Abstract
Four mitochondrial DNA segments, ND1, ND6, cyt b and D-loop, were analyzed by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) in 14 tench (Tinca tinca L.) populations located in Europe and Asia; data on 5 Italian populations previously analyzed for the same mtDNA segments were also included in the study. All the considered segments were polymorphic and originated a total of 9 composite haplotypes which were clustered into 2 haplogroups, A and B, possibly corresponding to the Western and Eastern phylogroups previously described in tench. Nine out of 19 populations showed polymorphism, with haplotype diversity ranging from 0.246 to 0.643 and nucleotide diversity from 0.009 to 0.078. Seventy-five percent of the pairwise comparisons were significant, indicating a high between-population variability. The Neighbour-Joining tree revealed the presence of 3 clusters, including pure populations, with only a A or B haplogroup, and mixed populations, with both haplogroups. The possibility of identifying populations with different haplotypes has practical implications for both conservation and supportive stocking.
Introduction
For the last twenty years, the genetic research in aquaculture has been exponentially increasing, but genetic information on tench (Tinca tinca L.) is still limited compared to other fish species. In fact, apart from some studies carried out over the past decades by means of protein markers (CitationValenta et al., 1978; CitationŠlechtova et al., 1995; CitationKohlmann and Kersten, 1998), only recently have tench specific microsatellite loci been described (CitationKohlmann and Kersten, 2006) and used to characterize many European and Asian populations (CitationKohlmann et al., 2007, Citation2010; CitationLo Presti et al., 2010b). Also, the complete mitochondrial DNA (mtDNA) sequence has only recently been made available (CitationSaitoh et al., 2006). The subsequent analysis of the mitochondrial cyt b gene, together with 3 nuclear genes, has helped clarify the molecular phylogeography of the tench with the discovery of 2 geographical clades (Eastern and Western), possibly developed in response to recurrent isolation in glacial refugia during the Pleistocene (CitationLajbner et al., 2007). Within the Eastern phylogroup, the analysis of cyt b also allowed the identification of populations distinct from the major Eastern clade in the Anzalee lagoon of the Caspian Sea in Iran and in the Iskar River of the Danube River drainage in Bulgaria (CitationLajbner et al., 2011). Only one study was devoted to analyzing the polymorphism of different mitochondrial segments as a tool to detect tench genetic variability (CitationLo Presti et al., 2010a).
The aim of this paper is to extend the study of the mtDNA polymorphisms to a larger number of tench populations distributed in a wide geographical area, in order to obtain a more comprehensive picture of the within-population and between-population variability in tench.
Materials and methods
A total of 126 individuals were analyzed. These belonged to 14 wild and cultured populations located in different European and Asian countries. Data on the 5 Italian populations previously studied for the same mtDNA segments (CitationLo Presti et al., 2010a) were also included in order to cover a larger geographical area (). All the populations had been already analyzed by microsatellite markers (CitationKohlmann et al., 2010; CitationLo Presti et al., 2010b).
Table 1 Description of the populations.
Total genomic DNA was extracted from muscle or fin using the NucleoSpin Tissue kit (Macheray-Nagel, Düren, Germany). PCR reactions to amplify ND1, ND6, cyt b and D-loop segments were performed as described by CitationLo Presti et al. (2010a). Each amplicon was digested with 4 restriction enzymes which were selected on the basis of the previous results (CitationLo Presti et al., 2010a) and in consideration of the expected restriction pattern derived from virtual digestion of the reference sequence with Webcutter 2.0 software (CitationHeiman, 1997). Some of the enzymes were used to digest different amplicons () so that a total of 7 endonucleases were used: AluI, Sau3A I (Sigma, St Louis, MO, USA), AseI, HaeIII, MspI (New England BioLabs, Beverly, MA, USA) HindIII, HinfI (Fermentas, Burlington, ON, Canada). The digested fragments were resolved on 2% agarose gels, stained with ethidium bromide and visualized under UV light. The size of the fragments was estimated by comparison with a 100 bp size ladder (Sigma, St Louis, MO, USA) and each different pattern produced by each enzyme was identified by a single letter code, with A assigned to the pattern expected on the basis of the reference sequence. Composite haplotypes were designed by a 16-letter code representing the pattern for each restriction enzyme.
Table 2 Approximate fragment size of the restriction morphs observed by digesting 4 mtDNA segments with 7 different endonucleases.
The relationships between composite haplotypes were analyzed by calculating the mean number of substitutions per site between all pairs of haplotypes from restriction site data (CitationNei and Li, 1979) which were used to construct a Neighbour-Joining tree as implemented in the PHYLIP version 3.5 package (CitationFelsenstein, 1993); the reliability of the tree topology was tested by 1,000 bootstrap replicates.
The ARLEQUIN version 3.1 program (CitationExcoffier et al., 2005) was used to evaluate the variability within populations by haplotype and nucleotide diversity (CitationNei and Tajima, 1981), as well as to test the population differentiation by the pairwise exact test (CitationRaymond and Rousset, 1995). Significance levels for multiple comparisons were adjusted using the sequential Bonferroni correction (CitationRice, 1989). The genetic distances between populations were also estimated as the pairwise net nucleotide divergence (CitationNei and Li, 1979), followed by the construction of the Neighbour-Joining tree, using the MEGA 4 software (CitationTamura et al., 2007).
Results
All the enzymes but Msp I detected restriction fragment length polymorphisms at some mtDNA segment (). The digestion of ND6 and ND1 revealed one and two variants, respectively, as previously reported (CitationLo Presti et al., 2010a), while additional variation was observed for cyt b and D-loop. At cyt b the Sau3A I endonuclease detected a new variant the pattern of which does not seem to derive directly from the loss or gain of a restriction site with respect to the reference pattern. A more complex situation could, therefore, be hypothesized, such as concomitant loss and gain of restriction sites. For the D-loop, two new variants were found, one with AseI and one with Hae III, respectively, due to the presence and absence of a restriction site. The polymorphisms at the 4 mtDNA segments gave rise to a total of 9 composite haplotypes, named H1 to H9, with H1 corresponding to the reference sequence (). An analysis of the overall frequencies indicated that H1 and H2 were the most frequent composite haplotypes with a cumulative frequency of 0.805, while the others were very rare, with frequencies lower than 0.05.
Table 3 Composite haplotypes and their overall frequency (P).
The analysis of the pairwise nucleotide divergence between composite haplotypes led to a phylogenetic tree where 2 highly divergent haplogroups were identified: one, designated as haplogroup A, included the H1, H3, H4, H5 and H6 composite haplotypes, while the other, designated as haplogroup B, included the H2, H7, H8 and H9 composite haplotypes (). The 2 haplogroups differed for polymorphisms at the ND1 and Cyt b segments: all the haplotypes belonging to the haplogroup A had the restriction morphs ND1/Alu I A, ND1/Hinf I A and Cyt b/Sau3AI A, while all the haplotypes of the haplogroup B had the restriction morphs B at the same sites (). The bootstrap value of 96% strongly supported the between-haplogroup differentiation, while the within-haplogroup relationships were less clear, with low to medium bootstrap values.
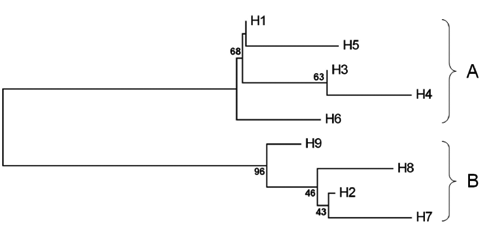
As for the composite haplotype distribution, H1 and H2 were present in 63 and 47% of the populations, respectively, whereas the others were limited to one or few populations (). H3 was observed only in the Central and Southern Italian populations (BOL, TRA, ALC); H4 and H6 were the rarest composite haplo-types, found in one individual only from Trasimeno (Italy) and Felchowsee (Germany) lakes, respectively. H5, H7 and H8 were private haplotypes for the wild VAL and TUR, and cultured MAL populations, respectively. Moreover, H8 seemed to be fixed in the latter population, so it might be used as a genetic tag if the data were to be confirmed on a larger sample (the present sample size is 10 individuals only). H9 was fixed in the GOL population (the golden color variety), but present also in one wild FEL individual.
Table 4 Within-population variability: haplotype frequency, haplotype (H) and nucleotide (π) diversity (mean value ± standard error).
Ten out of 19 populations showed no variability (). For ISE and BRA, the finding is possibly dependent on the low sample size (3 and 4 individuals, respectively), and, therefore, these 2 Italian wild populations were excluded from the subsequent analysis. On the contrary, the fixation of the haplotype H9 in GOL can be interpreted as a result of the founder effect (CitationKvasnika et al., 1993). It is worth underlining the fact that the other monomorphic populations are cultured, except for the German DÖL which is wild. The absence of polymorphism in DÖL and KÖW is quite unexpected considering that these populations when analyzed by microsatellite markers showed a high variability (CitationKohlmann et al., 2010).
In the polymorphic populations, the haplotype diversity ranged from 0.246 (PIA) to 0.643 (FEL), whereas the nucleotide diversity ranged from 0.009 (BOL) to 0.078 (FEL) (). As expected, the highest values for nucleotide diversity were observed in the populations where composite haplotypes of both evolutionary lineages were present. In particular, FEL showed the highest values for both indices, confirming its importance as a reservoir of genetic diversity, in agreement with the high variability detected by previous studies on microsatellite markers (CitationKohlmann et al., 2010).
Concerning the between-population differentiation, 75% of the pairwise comparisons were significant, indicating a high level of genetic variation at species level (). Going into more detail, GOL, MAL and VAL differed statistically from all the other populations, while most of the non-significant comparisons involved the Eastern populations (CHI, TUR, HUN, ROM, VEM and VOD) and the one from Spain (BAD). No differences were observed between the German populations (FEL, KÖW, DÖL) or between those of Central- Southern Italy (TRA, BOL, ALC).
Table 5 Nucleotide divergence within population (diagonal) and between populations (above the diagonal); significance (*) of the exact test of CitationRaymond and Rousset (1995) for population differentiation (below the diagonal).
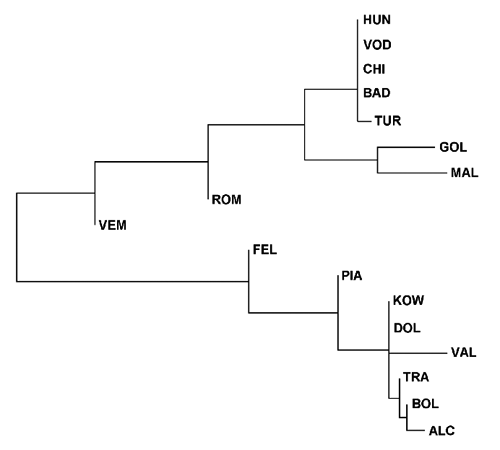
The Neighbour-Joining tree, constructed on the basis of the pairwise net nucleotide divergence, separated 2 clusters, one including the Italian and German populations and one including all the others (). The latter displayed lack of resolution, involving populations all fixed for the H2 composite haplotype (BAD, CHI, HUN and VOD). These populations had a nucleotide divergence of 0.248, while the Italian and German populations represented a more heterogeneous group with DA of 0.711. The divergence between the two branches was much higher (DA = 4.652) indicating a deep separation between the populations of the 2 groups. It is interesting to note that the populations with both haplogroups (FEL and PIA at one side, VEM and ROM at the other side) were located close to the principal node. Therefore, the tree can be subdivided into 3 clusters, corresponding to pure populations, with A or B haplogroup, and mixed populations, with both haplogroups.
Discussion
The PCR-RFLP analysis of ND1, ND6, cyt b and D-loop in 19 tench populations confirmed the effectiveness of these mtDNA markers for population genetic studies of this species. Of the 4 examined mtDNA segments, cyt b and D-loop showed the highest variability, with 4 and 3 variants, respectively. The quite high variability of the D-loop seems to be a peculiarity of the tench, which so far has not been observed in other teleosts; for example, no polymorphism was found in Danish brown trout (Salmo trutta L.) strains by digestion with 18 restriction enzymes (CitationHansen and Loeschke, 1996), nor in rainbow trout (Onchorhyncuhus mykiss) using 12 endonucleases (CitationSajedi et al., 2003). These findings underline the fact that mtDNA segments which are more appropriate for population studies have to be chosen for each species individually. Nine out of 19 populations examined showed considerable haplotype as well as nucleotide diversity. However, the mtDNA markers generally revealed a lower power than microsatellite markers in detecting the within-population variability. In fact, the average haplotype diversity level of mtDNA (Hmt) observed in the present study (0.215) was lower than the average heterozygosity level of microsatellites (Hms) deduced from the data of CitationKohlmann et al. (2010) and CitationLo Presti et al. (2010b) on the same populations (0.343). However, in some populations in which mtDNA was polymorphic, Hmt was even higher than Hms (TUR, FEL, ALC, TRA, BOL). The absence of relationships between nuclear and mitochondrial variability, already reported for other species (CitationPalumbi and Baker, 1994), is not surprising given the different genetic background and mode of evolution of the 2 types of markers. Therefore, for the different information they provide, the complementary analysis of nuclear and mitochondrial markers represent a powerful strategy to clarify the population genetic structure. On the other hand, mtDNA markers were shown to be an excellent tool for revealing the between-population variability. The identification of 2 mtDNA haplogroups in the present study, along with the recent discovery of 2 major growth hormone gene classes in tench (CitationKocour and Kohlmann, 2011) further support the results of CitationLajbner et al. (2007, Citation2011) who investigated the molecular phylogeography of tench by the analysis of the cyt b locus and some nuclear markers. They showed that the species is subdivided into deeply divergent Western and Eastern phylogroups which are not, however, distinct species (CitationLajbner et al., 2010). Also in other freshwater species, including the chub (Leuciscus cephalus) (CitationDurand et al., 1999), perch (Perca fluviatilis) (CitationNesbø et al., 1999) and barbel (Barbus barbus) (CitationKotlik and Berrebi, 2001), genetic lines related to the geographical location were observed which could indicate an evolutionary history common to different freshwater species.
On the basis of the haplogroup composition, composite haplotype distribution and population location, it can be inferred that the haplogroup A observed in the present study corresponds to the Western phylogroup reported by CitationLajbner et al. (2007, Citation2011) while the haplogroup B corresponds to the Eastern phylogroup.
It is worthy of note, for practical implications, that the mtDNA markers used in the present study allowed a good resolution for both phylogroups, compared to the markers used by CitationLajbner et al. (2011) and CitationLajbner and Kotlik (2011), that detected 3 subclades in the Eastern clade but showed only very little internal structure for the Western clade.
The Neighbour-Joining tree constructed on the basis of the pairwise net nucleotide divergence (CitationNei and Li, 1979) between populations showed some similarities with the tree obtained by CitationKohlmann et al. (2010) using the microsatellite markers: in both cases HUN, CHI, BAD and TUR clustered together and the German populations were located in the opposite branch. In particular, the results confirmed the genetic similarity between the Spanish and Chinese populations, which could be explained by human introduction of tench from the East to Spain.
Conclusions
The present PCR-RFLP based analysis of 4 segments of the tench mtDNA revealed considerable haplotype as well as nucleotide diversity in 9 out of 19 populations examined. Thus, these polymorphisms, which are easy and inexpensive to screen, might effectively be used for population genetic studies of this species, with implications for conservation and supportive stocking. For conservation purposes, these mtDNA markers might help to identify populations with different haplotypes within haplogroups and could thus help protect their genetic integrity. In case of stocking, donor and recipient populations should be as genetically similar as possible, i.e. they should at least belong to the same composite haplotype. On the other hand, mixed populations with higher mtDNA diversity (and also higher microsatellite variability) might be valuable baseline populations to start selective breeding programs, particularly if mtDNA and microsatellite information were to be combined with the recently described tench growth hormone gene polymorphisms.
References
- DurandJ.D. PersatH. BouvetY. 1999 Phylogeography and postglacial dispersion of the chub (Leuciscus cephalus) in Europe Mol. Ecol 8 989 997
- ExcoffierL. LavalG. SchneiderS. 2005 Arlequin ver. 3.0: An integrated software package for population genetics data analysis Evol. Bioinform 1 47 50
- FelsensteinJ. 1993 PHYLIP (Phylogeny Inference Package) version 3.5 Department of Genome Sciences University of Washington Publ. Seattle, USA
- HansenM.M. LoeschckeV. 1996 Genetic differentiation among Danish brown trout populations, as detected by RFLP analysis of PCR amplified mitochondrial DNA segments J. Fish Biol 48 422 436
- HeimanM. 1997 Webcutter 2.0 Available from: http//rna.lundberg.gu.se/cutter2
- KocourM. KohlmannK. 2011 Growth hormone gene polymorphisms in tench, Tinca tinca L Aquaculture 310 298 304
- KohlmannK. KerstenP. 1998 Enzyme variability in a wild population of tench (Tinca tinca L.) Pol. Arch. Hydrobiol 45 303 310
- KohlmannK. KerstenP. 2006 Microsatellite loci in tench: isolation and variability in a test population Aquacult. Int 14 3 7
- KohlmannK. KerstenP. FlajšhansM. 2007 Comparison of microsatellite variability in wild and cultured tench (Tinca tinca) Aquaculture 272 147 151
- KohlmannK. KerstenP. PaniczR. MemisD. FlajšhansM. 2010 Genetic variability and differentiation of wild and cultured tench populations inferred from microsatellite loci Rev. Fish Biol. Fisher 20 279 288
- KotlíkP. BerrebiP. 2001 Phylogeography of the barbel (Barbus barbus) assessed by mitochondrial DNA variation Mol. Ecol 10 2177 2185
- KvasnickaP. FlajšhansM. RabP. LinhartO. 1993 Inheritance studies of blue and golden varieties of tench (Pisces: Tinca tinca L.) J. Hered 89 553 556
- LajbnerZ. KotlìkP. 2011 PCR-RFLP assays to distinguish the Western and Eastern phylogroups in wild and cultured tench Tinca tinca Mol. Ecol. Resour 11 374 377
- LajbnerZ. LinhartO. KotlìkP. 2007 Molecular phylogeography of the tench Tinca tinca (Linnaeus, 1758) 35 (abstr.) Proc. 12th Eur. Congr. Ichthyology Cavtat, Croatia
- LajbnerZ. KohlmannK. LinhartO. KotlìkP. 2010 Lack of reproductive isolation between the Western and Eastern phylogroups of the tench Rev. Fish Biol. Fisher 20 289 300
- LajbnerZ. LinhartO. KotlìkP. 2011 Human-aided dispersal has altered but not erased the phylogeography of the tench Evol. Appl 4 545 561
- Lo PrestiR. GascoL. LisaC. ZoccaratoI. Di StasioL. 2010a PCR-RFLP analysis of mitochondrial DNA in tench (Tinca tinca L.) J. Fish Biol 76 401 407
- Lo PrestiR. KohlmannK. KerstenP. GascoL. Di StasioL. 2010b Tinca Gobba Dorata del Pianalto di Poirino: genetic characterization by microsatellite markers Ital. J. Anim. Sci 9 e85
- NeiM. LiW.H. 1979 Mathematical model for studying genetic variation in terms of restriction endonucleases P. Natl. Acad. Sci. USA 76 5269 5273
- NeiM. TajimaF. 1981 DNA polymorphism detectable by restriction endonucleases Genetics 97 145 163
- NesbøC.L. FossheimT. VøllestadL.A. JakobsenK.S. 1999 Genetic divergence and phylogeographic relationship among European perch (Perca fluviatilis) populations reflect glacial refugia and postglacial colonisation Mol. Ecol 8 1387 1404
- PalumbiS.R. BakerC.S. 1994 EPIC amplification of nuclear introns: opposing views of population structure using mitochondrial and nuclear sequences from humpback whales Mol. Biol. Evol 11 426 435
- RaymondM. RoussetF. 1995 An exact test for population differentiation Evolution 49 1280 1283
- RiceW.R. 1989 Analyzing tables of statistical tests Evolution 43 223 225
- SaitohK. SadoT. MaydenR.L. HanzawaN. NakamuraK. NishidaM. MiyaM. 2006 Mitogenomic evolution and interrelationships of the Cypriniformes (Actinopterygii: Ostariophysi): The first evidence toward resolution of higher-level relationships of the world's largest freshwater fish clade based on 59 whole mitogenome sequences J. Mol. Evol 63 826 841
- SajediR.H. AminzadehS. Naderi-ManeshH. SadeghizadehM. AbdolhayH. Naderi-ManeshM. 2003 Genetic variation within and among rainbow trout, Onchorhynchus mykiss, hatchery populations from Iran assessed by PCR-RFLP analysis of mitochondrial DNA segments J. Food Sci 68 870 873
- ŠlechtováV. ŠlechtáV. ValentaM. 1995 Genetic protein variability in tench (Tinca tinca L.) stocks in Czech Republic Pol. Arch. Hydrobiol 42 133 140
- TamuraK. DudleyJ. NeiM. KumarS. 2007 MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0 Mol. Biol. Evol 24 1596 1599
- ValentaM. ŠlechtováV. KàlalL. StratilA. JanatkovaJ. ŠlechtaV. RabP. PokornyJ. 1978 Polymorphic proteins of the common carp (Cyprinus carpio) and tench (Tinca tinca) and the possibility of using them in breeding Živočišná výroba 23 797 809