. Acknowledgements
We especially acknowledge C. Koeppel, D. S. Scharffe, S. Weber and all other members of the CARIBIC project. Lufthansa and Lufthansa Technik are gratefully acknowledged for enabling this scientific experiment. Financial support from the Swedish Research Council and the Swedish Research Council for Environments, Agricultural Sciences and Spatial Planning under grants 621-2007-4639 and 214-2009-613 is gratefully acknowledged.
Related Research Data
References
- Andersson S. M. , Martinsson B. G. , Friberg J. , Brenninkmeijer C. A. M. , Rauthe-Schöch A , co-authors . Composition and evolution of volcanic aerosol from eruptions of Kasatochi, Sarychev and Eyjafjallajökull in 2008–2010 based on CARIBIC observations. Atmos. Chem. Phys. 2013; 13: 1781–1796.
- Andreae M. O. , Andreae T. W. , Annegarn H. , Beer J. , Cachier H. , co-authors . Airborne studies of emissions from savanna fires in southern Africa. 2. Aerosol chemical composition. J. Geophys. Res. 1998; 103: 32119–32128.
- Andreae M. O. , Merlet P . Emission of trace gases and aerosols from biomass burning. Global Biogeocheml. Cycles. 2001; 15: 955–966.
- Appenzeller C. , Holton J. R. , Rosenlof K. H . Seasonal variation of mass transport across the tropopause. J. Geophys. Res. 1996; 101: 15071–15078.
- Baker A. K. , Slemr F. , Brenninkmeijer C. A. M . Analysis of non-methane hydrocarbons in air samples collected aboard the CARIBIC passenger aircraft. Atmos. Meas. Tech. 2010; 3: 311–321.
- Bazhenov O. E. , Burlakov V. D. , Dolgii S. I. , Nevzorov A. V . Lidar observations of aerosol disturbances of the stratosphere over Tomsk (56.5 N; 85.0 E) in volcanic activity period 2006–2011. Int. J. Opt. 2012; 2012
- Bourassa A. E. , Robock A. , Randel W. J. , Deshler T. , Rieger L. A , co-authors . Large volcanic aerosol load in the stratosphere linked to Asian monsoon transport. Science. 2012; 337: 78–81. [PubMed Abstract].
- Brenninkmeijer C. A. M. , Crutzen P. J. , Fischer H. , Güsten H. , Hans W. , co-authors . CARIBIC—Civil aircraft for global measurement of trace gases and aerosols in the tropopause region. J. Atmos. Oceanic Technol. 1999; 16: 1373–1383.
- Brenninkmeijer C. A. M. , Crutzen P. , Boumard F. , Dauer T. , Dix B , co-authors . Civil Aircraft for the regular investigation of the atmosphere based on an instrumented container: the new CARIBIC system. Atmos. Chem. Phys. 2007; 7: 4953–4976.
- Brewer A. W . Evidence for a world circulation provided by the measurements of helium and water vapour distribution in the stratosphere. Q. J. Roy. Meteorol. Soc. 1949; 75: 351–363.
- Brühl C. , Lelieveld J. , Crutzen P. J , Tost H . The role of carbonyl sulphide as a source of stratospheric sulphate aerosol and its impact on climate. Atmos. Chem. Phys. 2012; 12: 1239–1253.
- Bönisch H. , Engel A. , Curtius J. , Birner T. , Hoor P . Quantifying transport into the lowermost stratosphere using simultaneous in-situ measurements of SF6 and CO2. Atmos. Chem. Phys. 2009; 9: 5905–5919.
- Carn S. A. , Froyd K. D. , Anderson B. E. , Wennberg P. , Crounse J , co-authors . In situ measurements of tropospheric volcanic plumes in Ecuador and Colombia during TC4. J. Geophys. Res. 2011; 116: D00J24.
- Carn S. A. , Krueger A. J. , Krotkov N. A. , Yang K , Evans K . Tracking volcanic sulfur dioxide clouds for aviation hazard mitigation. Nat. Hazards. 2009; 51: 325–343.
- Carn S. A. , Prata F. J . Satellite-based constraints on explosive SO2 release from Soufrière Hills Volcano, Montserrat. Geophys. Res. Lett. 2010; 37: L00E22.
- Chin M. , Davis D. D . A reanalysis of carbonyl sulfide as a source of stratospheric background sulfur aerosol. J. Geophys. Res. 1995; 100: 8993–9005.
- Crutzen P. J . The possible importance of CSO for the sulfate layer of the stratosphere. Geophys. Res. Lett. 1976; 3: 73–76.
- Damoah R. , Spichtinger N. , Servranckx R. , Fromm M. , Eloranta E. W , co-authors . A case study of pyro-convection using transport model and remote sensing data. Atmos. Chem. Phys. 2006; 6: 173–185.
- Deshler T . A review of global stratospheric aerosol: measurements, importance, life cycle, and local stratospheric aerosol. Atmos. Res. 2008; 90: 223–232.
- Deshler T. , Anderson-Sprecher R. , Jäger H. , Barnes J. , Hofmann D. J , co-authors . Trends in the nonvolcanic component of stratospheric aerosol over the period 1971–2004. J. Geophys. Res. 2006; 111: D01201.
- Dessler A. E. , Hintsa E. J. , Weinstock E. M. , Anderson J. G. , Chan K. R . Mechanisms controlling water vapor in the lower stratosphere: “a tale of two stratospheres.”. J. Geophys. Res. 1995; 100: 23167–23172.
- Dobson G. M. B . Origin and distribution of the polyatomic molecules in the atmosphere. Proc. R. Soc. London, Ser. A. 1956; 236: 187–193.
- Dutton E. G. , Bodhaine B. A . Solar irradiance anomalies caused by clear-sky transmission variations above Mauna Loa: 1958–99. J. Clim. 2001; 14: 3255–3262.
- Flury T. , Wu D. L , Read W. G . Variability in the speed of the Brewer–Dobson circulation as observed by Aura/MLS. Atmos. Chem. Phys. 2013; 13: 4563–4575.
- Fortuin J. P. F. , Kelder H . An ozone climatology based on ozonesonde and satellite measurements. J. Geophys. Res. 1998; 103: 31709–31734.
- Fromm M. , Bevilacqua R. , Servranckx R. , Rosen J. , Thayer J. P , co-authors . Pyro-cumulonimbus injection of smoke to the stratosphere: observations and impact of a super blowup in northwestern Canada on 3–4 August 1998. J. Geophys. Res. 2005; 110: D08205.
- Fromm M. , Lindsey D. T. , Servranckx R. , Yue G. , Trickl T , co-authors . The untold story of pyrocumulonimbus. Bull. Am. Meteorol. Soc. 2010; 91: 1193–1209.
- Fromm M. , Torres O. , Diner D. , Lindsey D. , Vant Hull B , co-authors . Stratospheric impact of the Chisholm pyrocumulonimbus eruption: 1. Earth-viewing satellite perspective. J. Geophys. Res. 2008; 113: D08202.
- Gettelman A. , Holton J. R. , Rosenlof K. H . Mass fluxes of O3, CH4, N2O and CF2Cl2 in the lower stratosphere calculated from observational data. J. Geophys. Res. 1997; 102: 19149–19159.
- Gettelman A. , Hoor P. , Pan L. L. , Randel W. J. , Hegglin M. I , co-authors . The extratropical upper troposphere and lower stratosphere. Rev. Geophys. 2011; 49: RG3003.
- Guan H. , Esswein R. , Lopez J. , Bergstrom R. , Warnock A , co-authors . A multi-decadal history of biomass burning plume heights identified using aerosol index measurements. Atmos. Chem. Phys. 2010; 10: 6461–6469.
- GVP. Global Volcanism Program. 2011. Online at: http://www.volcano.si.edu/index.cfm .
- Hermann M. , Stratmann F. , Wilck M. , Wiedensohler A . Sampling characteristics of an aircraft-borne aerosol inlet system. J. Atmos. Ocean. Technol. 2001; 18: 7–19.
- Hoerling M. P. , Schaack T. K. , Lenzen A. J . Global objective tropopause analysis. Mon. Weather Rev. 1991; 119: 1816–1831.
- Hofmann D. , Barnes J. , O'Neill M. , Trudeau M , Neely R . 2009. Increase in background stratospheric aerosol observed with lidar at Mauna Loa Observatory and Boulder, Colorado. Geophys. Res. Lett. 36: L15808.
- Hoinka K. P . The tropopause: discovery, definition and demarcation. Meteorol. Z. 1997; 6: 281–303.
- Holton J. R. , Haynes P. H. , McIntyre M. E. , Douglas A. R. , Rood R. B. , co-authors . Stratosphere–troposphere exchange. Rev. Geophys. 1995; 33: 403–439.
- Hoor P. , Fischer H , Lelieveld J . Tropical and extratropical tropospheric air in the lowermost stratosphere over Europe: a CO-based budget. Geophys. Res. Lett. 2005; 32
- Hoskins B. J . Towards a PV-θ view of the general circulation. Tellus, Ser AB. 1991; 43: 27–35.
- Intergovernmental Panel on Climate Change. Solomon S. , etal. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, 2007. 2007; Cambridge, United Kingdom and New York, NY, USA: Cambridge University Press.
- Johansson S. A. E. , Campbell J. L . PIXE: A Novel Technique for Elemental Analysis. 1988; Hoboken, NJ: John Wiley.
- Jost H. J. , Drdla K. , Stohl A. , Pfister L. , Loewenstein M. , co-authors . In-situ observations of mid-latitude forest fire plumes deep in the stratosphere. Geophys. Res. Lett. 2004; 31: L11101.
- Junge C. E. , Chagnon C. W. , Manson J. E . A world-wide stratospheric aerosol layer. Science. 1961; 133: 1478–1479.
- Lelieveld J. , Bregman B. , Arnold F. , Bürger V. , Crutzen P. J , co-authors . Chemical perturbation of the lowermost stratosphere through exchange with the troposphere. Geophys. Res. Lett. 1997; 24: 603–606.
- Lin P , Fu Q . Changes in various branches of the Brewer–Dobson circulation from an ensemble of chemistry climate models. J. Geophys. Res. 2013; 118: 73–84.
- Martinsson B. G. , Brenninkmeijer C. A. M. , Carn S. A. , Hermann M. , Heue K. P , co-authors . Influence of the 2008 Kasatochi volcanic eruption on sulfurous and carbonaceous aerosol constituents in the lower stratosphere. Geophys. Res. Lett. 2009; 36: L12813.
- Martinsson B. G. , Nguyen H. N. , Brenninkmeijer C. A. M. , Zahn A. , Heintzenberg J , co-authors . Characteristics and origin of lowermost stratospheric aerosol at northern midlatitudes under volcanically quiescent conditions based on CARIBIC observations. J. Geophys. Res. 2005; 110: D12201.
- Martinsson B. G. , Papaspiropoulos G. , Heintzenberg J. , Hermann M . Fine mode particulate sulphur in the tropopause region measured from intercontinental flights (CARIBIC). Geophys. Res. Lett. 2001; 28: 1175–1178.
- Massie S. T . AURA Cloud/Aerosol/SO2Working Group. 2014. Online at: http://avdc.gsfc.nasa.gov/PDF/volcano.pdf .
- McCormick M. P. , Thomason L. W. , Trepte C. R . Atmospheric effects of the Mt Pinatubo eruption. Nature. 1995; 373: 399–404.
- Murphy D. M. , Thomson D. S , Mahoney M. J . In situ measurements of organics, meteoritic material, mercury, and other elements in aerosols at 5 to 19 kilometers. Science. 1998; 282: 1664–1669. [PubMed Abstract].
- Nagai T. , Liley B. , Sakai T. , Shibata T , Uchino O . Post-Pinatubo evolution and subsequent trend of the stratospheric aerosol layer observed by mid-latitude lidars in both hemispheres. Sola. 2010; 6: 69–72.
- Neely III, R. R. , Toon O. B. , Solomon S. , Vernier J. P. , Alvarez C , co-authors . Recent anthropogenic increases in SO2 from Asia have minimal impact on stratospheric aerosol. Geophys. Res. Lett. 2013; 40: 999–1004.
- Nguyen H. N. , Gudmundsson A , Martinsson B. G . Design and calibration of a multi-channel aerosol sampler for tropopause region studies from the CARIBIC platform. Aerosol Sci. Technol. 2006; 40: 649–655.
- Nguyen H. N , Martinsson B. G . Analysis of C, N and O in aerosol collected on an organic backing using internal blankmeasurements and variable beam size. Nucl. Instrum. Methods Phys. Res. Sect. B. 2007; 264: 96–102.
- Nguyen H. N. , Martinsson B. G. , Wagner J. B. , Carlemalm E. , Ebert M , co-authors . Chemical composition and morphology of individual aerosol particles from a CARIBIC flight at 10 km altitude between 50 N and 30 S. J. Geophys. Res. 2008; 113: D23209.
- Oram D. E. , Mani F. S. , Laube J. C. , Newland M. J. , Reeves C. E. , co-authors . Long-term tropospheric trend of octafluorocyclobutane (c-C4F8 or PFC-318). Atmos. Chem. Phys. 2012; 12: 261–269.
- Papaspiropoulos G. , Mentes B. , Kristiansson P. , Martinsson B. G . A high sensitivity elemental analysis methodology for upper tropospheric aerosol. Nucl. Instrum. Methods Phys. Res. Sect. B. 1999; 150: 356–362.
- Prata A. J. , Bernardo C . Retrieval of volcanic SO2 column abundance from atmospheric infrared sounder data. J. Geophys. Res. 2007; 112: D20204.
- Rauthe-Schöch A. , Weigelt A. , Hermann M. , Martinsson B. G. , Baker A. K. , co-authors . CARIBIC aircraft measurements of Eyjafjallajökull volcanic clouds in April/May 2010. Atmos. Chem. Phys. 2012; 12: 879–902.
- Robock A . Volcanic eruptions and climate. Rev. Geophys. 2000; 38: 191–219.
- Rosen J. M . The boiling point of stratospheric aerosols. J. Appl. Meteorol. 1971; 10: 1044–1046.
- Rudnick R. L. , Fountain D. M . Nature and composition of the continental crust: a lower crustal perspective. Rev. Geophys. 1995; 33: 267–309.
- Schmale J. , Schneider J. , Jurkat T. , Voigt C. , Kalesse H , co-authors . Aerosol layers from the 2008 eruptions of Mount Okmok and Mount Kasatochi: in situ upper troposphere and lower stratosphere measurements of sulfate and organics over Europe. J. Geophys. Res. 2010; 115: D00L07.
- Schuck T. J. , Brenninkmeijer C. A. M. , Slemr F. , Xueref-Remy I. , Zahn A . Greenhouse gas analysis of air samples collected onboard the CARIBIC passenger aircraft. Atmos. Meas. Tech. 2009; 2: 449–464.
- Schwarz J. P. , Spackman J. R. , Gao R. S. , Watts L. A. , Stier P , co-authors . Global-scale black carbon profiles observed in the remote atmosphere and compared to models. Geophys. Res. Lett. 2010; 37: L18812.
- Smith J. B. , Hintsa E. J. , Allen N. T. , Stimpfle R. M. , Anderson J. G . Mechanisms for midlatitude ozone loss: heterogeneous chemistry in the lowermost stratosphere?. J. Geophys. Res. 2001; 106: 1297–1309.
- Solomon S. , Borrmann S. , Garcia R. R. , Portmann R. , Thomason L , co-authors . Heterogeneous chlorine chemistry in the tropopause region. J. Geophys. Res. 1997; 102: 21411–21429.
- Solomon S. , Daniel J. S. , Neely R. R. , Vernier J. P. , Dutton E. G , co-authors . The persistently variable “background” stratospheric aerosol layer and global climate change. Science. 2011; 333: 866–870. [PubMed Abstract].
- Spracklen D. V. , Arnold S. R. , Sciare J. , Carslaw K. S. , Pio C . Globally significant oceanic source of organic carbon aerosol. Geophys. Res. Lett. 2008; 35: L12811.
- Sprenger M , Wernli H . A northern hemispheric climatology of cross-tropopause exchange for the ERA15 time period (1979–1993). J. Geophys. Res. 2003; 108(D12): 8521.
- Sprung D , Zahn A . Acetone in the upper troposphere/lowermost stratosphere measured by the CARIBIC passenger aircraft: distribution, seasonal cycle, and variability. J. Geophys. Res. 2010; 115: D16301.
- Stramska M . Particulate organic carbon in the global ocean derived from SeaWiFS ocean color. Deep Sea Res. Part I. 2009; 56: 1459–1470.
- Tang Q. , Prather M. J , Hsu J . Stratosphere–troposphere exchange ozone flux related to deep convection. Geophys. Res. Lett. 2011; 38: L03806.
- Thomas H. E. , Watson I. M. , Carn S. A. , Prata A. J , Realmuto V. J . A comparison of AIRS, MODIS and OMI sulphur dioxide retrievals in volcanic clouds. Geomatics Nat. Hazards Risk. 2011; 2(3): 217–232.
- Trickl T. , Giehl H. , Jäger H , Vogelmann H . 35 yr of stratospheric aerosol measurements at Garmisch-Partenkirchen: from Fuego to Eyjafjallajökull, and beyond. Atmos. Chem. Phys. 2013; 13: 5205–5225.
- Vernier J. P. , Pommereau J. P. , Garnier A. , Pelon J. , Larsen N , co-authors . Tropical stratospheric aerosol layer from CALIPSO lidar observations. J. Geophys. Res. 2009; 114: D00H10.
- Vernier J. P. , Thomason L. W. , Kar J . CALIPSO detection of an Asian tropopause aerosol layer. Geophys. Res. Lett. 2011a; 38: L07804.
- Vernier J. P. , Thomason L. W. , Pommereau J. P. , Bourassa A. , Pelon J , co-authors . Major influence of tropical volcanic eruptions on the stratospheric aerosol layer during the last decade. Geophys. Res. Lett. 2011b; 38: L12807.
- Weisenstein D. K. , Yue G. K. , Ko M. K. W. , Sze N. D. , Rodriguez J. M , co-authors . A two-dimensional model of sulfur species and aerosols. J. Geophys. Res. 1997; 102: 13019–13035.
- Yang K. , Liu X. , Bhartia P. K. , Krotkov N. A. , Carn S. A. , co-authors . Direct retrieval of sulfur dioxide amount and altitude from spaceborne hyperspectral UV measurements: theory and application. J. Geophys. Res. 2010; 115: D00L09.
- Zahn A. , Brenninkmeijer C. A. M . New directions: a chemical tropopause defined. Atmos. Environ. 2003; 37: 439–440.
- Zahn A. , Brenninkmeijer C. A. M. , Asman W. A. H. , Crutzen P. J. , Heinrich G , co-authors . Budgets of O3 and CO in the upper troposphere: CARIBIC passenger aircraft results 1997–2001. J. Geophys. Res. 2002; 107(D17): 4337.
- Zahn A. , Weppner J. , Widmann H. , Schlote-Holubek K. , Burger B , co-authors . A fast and precise chemiluminescence ozone detector for eddy flux and airborne application. Atmos. Meas. Tech. 2012; 5