Abstract
For cost-effective, economically competitive production of bioethanol from cellulosic plant matter improvements in the production of enzymes to depolymerize the plant biomass are necessary. The fungus Trichoderma reesei is a prolific producer of cellulases and hemicellulases, and intensive research efforts are ongoing to further increase strain efficiency by maximizing enzyme production levels and optimizing the produced enzyme cocktail. With the genome sequencing of T. reesei QM6a cellulase research has entered a new era. Whole-genome comparisons of hyperproducing strains provide new insights into the mechanisms relevant for cellulase gene expression. The recent discovery that this fungus is also susceptible to sexual crossing opens new possibilities for strain improvement by combining beneficial properties or crossing out deleterious ones. In this review we outline new strategies, tools and recent developments based on genomic and proteomic approaches that are now available to gain better insights into the cellulolytic enzyme machinery of T. reesei.
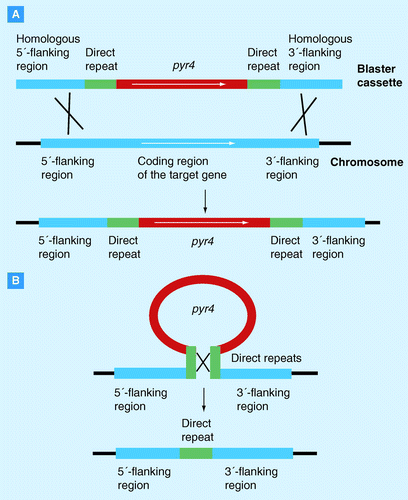
(A) The blaster cassette is composed of a genetic marker ( T. reesei pyr4) flanked by two direct repeats of any sequence and homologous flanking regions of the target gene. By homologous recombination this blaster cassette replaces the coding region of the target gene in the fungal chromosome. The respective strain is now uridine prototrophic. (B) To select for the excision of the blaster cassette the transformed strains are grown in the presence of 5-fluoroorotic acid and uridine. Homologous recombination between the two direct repeats eliminates the marker gene and one direct repeat from the chromosome. The resulting strain is therefore resistant against 5-fluoroorotic acid and uridine auxotrophic, which allows a new round of genetic transformation.
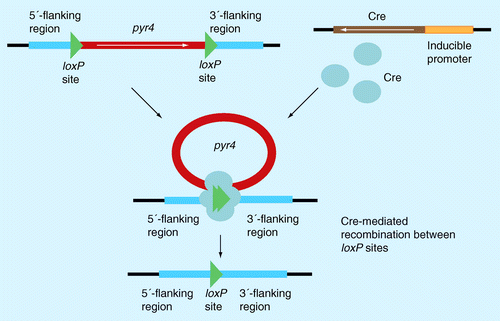
Similar to the blaster cassette-mediated gene deletion the target gene was replaced by homologous recombination by the pyr4 marker. In this case, the pyr4 gene is flanked by two loxP sites. In addition, a Cre recombinase expressed under an inducible promoter was introduced into the genome. The expressed Cre recombinase assembles at each loxP site as a dimer and induces recombination between loxP sites as a tetramer. Upon successful excision of the pyr4 marker, the resulting strain is again uridine auxotrophic.
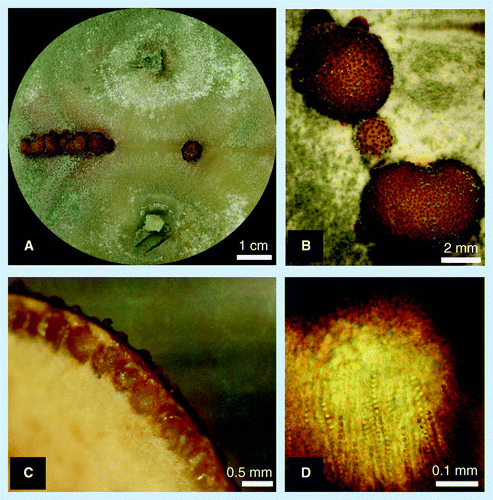
(A) Formation of stromata (fruiting bodies) occurs when fungal strains of opposite mating types are grown on agar plates. (B) Into the upper surface of the stromata perithecia are embedded, which can be seen macroscopically as dark brown dots. (C) Perithecia have the shape of a round bodied bottle. (D) contain asci, which are tube-like structures containing 16 sexual spores (ascospores).
Bioethanol production from lignocellulosic biomass is a promising, environmentally compatible alternative for supplementation and replacement of petroleum-based fuels. Geopolitical aspects such as the change of climate and other environmental problems, and industrial concerns due to fossil fuel limitations in the near future have triggered a boost in research programs investigating the potential of renewable energy sources. The aims are to achieve a reduction of total carbon dioxide emissions through the use of biofuels and carbon dioxide recycling and to advance economically feasible technologies for biofuel production. In bioethanol production processes, monomeric sugars need to be released from plant material and subsequently used in a fermentation step to obtain ethanol. The sugars are either derived from starch-based biomass (e.g., corn starch, sugarcane and sugar beet) or from inedible plant waste material (e.g., corn stover and wheat straw) and nonfood energy crops that can be grown on marginal land. Starch-based first generation bioethanol relies on the use of crops that are also utilized as food and animal feed, which influences their availability and price Citation[1]. Furthermore, these plants require a high agricultural input and the use of fertilizers, which limit the reduction of total carbon dioxide emissions that can be achieved. Due to these limitations the goal of second-generation biofuels is to rely on sustainable bioethanol production from lignocellulosic biomass, thereby reducing carbon dioxide emissions. Improvements in different areas will be necessary for economically compatible second-generation bioethanol production, including optimizing pretreatment of the plant material, genetically modifying the composition of the plant feedstock, and enhancing the efficiency of enzyme mixtures and fermentation steps.
One of the major limitations that we are currently facing in the production of second-generation biofuels is the recalcitrance of lignocellulosic plant matter Citation[2]. The roles of leaves, stalks and stems are to provide structure and rigidity to the plants and therefore they are less susceptible to chemical and enzymatic degradation than any starch-based storage parts of the plants, for example. Biomass recalcitrance is usually reduced by a pretreatment step, exposing cellulose microfibrils for degradation by cellulases. Cellulose degradation is, however, compared with storage polysaccharides, less effective and results in the need for large amounts of enzymes to break down the crystalline cellulose parts of the plants to fermentable monomers. This makes the enzyme mixtures still a critical factor for cost-competitive biofuel production from lignocellulosic material in comparison to starch-based biomass Citation[1].
Minimization of production costs of the respective enzymes, mainly cellulases, and optimization of the efficiency and stability of the generated enzyme mixtures present important goals for economically feasible biofuel production from cellulosic waste material. Today, the ascomycete Trichoderma reesei is the main industrial source of cellulases and hemicellulases Citation[1]. After its isolation on the Solomon Islands in 1944 by the US army, its cellulolytic potential was soon recognized Citation[3] and its high production levels of cellulases and hemicellulases are nowadays industrially exploited in the pulp and paper and food and textile industries. Academic and industrial research programs employing random mutagenesis strongly improved enzyme yields and led to a several-fold increase in the amount of secreted enzymes in comparison to the original isolate T. reesei QM6a Citation[4,5]. Industrial strains are today capable of producing more than 100 g/l of cellulases Citation[6]. Despite these efforts there is still the need to reduce the costs and maximize the yield and efficiency of the produced enzyme mixtures even further and intensive research efforts in this area are ongoing Citation[7].
In this review we will outline new strategies that are currently followed to gain better insights into the transcriptional regulation of the cellulolytic enzyme machinery of T. reesei and we will highlight recent breakthroughs and developments in strain improvement toward increased and optimized enzyme production in T. reesei.
T. reesei strains: past & present
All industrial strains that are nowadays used derive from the original isolate from the Solomon Islands, T. reesei QM6a. Initial strain improvement approaches to increase cellulase yields were based on classical mutagenesis techniques using UV light and mutagenic chemicals followed by growth on selective media Citation[5]. The main target in this selection process was to improve cellulase yields under carbon catabolite repressing conditions. One of the most prominent producer strains, RUT-C30, was obtained by three rounds of mutagenesis Citation[5]. First, UV mutagenesis and selection for the ability to hydrolyze cellulose under carbon catabolite repressing conditions was employed. Strain M7 obtained under these conditions was subjected to mutagenesis by N-nitrosoguanidine under the same selection pressure, which led to the isolation of strain NG14, which already showed a strong increase in secreted proteins and cellulase activity. NG14 was subsequently used for another round of UV mutagenesis and screening for cellulose hydrolysis and resistance to 2-deoxyglucose to eliminate carbon catabolite repression. The resulting strain, RUT-C30, is nowadays still one of the best cellulase-producing strains in the public domain. Other strains derived from mutagenesis programs and widely used in academic research are the early cellulase mutant QM9414 and strain CL847, which is reportedly the best producer strain, although is not publicly available. This strain produces around 40 g/l cellulases during growth on lactose Citation[4].
Despite considerable progress in understanding the transcriptional regulation of cellulase gene expression in the last years, the efforts to improve cellulase production by recombinant DNA technology did not, so far, succeed in matching the levels of RUT-C30 or CL847. The current state of knowledge on the regulatory aspects of cellulase genes has recently been reviewed in detail and is only highlighted here Citation[8–10]. Cellulase gene expression is mainly regulated at the transcription level, and most of the cellulase genes are coordinately expressed, independent of the inducing sugar. The quest for key cellulase gene regulators was mainly focused on the isolation of different transcriptional activators and repressors and has so far unraveled several positive (XYR1, ACE2, and HAP2/3/5) and negative (CRE1 and ACE1) regulators of T. reesei (hemi) cellulases. Although deletion of positive regulators has led to (partially) drastic reductions of (hemi)cellulase gene-expression levels, there are no conclusive studies available, which show that overproduction of one of the transcriptional activators or deletion of repressors alone will lead to groundbreaking improvements in terms of enzyme yields. The most promising results, with respect to their effect on cellulase gene expression, were obtained by the deletion of the general carbon catabolite repressor CRE1, which was also lost during mutagenesis in strain RUT-C30 (see section ‘Elucidating the roots of cellulase hyperproduction section’). Although some of the key players in the transcriptional regulation have now been identified, our understanding of the multiple cellular processes involved in cellulase production is still far from complete. A coordinated understanding of these processes will, however, be necessary to facilitate the identification of genetic targets for modification to improve strains. Transcription of the major cellulase genes is not only induced during growth on cellulose but also by a variety of disaccharides including lactose, cellobiose and sophorose (glycosyl-β-1,2-glucose) or the monosaccharide L-sorbose. Induction by these molecules is antagonized by the presence of readily available carbon sources including D-glucose or D-fructose, which can repress cellulase gene transcription directly by the binding of CRE1 to the promoter regions or indirectly by preventing the uptake of the inducer. Besides cellulose-based substrates, the dissaccharide lactose is used as an inducing carbon source in large-scale fermentations, especially for recombinant protein production. It accumulates as a waste product in cheese production and is therefore the only economically available soluble carbon source for T. reesei fermentations Citation[11]. More recently, the influence of different signal transduction pathways on cellulase gene expression in T. reesei was also addressed Citation[9,12].
The complexity of cellulase regulation makes it difficult to obtain a complete understanding of the whole process based on the knowledge obtained from single-gene studies and, on the other hand, offers innumerable possibilities for modifying different components of the cellular machinery responsible for cellulase production. The advent of the (post-)genomic age of T. reesei research provided us with a number of new high-throughput technologies to accelerate our understanding of the cellular mechanisms governing cellulase production. In addition, new tools were developed to facilitate strain improvement by classical genetics and genetic engineering approaches. Novel insights that are addressed in this review are:
▪ The genome sequence of T. reesei QM6a; | |||||
▪ Genomic and proteomic comparisons of hyperproducing strains; | |||||
▪ Generation of optimized enzyme mixtures; | |||||
▪ Combining advantageous strain properties by sexual crossings; | |||||
▪ Enhancing gene targeting and knockout efficiency in fungal transformations. | |||||
Lessons from the T. reesei genome
The first genome-wide approaches to studying cellulase gene expression in T. reesei were based on cDNA sequencing and cDNA subtraction Citation[13–16]. Although these studies revealed interesting new facts about cellulase regulation, they also highlighted that there were still many unknown players involved in cellulosic biomass degradation in T. reesei and that even our knowledge about the secreted enzymes and proteins involved in this process was far from complete. Foreman et al. generated a cDNA library and subsequently used microarray analysis to study the expression levels of the obtained sequences under different conditions known to induce cellulolytic and hemicellulolytic enzyme synthesis Citation[15]. They found 12 genes encoding new enzymes involved in biomass degradation including, amongst others, three so far unknown endoglucanases (CEL5B, CEL74A and CEL61B), five β-glucosidases (CEL1B, CEL3B, CEL3C, CEL3D and CEL3E; numbers in protein names of endoglucanases and β-glucosidases correspond to the respective glycoside hydrolase family), and two secreted proteins solely containing carbohydrate-binding domains (CIP1 and CIP2). Interestingly, genes whose products are involved in extracellular protein processing and secretion were not highly expressed during cellulase induction. In the same study, gene-expression patterns of QM6a and RL-P37, a strain derived from QM6a and selected for its increased cellulase production levels, were compared. Different sets of genes were identified that were either down- or upregulated during induction in RL-P37 relative to QM6a. They represent potentially important candidates for cellulase gene regulation, but despite similarities to sugar transporters, for example, many of these sequences could not be assigned a function due to the limited sequence information available at that time.
This already illustrated that in order to fully comprehend the cellulase machinery of T. reesei, an indispensable step was to sequence the whole genome of T. reesei. Strain QM6a was therefore shotgun sequenced by the US Department of Energy Joint Genome Institute and the genome sequence was published in 2008 Citation[17,103]. Analysis of the T. reesei genome sequence enabled us to gain a complete picture of its cellulolytic potential, capacity for protein secretion and its regulatory machinery. The results from the analysis of the approximately 34 Mb genome came with a few surprises and finally opened the doors to whole-genome-based genomic and proteomic approaches.
Unexpectedly, considering that T. reesei is an efficient (hemi)cellulase producer, its genome shows an under-representation of genes encoding specific groups of glycoside hydrolases (GHs)Citation[101,102] involved in plant cell-wall degradation when compared with the genomes of other fungi. Among the analyzed fungal genomes, T. reesei had the fewest cellulases (seven total, two exoglucanases and five endoglucanases) and the smallest set of hemicellulases and pectin degrading enzymes. Genome analysis also provided little mechanistic insight into the reasons for its extraordinary capacity for protein secretion. So, what makes T. reesei such an efficient cellulolytic fungus? One apparent feature, and certainly one of the highlights of the analysis of the genome was the finding that 46% of the GH-encoding genes in the genome, including the main and important cellulases cellobiohydrolase 1 (CBH1/CEL6A) and cellobiohydrolase 2 (CBH2/CEL7A), were nonrandomly distributed in the genome and were found in distinct clusters, which were in gaps of synteny in comparison to other closely related ascomycetes. In such nonsyntenic regions, typically species-specific characteristics can be found. Analysis of the Aspergillus oryzae genome revealed that these blocks are enriched for genes involved in metabolism, particularly those for the synthesis of secondary metabolites Citation[18]. In T. reesei, analysis of the GHs found in these nonsyntenic clusters showed that they are usually from different GH families and only a few paralogs were found, which indicates that gene relocation rather than duplication was mainly responsible for cluster formation. It was conspicuous that these clusters also contained a relatively high number of genes encoding proteins involved in secondary metabolism. One obvious conclusion from this gene clustering – although missing experimental evidence as yet – would be that T. reesei is able to regulate GHs and secondary metabolites in a coordinate manner. As a consequence, T. reesei could possibly be able to fend off competition for nutrients by producing toxic substances in its natural environment while decomposing the plant material. In contrast to A. oryzae, where these clustering are species specific, many of these clusters are also present in the two other sequenced Trichoderma species Trichoderma atroviride and Trichoderma virens [Seiboth B, Seidl V, Unpublished Data]. The T. reesei genome opened a whole new roadmap for constructing improved T. reesei strains for industrial applications and shed light onto completely new aspects of this field, which will help to identify bottlenecks to make T. reesei strains even more efficient. It has been long known that cellulases and hemicellulases are expressed in a coordinate manner Citation[8,15] and the question arises if and how these GH/secondary metabolite clusters could be coordinately regulated and if their accessibility on a transcriptional level could be enhanced even further? One possibility would be that the regulation is controlled by wide domain regulators, which would in turn control the accessibility of these regions by chromatin remodeling for more specific gene regulators, such as the cellulase- and xylanase-regulator XYR1. The influence of the location of genes on their expression levels relative to these clusters should be considered in future studies when new or altered genes are inserted in the T. reesei genome. These issues can now be addressed with genome-wide high-throughput approaches on the transcriptional and protein level.
Elucidating the roots of cellulase hyperproduction
One approach to better understand the biology underlying cellulase hyperproduction is the analysis of improved producer strains derived from classical mutagenesis. In the last years three different mutations were uncovered in T. reesei RUT-C30. The first was a truncation of the cre1 gene, the key transcription factor for carbon catabolite repression Citation[19]. The strain selection procedure for RUT-C30 had included growth on 2-deoxyglucose to eliminate carbon catabolite repression and it is therefore not surprising that the cre1 truncation had arisen during this round of mutagenesis Citation[20]. It was recently shown that the deletion of either the complete cre1 gene or replacing it with the truncated version that was found in RUT-C30 had largely the same physiological effects, indicating that the truncated cre1 gene is practically a null allele Citation[21]. Such a nonfunctional cre1 gene results in a drastically altered phenotype with smaller colonies and fewer aerial hyphae and spores. Furthermore, cre1 deleted or truncated strains did not only show a derepression of cellulase and hemicellulase genes in a medium containing glucose, but they also produced significantly elevated levels of these enzymes under inducing conditions. This suggests that cre1 acts as a modulator of (hemi)cellulase gene expression under both noninducing and inducing conditions. The second mutation that was detected in RUT-C30 was a frameshift mutation in the gene gls2α, encoding the glucosidase α-subunit, resulting in a truncated gene product Citation[22]. This subunit is the catalytic part of the glucosidase II heterodimeric enzyme which is involved in the structural modification of N-linked oligosaccharides present on glycoproteins and the quality control of polypeptides within the endoplasmic reticulum. Replacement with the wild-type allele seemed to decrease protein secretion. The third mutation that was detected in RUT-C30 was a large genomic lesion of 85 kb, affecting 29 genes Citation[20], including transcription factors, enzymes of the primary metabolism. Some of the genes lacking in RUT-C30 could be correlated with pronounced alterations in its phenotype, such as poor growth on α-linked oligosaccharides. However, this mutation was shown to be unrelated to the cre1 locus and was found to be already present in NG14, the ancestor of RUT-C30. With the availability of the genome sequence and high-throughput methods, such as massively parallel DNA sequencing, it was recently possible to identify many additional mutations found in the mutant line consisting of NG-14 and RUT-C30 (see section titled T. reesei strains: past and present) in comparison to QM6a Citation[23]. The authors found in addition to the above described mutations a surprisingly high number of mutagenic events: 223 single nucleotide variants (SNVs), 15 small deletions and insertions, and 18 larger deletions, leading to the loss of more than 100 kb of genomic DNA. These SNVs resulted in mutations in more than 50 genes, which are mainly involved in nuclear transport, transcription and mRNA stability, and in protein secretion and vacuolar targeting. In both strains, NG14 and RUT-C30, a number of genes belonging to these categories were mutated. Interestingly, with the exception of CRE1, none of the above described transcriptional regulators were affected by a mutagenic event. These results highlight additional mechanisms of possibly major importance for cellulase hyperproduction, but also points out the complexity of the problem to distinguish which mutations are detrimental, neutral or beneficial for cellulase production. It also becomes clear by these results that many mutations and not just one or two clear-cut modifications are probably necessary to make a significantly improved cellulase-producer strain. In addition, the carbon source assimilation profiles of the investigated strains were also measured and in contrast to its importance as an alternative inducer of cellulase formation, the utilization of lactose and its constituent D-galactose decreased with increasing cellulase production. This suggests that the rate-limiting steps in cellulase induction by cellulose might be different from lactose or induction by the latter is indirectly correlated with its utilization as a carbon source. Based on the results from this genomic study, new hypotheses about the mechanisms underlying cellulase and hemicellulase production and secretion in T. reesei including novel areas, such as nuclear transport, mRNA turnover and vacuolar protein trafficking, can be tested to enable directed strain improvement for even further improved enzyme yields.
As a complementary tool to genomics, proteomic approaches can now, having the annotated genome available, be employed more efficiently to study the differences between hyperproducing strains. A comparison of the secretomes of T. reesei RUT-C30 with the industrial hypersecretory strain CL847 revealed different enzyme cocktail compositions Citation[24]. While CL847 has a more diversified secretome and seems to be a general hypersecretory strain, the secretome of RUT-C30 is more cellulase oriented.
Optimizing the enzyme mixture
To obtain an optimal degradation of cellulosic biomass, different aspects of the enzyme mixture need to be optimized and adapted to the respective substrate. One aspect is the improvement of the catalytic efficiency of individual enzymes on insoluble substrates and their thermal tolerance. Respective engineering approaches include rational design, based on increasing knowledge regarding the substrate binding and catalytic mechanisms of cellulases, and directed evolution, based on random mutagenesis and/or molecular recombination followed by screening for improved enzymes Citation[25]. Another aspect is the optimal combination of enzymes with different catalytic properties and addition of carbohydrate-binding proteins that make the substrate more accessible. It has long been recognized that an efficient cellulase system requires sufficient β-glucosidase to hydrolyze cellobiose produced by the action of the exoglucanases CBH1 and CBH2, to prevent product inhibition Citation[26]. However, the generation of more complex enzyme mixtures is probably necessary to obtain the desired several-fold improvement of cellulose degradation. In nature, biomass decomposition is achieved by the synergistic action of enzymes from different microbes and, therefore, in addition to optimizing the composition of the cocktail of available T. reesei enzymes Citation[27], a promising approach is the screening of the hydrolytic enzyme mixtures of other microorganisms for novel, synergistically acting components to improve the T. reesei enzyme mixture. The large collection of bacterial and fungal genomes of the US Department of Energy Joint Genome Institute Citation[104] is a valuable resource for detecting novel proteins with properties that are possibly beneficial for the T. reesei enzyme mixture. Potential targets are hemicellulases and pectinases, considering the relative paucity of several enzyme groups in the T. reesei genome.
Another interesting group of extracellular proteins that could possibly aid in enhancing biomass degradation efficiency of T. reesei are accessory proteins. These secreted proteins consist solely of carbohydrate-binding domains. Recently, accessory proteins gained attention as potential facilitators of carbohydrate polymer degradation because they increase the substrate accessibility of enzymes by loosening up the rigid structure of the biomass fibers Citation[28]. In T. reesei the carbohydrate-binding proteins CIP1 and CIP2 have already been reported to be expressed under cellulase-inducing conditions Citation[15]. These two proteins consist of signal peptides, targeting them to the secretory pathway, and a cellulose-binding domain, but no catalytic domains could be annotated. Swollenin (SWO1) is another protein that has been reported to be involved in loosening up of the cell wall Citation[29]. It consists of a cellulose-binding domain and a domain similar to expansins, which are proteins with a cell wall loosening action in plants. The T. reesei genome contains three further genes encoding proteins with expansin-like domains, but no strong upregulation under cellulase-inducing conditions of the respective genes was found Citation[30]. Furthermore, some members of family GH61 possibly also possess carbohydrate-binding functions but no catalytic activity. While CEL61A T. reesei has a cellulose-binding domain and endoglucanase activities have been reported for this protein, CEL61B has no cellulose-binding domain and only 49% sequence identity to CEL61A, although they are both members of family GH61. The gene encoding CEL61B was shown to be expressed under cellulase-inducing conditions Citation[15]. Interestingly, addition of a family GH61 protein from Thielavia terrestris also significantly enhanced the activity of T. reesei cellulases in synergism assays Citation[1]. Structural analysis of CEL61B showed no easily identifiable carbohydrate-binding cleft, pocket or catalytic center of the types normally seen in GHs Citation[31]. Structural comparison revealed that the protein most similar to CEL61B with a known structure is CBP21 from the Gram-negative soil bacterium Serratia marcescens. CBP21 is a chitin-binding protein and a member of the carbohydrate-binding module family 33. It has been shown to enhance chitinase activities on insoluble substrates Citation[32]. Due to the obvious limitations of substrate accessibility in plant cell walls, carbohydrate-binding and accessory proteins will therefore undoubtedly play an important role for enzyme cocktail optimization.
To study this important aspect of substrate accessibility, the penetration of T. reesei cellulases into the plant cell walls was recently visualized in a study by immune transmission electron microscopy Citation[33]. The effect of different dilute acid pretreatments on altering the condensed ultrastructure of biomass cell walls and on cellulose accessibility was evaluated and it was found that more severe pretreatments enhanced enzyme penetration. The authors could also show that not all available surfaces were equally accessible for enzymes, reflected by the finding that some delamination surfaces did not display bound enzyme. Furthermore, the progress of enzymes through the cell walls was in the form of a planar front rather than by ‘drilling down’ in focused regions. This might be due to biomass recalcitrance, which makes it more resistant to the focused pinhole-type attack that is thought to occur during the enzymatic digestion of starch granules Citation[34].
Different pretreatment methods also need to be considered when enzyme cocktails are adjusted to various substrates. Acid pretreatments will hydrolyze the majority of the (hemi)celluloses, largely leaving cellulose and lignin, whereas alkali pretreated biomass contains more (hemi)celluloses Citation[2]. It has been shown that during acid pretreatments the lignin sheath surrounding cellulose fibrils coalesces extensively and migrates within and out of the cell wall, leading to lignin relocalization Citation[35]. The lignin content of the plant biomass influences the yields for bioethanol production directly as it cannot be converted to fermentable sugars and indirectly as it influences the accessibility of cellulases and hemicellulases to their substrate. Microbial lignin degradation is not yet well understood. White-rot fungi produce a complex ligninolytic system that rely on oxidative enzymes such as peroxidases and laccases, and are able to degrade lignin to CO2 and H2O. They accomplish this via Fenton chemistry, involving hydroxyl radicals. In contrast to white-rot fungi, brown-rot fungi modify lignin only weakly but depolymerize only cellulose and (hemi)cellulose Citation[36]. Analysis of genome sequence of the brown-rot fungus Postia placenta in combination with transcriptomics and proteomics demonstrated that it has an unusual mechanism of cellulose conversion. It has only two putative endoglucanases, but a number of oxidative enzymes, suggesting that hydroxyl radicals generated via Fenton reactions are capable of depolymerizing cellulose Citation[36]. It remains to be elucidated whether such enzyme systems are compatible with T. reesei, which modifies lignin only weakly or if T. reesei itself possesses such properties that can be enhanced by genetic engineering. Fungal oxidoreductases potentially involved in lignin catabolism are compiled in the Fungal Oxidative Lignin enzymes (FOLy) database Citation[37,105].
Tools for genetic engineering
Expression studies and comparative genomics of different cellulase-producing strains have provided us with large lists of genes whose implications on the production of cellulases deserve further attention. Although some gene functions might be predictable from the identification of well-characterized orthologs, the function of a large fraction of the newly discovered genes remains elusive, especially since a high number of these proteins lack any significant similarity to other gene products from public databases. Therefore, advanced molecular biology tools to assign gene functions are necessary. Gene function can be addressed by various methods, but provided that adequate screening procedures for phenotypes are available, gene deletion has become the method of choice in elucidating gene functions. These studies depend on the construction of defined recombinant strains by an efficient gene targeting system. Genetic manipulation of many industrially applied fungal strains is restricted due to the absence of sexual propagation (see section ‘A long missing link: sexual crossing of T. reesei strains’). The lack of research with the sexual form prevented characterization of auxotrophic mutants, and therefore transformation strategies that involve the conversion of auxotrophic mutants to prototrophy are only poorly developed in T. reesei, while well established in sexually propagating fungi including Aspergillus nidulans or Neurospora crassa.
In T. reesei, DNA-mediated transformation relies on a small number of dominant and auxotrophic markers, which generally limits the number of genetic manipulations. To circumvent this problem, transformation systems independent of the number of available markers were recently developed that are based on the removal of the marker gene after each transformation step. A system that makes use of recyclable markers is the blaster cassette system, which is dependent on marker genes that can be bidirectionally selected. One such example is the T. reeseipyr4 (encoding orotidine 5´-phosphate carboxylase), which confers uracil/uridine prototrophy whereas its absence results in resistance to 5-fluoroorotic acid (5-FOA). Therefore, transformants where the blaster cassette was integrated into their genomic DNA are selected via pyr4 function, in that they grow on medium that is not supplemented with uridine anymore. Excision of the pyr4 marker is then forced in the presence of 5-FOA, which is toxic for the fungus in the presence of the pyr4 gene product . To facilitate the removal of the pyr4 blaster cassette, the marker gene is located between two direct repeats, which lead to an enhanced excision by homologous recombination between them. As a consequence, this blaster cassette can be reused for multiple rounds of gene deletions as exemplified by the deletion of the gluco- and hexokinase encoding genes Citation[38].
An alternative strategy for marker recycling is induced recombination, which is illustrated here by the bacteriophage P1 bipartite Cre/loxP recombination system Citation[39]. The system consists of the site-specific DNA recombinase Cre, which is able to catalyze the recombination of DNA between specific loxP sites. These 34 bp DNA sequences contain specific binding sites for Cre that surround a directional core sequence where recombination can occur. The Cre recombinase is transiently expressed by placing it under an inducible promoter. The genetic marker is flanked similar to the blaster cassette by loxP sites at either side. After successful gene deletion and selection of transformants due to the used marker gene, the Cre recombinase expression is induced and the marker gene will be eliminated by Cre-catalyzed recombination between the loxP sites. Consequently, the marker gene is looped out and can be reused in another round of transformation. This system has been shown to function in Saccharomyces cerevisiaeCitation[40] and higher eukaryotes and can also be applied to different Trichoderma spp. [Zach, V Seidl, Unpublished Data].
Besides a number of auxotrophic markers, other dominant systems for selection and counterselection are now available and were tested for Aspergillus fumigatus, for example Citation[41]. These dominant systems are based on antibiotic resistance genes from other organisms as selection markers, which might be useful for excluding effects that are based on only partial complementation of genetic lesions of auxotrophic marker genes.
Gene deletions on a large-scale basis have proven to be difficult since many fungi exhibit a low frequency of homologous recombination (HR). This homologous integration frequency is usually found to be in the range of 10% in T. reesei depending on the locus, the genetic marker and the flanking regions Citation[42]. Therefore, a large number of transformants have to be screened to identify strains in which gene deletion has actually occurred. The reason for this low frequency is that many organisms seem to preferentially use the nonhomologous endjoining (NHEJ) pathway in double-strand break repair (DSB). To improve homologous recombination frequencies it is necessary to eliminate ectopic (random) integration of the deletion cassette. This can be achieved either by improving HR or by impairing NHEJ. Today, impairment of the NHEJ seems to be the method of choice. The NHEJ process is mediated by different proteins including the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), the Ku70/Ku80 heterodimer and the DNA ligase IV–XRCC4 complex Citation[43,44]. Thereby the free DNA ends are bound by the Ku proteins, which in turn recruit the other proteins involved in NHEJ repair. In filamentous fungi the first study came from N. crassa, where ku-disruption strains exhibited integration at the homologous site close to 100% compared with 10 to 30% for a wild-type recipient Citation[45]. Ku-deleted strains are now available from various fungi Citation[41]. All these fungi show improved gene targeting and allow gene-knockout studies on a high-throughput basis Citation[46,106]. For T. reeseitku70 deleted strains the efficiency of gene targeting for two tested genes was higher than 95% when 1 kb homologous flanking regions were used for the deletion Citation[42].
Although this system represents a considerable breakthrough for the study of gene functions in filamentous fungi, long-term studies that deal with the potential side effects of the impairment of the NHEJ repair system on genome stability and integrity are not available. To minimize the potentially deleterious effects of this genetic lesion it can be crossed out in sexually propagating fungi or removed by retransformation of the ku70 deleted strains, thereby taking advantage of the high homologous recombination frequency, or by the construction of transiently inactivated ku70 genes Citation[47].
A long missing link: sexual crossing of T. reesei strains
Sexual crossing approaches yield considerable advantages for strain improvement as a fast and efficient means to combine different strain properties, but – similar to other industrially applied filamentous fungi – classical genetic approaches using sexual crossings were not available for a long time in T. reesei. The genus Trichoderma/Hypocrea contains several hundred species, some of which only occur as teleomorphs (i.e., in their sexual form), whereas others have so far only been observed as asexually propagating anamorphs Citation[48]. Using molecular characters (sequence analyses of the nuclear ribosomal DNA region and randomly amplified polymorphic DNA fingerprinting), Kuhls et al. found that T. reesei is indistinguishable from the pantropical ascomycete Hypocrea jecorina and suggested that the latter species is its teleomorph Citation[49]. However, despite this in silico evidence, initial attempts to cross T. reesei with wild-type strains of H. jecorina failed, giving rise to the assumption that T. reesei QM6a would be an asexual clonal lineage of H. jecorina.
In a recent study Seidl et al. reported that based on its genome sequence T. reesei QM6a has a MAT1–2 mating type locus, indicating that it is a heterothallic species and, consequently, that a strain of the opposite mating type would be required for successful sexual reproduction Citation[50]. The respective counterpart, a strain with a MAT1–1 mating-type locus, was obtained and characterized from a natural isolate of H. jecorina that was known to mate under laboratory conditions. The purified strain could be successfully crossed with T. reesei QM6a and notably also with several cellulase hyperproducing mutants including RUT-C30, thereby showing for the first time that T. reesei QM6a is not completely asexual . This discovery now enables the combination of different properties of strains by sexual crossing, thereby strongly accelerating strain improvement. Further analysis showed that QM6a and its derived strains can only act as the male mating partner: when the MAT1–2 locus was exchanged for MAT1–1 in the QM6a strain by genetic engineering, no formation of fruiting bodies was observed upon crossing with the QM6a wild-type strain ( MAT1–2). This suggested that QM6a is unable to form fruiting bodies and is thus female sterile. By contrast, when this inversion of the mating type locus was performed in the H. jecorina wild-type isolate, the strains could be crossed. Genome analysis of genes with a reported role in sexual development in other fungi did not reveal any obvious candidates to explain the female sterility of QM6a. For identification of altered loci in the QM6a isolate and to consequently overcome its deficiency of female sterility by genetic engineering, a comparison of the complete genome sequence of QM6a with that of the fertile H. jecorina strains will be necessary. However, in the meantime, to overcome this obstacle for strain improvement of industrial strains, the progeny from crossings of H. jecorina strains with a MAT1–1 locus with industrial QM6a-derived strains could be screened for the capability of sexual development and retention of enzyme production efficiency. At the same time, elimination of unintended random mutations that occurred during mutagenesis in the industrial mutants detrimental to, for example, growth, by complementation with the wild-type background can be beneficial to improve the performance of these strains in industrial fermentations.
Future perspective
With the sequencing of T. reesei QM6a the genomic age has opened a new toolbox for strain improvement strategies. A number of so far unknown or disregarded aspects now need to be evaluated. There are two main areas that need to be improved: further increasing enzyme expression levels and increasing the efficiency of the enzyme mixture. With respect to increasing enzyme-expression levels the regulation of the gene clusters that contain polysaccharide degrading enzymes needs further attention. When new genes are introduced, placing them into these clusters in the vicinity of cellulases, for example, with high-expression levels might turn out to be beneficial. Increasing our understanding of cellulase regulation on the transcriptional level and considering the novel aspects of mRNA stability and nuclear transport is a promising approach with respect to industrial strain improvement. A better understanding of cellulase regulation as a whole might also lead to strains that produce cellulases that are independent of inducers. In regard to increasing the efficiency of the enzyme mixture, the accessibility of the substrate is a key issue, which can be improved by making use of other (fungal) systems and proteins, particularly oxidative mechanisms, such as proteins that are able to disintegrate lignin and carbohydrate accessory proteins that improve cellulase efficiency.
Cellulose (∼50%), (hemi)cellulose (∼25%), lignin (∼25%)
Fungal reference strain isolated from the Solomon islands from which all industrial, currently used cellulase-producing strains are derived
Generation of stable changes in the genome of an organism. Classical mutagenesis comprises physical causes (UV light) and chemicals, while modern methods involve genetic engineering
Globally acting control system that allows organisms to adapt to their preferred (rapidly metabolizable) carbon and energy source by inhibition of enzymes involved in their metabolism. When glucose or other readily available monosaccharides whose catabolism provides a high yield of ATP are present, gene expression of carbohydrate-degrading enzymes or other metabolic pathways are usually repressed
Modification of the genome of an organism by directly manipulating the DNA sequence of specific genes (e.g., coding for a glycoside hydrolase), in order to obtain desired traits (e.g., enhanced production of enzymes)
Alteration of genetic and regulatory processes within cells to increase the production of a selected substance by improving the flux through the product-forming pathway and reducing by-product formation and the cellular energy use
Ascomycota are homothallic when no partner is required for sexual reproduction and heterothallic when a strain of an opposite mating type is required for sexual reproduction. In heterothallic species both mating types can act as the male or female reproduction partner
Enzymes which hydrolyse glycosidic bonds. A glycosidic bond is formed when the anomeric OH-group of a carbohydrate reacts with the alcohol-group of another molecule. In the CAZy (Carbohydrate Active EnZYmes) classification GHs are grouped into families based on amino acid sequence similarities
Gene loci on the same chromosome, regardless of whether they are genetically linked by classic linkage analysis or not. Today, the termis also used in genomics also used to refer to gene loci in different organisms located on a chromosomal region of common evolutionary ancestry. It describes the precise preservation of the order of genes on a chromosome of related species. During species evolution, rearrangements within the genome may separate two loci, resulting in the loss of synteny
Domain-Eukaryota, Kingdom (five total)-Fungi, Phylum (seven total)-Ascomycetes (Ascomycota)
Degrade lignin, cellulose and hemicellulose
Degrade only cellulose and hemicellulose
Degrade cellulose and hemicellulose, modify lignin only weakly
Trichoderma reesei strains: past & present
▪ Strain improvement strategies in the 1970s and 1980s, based on random mutagenesis, resulted in cellulase hyperproducing strains, such as RUT-C30. | |||||
▪ Using recombinant DNA methods a number of positive and negative transcriptional regulators of cellulase gene expression were identified, in single-gene studies. | |||||
Lessons from the T. reesei genome
▪ T. reesei has surprisingly few cellulases (seven total) and also only small sets of hemicellulases and pectinases. | |||||
▪ Many of the polysaccharide degrading enzymes of T. reesei are organized in clusters in genomic regions that are not syntenic to closely related fungi, possibly explaining their efficient expression. | |||||
Elucidating the roots of cellulase hyperproduction
▪ Comparison of the genomes of the mutagenized strains RUT-C30 and its ancestor NG14 in comparison with the original isolate QM6a revealed a high number of mutagenic events. | |||||
▪ RUT-C30 has a truncated version of the carbon catabolite repressor CRE1 and a large genomic lesion of 85 kb. | |||||
▪ Additional mutations affect genes predominantly associated with nuclear transport, mRNA turnover and vacuolar protein trafficking, which opens new possibilities for directed strain improvement to improve enzyme yields. | |||||
Optimizing the enzyme mixture
▪ To improve biomass degradation efficiency, T. reesei enzyme mixtures need to be supplemented with synergistically acting proteins from other microorganisms and by improving the properties of the existing enzymes. | |||||
▪ This includes hydrolytic enzymes, as well as carbohydrate-binding proteins, which loosen up the plant cell walls and thereby enable better access for T. reesei cellulases. | |||||
▪ The large number of fungal and bacterial genomes available can supplement screening approaches for novel proteins and aid in finding optimal candidates. | |||||
Cool tools for genetic engineering techniques
▪ Blaster cassettes and inducible recombination systems (Cre/loxP) facilitate multiple genetic transformations. | |||||
▪ tku70 deleted strains show improved homologous recombination which allows gene replacement on a high-throughput basis. | |||||
A long missing link: sexual crossing of T. reesei strains
▪ Sexual recombination is a fast and simple way to combine beneficial strain properties. | |||||
▪ Hypocrea jecorina is the sexual form of T. reesei. ▪ T. reesei QM6a-derived strains have a MAT1–2 mating-type locus and need strains with the opposite mating type ( MAT1–1) for successful sexual reproduction. | |||||
▪ A strain with a MAT1–1 locus was purified from a H. jecorina wild-type isolate and could be crossed with QM6a under laboratory conditions. This opens new opportunities for industrial strain improvement using mating approaches. | |||||
Financial & competing interests disclosure
The author’s research is supported by the FWF (Austrian Science Fund) with grant P19421 to Bernhard Seiboth and P20559 and a Hertha-Firnberg grant (T390) to Verena Seidl. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Related Research Data
Bibliography
- Merino ST, Cherry J. Progress and challenges in enzyme development for biomass utilization. Adv. Biochem. Eng. Biotechnol.108,95–120 (2007).
- Himmel ME, Ding SY, Johnson DK et al. Biomass recalcitrance. engineering plants and enzymes for biofuels production. Science315(5813),804–807 (2007).
- Reese ET. History of the cellulase program at the US army Natick Development Center. Biotechnol. Bioeng. Symp.6,9–20 (1976).
- Durand H, Clanet M, Tiraby G. Genetic improvement of Trichoderma reesei for large scale cellulase production. Enzyme Microbiol. Technol.10,341–346 (1988).
- Eveleigh DE, Montenecourt BS. Increasing yields of extracellular enzymes. Adv. Appl. Microbiol.25,57–74 (1979).
- Cherry JR, Fidantsef AL. Directed evolution of industrial enzymes: an update. Curr. Opin. Biotechnol.14(4),438–443 (2003).
- Stephanopoulos G. Challenges in engineering microbes for biofuels production. Science315(5813),801–804 (2007).
- Aro N, Pakula T, Penttila M. Transcriptional regulation of plant cell wall degradation by filamentous fungi. FEMS Microbiol. Rev.29(4),719–739 (2005).
- Kubicek CP, Mikus M, Schuster A, Schmoll M, Seiboth B. Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol. Biofuels2,19 (2009).
- Stricker AR, Mach RL, De Graaff LH. Regulation of transcription of cellulases- and hemicellulases-encoding genes in Aspergillus niger and Hypocrea jecorina ( Trichoderma reesei). Appl. Microbiol. Biotechnol.78(2),211–220 (2008).
- Seiboth B, Pakdaman B, Hartl L, Kubicek CP. Lactose metabolism in filamentous fungi: how to deal with an unknown substrate. Fungal Biol. Rev.21,42–48 (2007).
- Schmoll M. The information highways of a biotechnological workhorse – signal transduction in Hypocrea jecorina. BMC Genomics9,430 (2008).
- Arvas M, Pakula T, Lanthaler K et al. Common features and interesting differences in transcriptional responses to secretion stress in the fungi Trichoderma reesei and Saccharomyces cerevisiae. BMC Genomics7,32 (2006).
- Chambergo FS, Bonaccorsi ED, Ferreira AJ et al. Elucidation of the metabolic fate of glucose in the filamentous fungus Trichoderma reesei using expressed sequence tag (EST) analysis and cDNA microarrays. J. Biol. Chem.277(16),13983–13988. (2002).
- Foreman PK, Brown D, Dankmeyer L et al. Transcriptional regulation of biomass-degrading enzymes in the filamentous fungus Trichoderma reesei. J. Biol. Chem.278(34),31988–31997 (2003).
- Schmoll M, Zeilinger S, Mach RL, Kubicek CP. Cloning of genes expressed early during cellulase induction in Hypocrea jecorina by a rapid subtraction hybridization approach. Fungal Genet. Biol.41(9),877–887 (2004).
- Martinez D, Berka RM, Henrissat B et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat. Biotechnol.26(5),553–560 (2008).
- Machida M, Asai K, Sano M et al. Genome sequencing and analysis of Aspergillus oryzae. Nature438(7071),1157–1161 (2005).
- Ilmen M, Thrane C, Penttilä M. The glucose repressor gene cre1 of Trichoderma: isolation and expression of a full-length and a truncated mutant form. Mol. Gen. Genet.251(4),451–460 (1996).
- Seidl V, Gamauf C, Druzhinina IS, Seiboth B, Hartl L, Kubicek CP. The Hypocrea jecorina ( Trichoderma reesei) hypercellulolytic mutant RUT C30 lacks a 85 kb (29 gene-encoding) region of the wild-type genome. BMC Genomics9,327 (2008).
- Nakari-Setälä T, Paloheimo M, Kallio J, Vehmaanpera J, Penttilä M, Saloheimo M. Genetic modification of carbon catabolite repression in Trichoderma reesei for improved protein production. Appl. Environ. Microbiol.75(14),4853–4860 (2009).
- Geysens S, Pakula T, Uusitalo J, Dewerte I, Penttila M, Contreras R. Cloning and characterization of the glucosidase II α subunit gene of Trichoderma reesei: a frameshift mutation results in the aberrant glycosylation profile of the hypercellulolytic strain Rut-C30. Appl. Environ. Microbiol.71(6),2910–2924 (2005).
- Le Crom S, Schackwitz W, Pennacchio L et al. Tracking the roots of cellulase hyperproduction by the fungus Trichoderma reesei using massively parallel DNA sequencing. Proc. Natl Acad. Sci. USA106(38),16151–16156 (2009).
- Herpoel-Gimbert I, Margeot A, Dolla A et al. Comparative secretome analyses of two Trichoderma reesei RUT-C30 and CL847 hypersecretory strains. Biotechnol. Biofuels1(1),18 (2008).
- Zhang PY-H, Himmel ME, Mielenz JR. Outlook for cellulase improvement: screening and selection strategies. Biotechnol. Adv.24,452–481 (2006).
- Sternberg D, Vijayakumar P, Reese ET. β-glucosidase: microbial production and effect on enzymatic hydrolysis of cellulose. Canad. J. Microbiol.23,139–147 (1977).
- Meyer AS, Rosgaard L, Sorensen HR. The minimal enzyme cocktail concept for biomass processing. J. Cereal Sci.50(3),337–344 (2009).
- Eijsink VG, Vaaje-Kolstad G, Varum KM, Horn SJ. Towards new enzymes for biofuels: lessons from chitinase research. Trends Biotechnol.26(5),228–235 (2008).
- Saloheimo M, Paloheimo M, Hakola S et al. Swollenin, a Trichoderma reesei protein with sequence similarity to the plant expansins, exhibits disruption activity on cellulosic materials. Eur. J. Biochem.269(17),4202–4211 (2002).
- Verbeke J, Coutinho P, Mathis H et al. Transcriptional profiling of cellulase and expansin-related genes in a hypercellulolytic Trichoderma reesei. Biotechnol. Lett.31(9),1399–1405 (2009).
- Karkehabadi S, Hansson H, Kim S, Piens K, Mitchinson C, Sandgren M. The first structure of a glycoside hydrolase family 61 member, Cel61B from Hypocrea jecorina, at 1.6 A resolution. J. Mol. Biol.383(1),144–154 (2008).
- Vaaje-Kolstad G, Houston DR, Riemen AH, Eijsink VG, van Aalten DM. Crystal structure and binding properties of the Serratia marcescens chitin-binding protein CBP21. J Biol. Chem.280(12),11313–11319 (2005).
- Donohoe BS, Selig MJ, Viamajala S, Vinzant TB, Adney WS, Himmel ME. Detecting cellulase penetration into corn stover cell walls by immuno-electron microscopy. Biotechnol. Bioeng.103(3),480–489 (2009).
- Sreenath HK. Studies on starch granules digestion by α-amylase. Starch44(2),61–63 (1992).
- Donohoe BS, Decker SR, Tucker MP, Himmel ME, Vinzant TB. Visualizing lignin coalescence and migration through maize cell walls following thermochemical pretreatment. Biotechnol. Bioeng.101(5),913–925 (2008).
- Martinez D, Challacombe J, Morgenstern I et al. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc. Natl Acad. Sci. USA106(6),1954–1959 (2009).
- Levasseur A, Piumi F, Coutinho PM et al. FOLy: an integrated database for the classification and functional annotation of fungal oxidoreductases potentially involved in the degradation of lignin and related aromatic compounds. Fungal Genet. Biol.45(5),638–645 (2008).
- Hartl L, Seiboth B. Sequential gene deletions in Hypocrea jecorina using a single blaster cassette. Curr. Genet.48(3),204–211 (2005).
- Thomson JG, Ow DW. Site-specific recombination systems for the genetic manipulation of eukaryotic genomes. Genesis44(10),465–476 (2006).
- Sauer B. Functional expression of the cre-lox site-specific recombination system in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol.7(6),2087–2096 (1987).
- Krappmann S. Gene targeting in filamentous fungi: the benefits of impaired repair. Fungal Biol. Rev.21,25–29 (2007).
- Guangtao Z, Hartl L, Schuster A et al. Gene targeting in a nonhomologous end joining deficient Hypocrea jecorina. J. Biotechnol.139(2),146–151 (2009).
- Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature412(6847),607–614 (2001).
- Critchlow SE, Jackson SP. DNA end-joining: from yeast to man. Trends Biochem. Sci.23(10),394–398 (1998).
- Ninomiya Y, Suzuki K, Ishii C, Inoue H. Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc. Natl Acad. Sci. USA101(33),12248–12253 (2004).
- Colot HV, Park G, Turner GE et al. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl Acad. Sci. USA103(27),10352–10357 (2006).
- Nielsen JB, Nielsen ML, Mortensen UH. Transient disruption of non-homologous end-joining facilitates targeted genome manipulations in the filamentous fungus Aspergillus nidulans. Fungal Genet. Biol.45(3),165–170 (2008).
- Druzhinina IS, Kopchinskiy AG, Kubicek CP. The first 100 Trichoderma species characterized by molecular data. Mycoscience47,55–64 (2006).
- Kuhls K, Lieckfeldt E, Samuels GJ et al. Molecular evidence that the asexual industrial fungus Trichoderma reesei is a clonal derivative of the ascomycete Hypocrea jecorina. Proc. Natl Acad. Sci. USA93(15),7755–7760 (1996).
- Seidl V, Seibel C, Kubicek CP, Schmoll M. Sexual development in the industrial workhorse Trichoderma reesei. Proc. Natl Acad. Sci. USA106(33),13909–13914 (2009).
▪ Websites
- SwissProt. ExPASy Proteomics Server – Enzyme Nomenclature Database www.expasy.org/enzyme/
- Carbohydrate Active Enzymes Database www.cazy.org
- Department of Energy Joint Genome Institute – Trichoderma reeseihttp://genome.jgi-psf.org/Trire2/Trire2.home.html
- Joint Genome Institute Genome Portal http://genome.jgi-psf.org/
- Fungal Oxidative Lignin Enzymes http://foly.esil.univ-mrs.fr
- Functional Analysis of a Model Filamentous Fungus www.dartmouth.edu/∼neurosporagenome/index.html