
Abstract
Aim: C1-INH-HAE is caused by activation of plasma kallikrein which subsequently cleaves high-molecular-weight kininogen (HMWK) to generate bradykinin and cHMWK. Materials & methods: A novel ion-pair 2D LC–MS/MS assay was developed to measure the 46 kDa cHMWK in plasma as a biomarker for C1-INH-HAE. The sample preparation included sodium dodecyl sulfate denaturation, methanol crash, chymotryptic digestion and peptide enrichment by solid phase extraction. Results: The LLOQ was 200 ng/ml. The overall cHMWK recovery combining crash and digestion was 57.5%. The precision of the method was ≤12.7% and accuracy ≤-13.8%. Conclusion: A reagent-free LC–MS assay has been developed for the quantitation of 46 kDa cHMWK, which was shown to be elevated in plasma of C1-INH-HAE patients due to C1-INH deficiency relative to that of healthy subjects.
Hereditary angioedema (HAE) due to a genetic deficiency in total or functional C1-INH (type I and type II C1-INH-HAE, respectively) leads to dysfunctional regulation of contact system activation and excess bradykinin generation. Type I C1-INH-HAE constitutes approximately 85% of these patients with the remainder being type II [Citation1]. C1-INH is a serpin that inhibits a number of proteases including the C1 complex in the classical complement pathway as well as contact system proteases, coagulation factor XIIa and plasma kallikrein [Citation2,Citation3]. While C1-INH-HAE type I or type II is attributed to mutations in one of the two C1-INH alleles, other forms of angioedema exist including HAE with normal C1-INH, recurrent idiopathic angioedema and acquired angioedema [Citation4–6]. Clinical symptoms of C1-INH-HAE include episodic swelling of subcutaneous tissues, bowel walls, upper airways and the recurrent cutaneous edema affecting the face, extremities, trunk and genitals [Citation7].
High-molecular-weight kininogen (HMWK) is a single-chain glycoprotein with a molecular weight of approximately 120 kDa and present in systemic circulation at a concentration of about 80 μg/ml and plays a central role in the activation of contact system [Citation8]. Recurrent activation of the contact system generates excess plasma kallikrein, a proteolytic enzyme that primarily cleaves HMWK between Lys380-Arg381 and Arg389-Ser390 residues to release the nine amino acid peptide bradykinin. The resulting cleaved kininogen (cHMWK) contains two chains linked together by an interchain disulfide bond, the heavy chain (66 kDa) containing domains 1–3 and the light chain (56 kDa) containing domains 5–6. The 56 kDa light chain in cHMWK (denoted 56 kDa cHMWK) can be further cleaved by plasma kallikrein at the N-terminal portion of the polypeptide to generate a 46 kDa light chain (denoted 46 kDa cHMWK) as the final cleavage product () [Citation9–12]. The bradykinin released from HMWK acts as a key mediator of pain, inflammation, edema and angiogenesis [Citation13]. Dysregulated plasma kallikrein proteolysis of HMWK can result in accumulation of bradykinin, which induces vasopermeability resulting in the edematous attacks associated with C1-INH-HAE [Citation12,Citation14–17]. Therapeutic intervention by inhibition of plasma kallikrein has led to acute and prophylactic treatments of C1-INH-HAE [Citation18–21].
Proteolytic cleavage by circulating kallikrein releases bradykinin (Arg381-Arg389, green), and the cHMWK that is composed of a heavy chain (Gln19-Lys380, blue) and a 56 kDa light chain (Ser390-Ser644, red) linked by an interchain disulfide bond (Cys28 and Cys614). Further cleavage of the 56 kDa light chain by kallikrein gives rise to a 46 kDa light chain (Lys438-Ser644) and exposure of a unique C-terminus with the SP generated following chymotrypsin digestion underlined from Lys438 to His445.
SP: Surrogate peptide.

Biomarkers of contact system activation, such as plasma cHMWK and bradykinin, can provide insight into the pathobiology of bradykinin-mediated angioedema and potentially improve diagnosis and patient management. Such biomarkers could also facilitate the evaluation of treatment efficacy [Citation21]. Previous reports using plasma biomarkers of contact system activation, including cHMWK, have shown that plasma kallikrein is generated during C1-INH-HAE attacks [Citation6,Citation9,Citation22–24]. As a result, there is considerable interest in developing sensitive and reliable assays to measure the level of cHMWK as a potential biomarker for C1-INH-HAE [Citation10,Citation24–25]. Due to the lack of appropriate immunoassay reagents to specifically detect cleaved products, western blotting has currently been utilized to measure the cHMWK concentration for a variety of angioedema conditions and correlate it with drug efficacy in the early stage of clinical study [Citation12,Citation18–20,Citation24]. It represents a challenge to validate the western assays since they are generally semiquantitative, variable and of low sample throughput. As such, an immunoassay for cHMWK based on the discovery of a proprietary antibody capable of selectively measuring cHMWK in the presence of high abundance of its precursor HMWK protein has recently been developed (manuscript in preparation).
LC–MS/MS has become an attractive technique to measure protein biomarkers in biological samples due to its superior selectivity and specificity [Citation26–33]. The LC–MS/MS approach quantifies a protein by selectively analyzing an appropriate surrogate peptide (SP) derived from the target analyte using multiple reaction monitoring (MRM). A great deal of progress has been made to enhance the specificity and sensitivity for the detection of low abundance proteins by reducing sample complexity through immunoprecipitation (IP) or multidimensional chromatographic fractionation at protein and/or peptide levels prior to LC–MS/MS [Citation34–36]. Recently, we described an online 2D-LC–MS/MS platform for therapeutic protein quantitation in the presence of antidrug antibodies [Citation37]. The orthogonal separation of peptides by the 2D-LC substantially enhances sensitivity of the assay, allowing for low-nanogram quantitation of protein therapeutics in clinical samples. We present here a novel ion-pair 2D-LC–MS/MS assay combined with specific pellet digestion using chymotrypsin to measure 46 kDa cHMWK in human plasma. The assay was reliable, robust and was validated following the regulatory guideline [Citation38]. We demonstrate that the 46 kDa cHMWK isoform is able to differentiate C1-INH-HAE patients and healthy individuals and could potentially be used as a disease biomarker after further investigation.
Materials & methods
Materials
The human cHMWK and HMWK (Uniprot version 186, accession number P01042; molecular weight ∼120 kDa; 95% pure) were purchased from Enzyme Research Laboratories (IN, USA). The proteins were supplied as a stock solution of 1.40 mg/ml (cHMWK) and 1.91 mg/ml (HMWK), respectively. SDS-PAGE gel of the reduced cHMWK confirmed that the 46 kDa fragment was the predominant light chain, whereas 56 kDa cHMWK isoform was minimally present in the commercial preparation. Tosyl-L-lysine chlorometyl ketone (TLCK)-treated chymotrypsin was from Sigma-Aldrich (MO, USA). Lys-C was purchased from Wako (Osaka, Japan). Mass spec grade proteinases Asp-N and Glu-C were obtained from Promega (WI, USA). Dithiothreitol (DTT), iodoacetamide (IAA), ammonium bicarbonate (ABC), ammonium hydroxide, urea, formic acid (FA), trifluoroacetic acid (TFA), heptafluorobutyric acid (HFBA), HPLC grade methanol (CH3OH) and acetonitrile (CH3CN), sodium dodecyl sulfate (SDS) solution (10%) and phosphate-buffered saline (PBS) were all purchased from Sigma-Aldrich (MO, USA). The MCX 96-well plate (30 μm, 10 mg) was obtained from Waters (MA, USA). The unlabeled SP, KHNLGHGH and 13C-labeled internal standard (IS), KHNL[13C6]GHGH, were custom-synthesized at New England Peptide (MA, USA) (>95% purity). A 10× protease inhibitor cocktail (SCAT169) solution was prepared in house and contains 100 mM benzamidine, 400 μg/ml polybrene, 2 mg/ml soybean trypsin inhibitor, 20 mM EDTA, 263 μM leupeptin, 20 mM AEBSF, 100 mM trisodium citrate and 2% dextrose. Evacuated, plastic blood collection tubes (SCAT169–4.5/5) containing the above 10× protease inhibitor cocktail were provided by Haematologic Technologies Inc. (VT, USA). Control human plasma collected in SCAT169 tubes was obtained from Bioreclamation Inc. (NY, USA).
SP selection
To probe the appropriate SP to distinguish cHMWK and HMWK, the two proteins were denatured in 8 M urea, reduced with 5 mM DTT at 37°C for 60 min, alkylated with 5 mM IAA for 30 min at ambient temperature and subsequently digested overnight at 37°C by chymotrypsin (1:50 enzyme/substrate [E/S]). The resulting peptide mixture was analyzed on an Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, MA, USA). The instrument was operated in the data-dependent acquisition mode to collect MS and MS/MS spectra of the peptides with ion intensities exceeding the predefined threshold (Automated Gain Control target 106). The mass spectra were searched against the human HMWK sequence database (Uniprot version 186) using Proteome Discoverer software (Thermo Scientific) to identify peptides and calculate their relative ion intensities.
2D-LC–MS/MS conditions
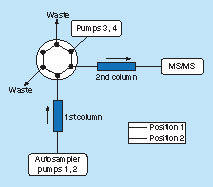
The 2D-LC–MS/MS system include a Waters UPLC as the first dimension (Waters, MA, USA) connected to an Agilent 1260 Infinity UHPLC (Agilent, Santa Clara, CA, USA) as the second dimension through a six-port, two-position valve built into a QTrap6500 mass spectrometer (Sciex, ON, Canada) with a TurboIonspray source (Sciex) utilizing a positive-ion MRM mode. The first-dimension and second-dimension chromatography utilized the same type of Agilent Metasil AQ C18 Column (Agilent, 2.0 mm × 100 mm, 5 μm; CA, USA) but different gradient elution conditions (). For the first dimension, mobile phase A (0.1% TFA) and mobile phase B (0.1% TFA in methanol) were operated at a flow rate of 0.4 ml/min with the following gradient: 0 min/9.0% B, 7.95 min/9.0% B, 8.0 min/75.0% B, 12.9 min/75.0% B, 13.0 min/9.0% B and 15.0 min/9.0% B. For the second dimension, mobile phase A (0.1% TFA) and mobile phase B (0.1% TFA in methanol) were operated at a flow rate of 0.35 ml/min with a gradient: 0 min/8.0% B, 7.6 min/8.0% B, 13.0 min/45.0% B, 13.5 min/70.0% B, 14.8 min/70.0% B, 14.9 min/8.0% B and 15.0 min/8.0% B. Column switch was achieved by the six-port valve, which was placed initially in position 1 during the sample injection and gradient elution of the first LC at which the column eluent was diverted to waste. The LC fraction containing the SP and IS, eluting between 6.0 and 7.3 min from the first column, was directed to the second column by switching the valve to position 2 at 6.0 min. Shortly after complete elution of the analyte from the first column and transfer to the second column, the valve was switched back to position 1 at 7.3 min, and gradient elution of the trapped peptides from the second column began. All of the original data acquisition and process were performed with Sciex Analyst software, version 1.6.3.
The six-port valve was placed initially in position 1 during the sample injection at which the column eluent was diverted to waste and peptides were gradient eluted from the first column through pumps 1 and 2. The valve was switched to position 2 at 6.0 min, and the LC fraction containing the SP and IS was directed to the second column. Shortly after complete elution of the analyte from the first column and transfer to the second column, the valve was switched back to position 1 at 7.3 min. The trapped peptides were eluted from the second column by pumps 3 and 4, and were subsequently analyzed by mass spectrometry.
IS: Internal standard; SP: Surrogate peptide.
The system was operated in a positive ion MRM mode, and the transition ions were optimized as m/z 450.2 → 634.3 for cHMWK SP (KHNLGHGH) and 453.1 → 640.6 for IS (KHNL[13C6]GHGH) in terms of the formation of the most abundant product ion. The detailed MRM parameters utilized in the QTrap6500 system were set as follows: collision energy of 25 eV, collision exit potential of 15 V, declustering potential of 60 V, entrance potential of 10 V, curtain gas of 20 units, ion source gas 1 (GS1) of 50 units, ion source gas 2 (GS2) of 45 units, collision-activated dissociation gas setting medium, dwell time 100 ms, ion spray voltage of 5000 V and temperature of 550°C.
Preparation of calibration standards & quality control samples
The calibration standards were prepared by spiking appropriate amounts of the cHMWK stock solution or subsequent calibration standards to beagle plasma containing 10% (v/v) of SCAT169 solution (10×) as a surrogate matrix. Nominal concentrations of the calibrators were 200, 400, 1000, 2500, 10,000, 15,000, 18,000 and 20,000 ng/ml, respectively. As a standard practice for validation of assay accuracy and precision, ten individual lots of human plasma (sodium citrate) from healthy subjects (collected and stored without treatment with SCAT 169) were pooled, and were immediately treated with the 10% (v/v) SCAT169 solution prior to the preparation of the quality control (QC) samples. The endogenous level (EL) of cHMWK in the pooled healthy plasma was 2080 ng/ml, which was significantly higher than healthy plasma treated with SCAT 169 during sample collection and obviously due to the kallikrein-induced cleavage during sample collection and storage. The above pooled plasma sample was only used for the assay validation. It is difficult to distinguish EL from lower limit of quantitation (LLOQ) in regard to the high EL samples. The endogenous cHMWK concentration was taken into consideration during the calculation of cHMWK concentration of QC samples. The similar procedure was also utilized for the preparation of QC samples at five concentration levels, denoted as EL, LLQC (EL + 200 ng/ml), low quality control (LQC; EL + 600 ng/ml), middle quality control (MQC; EL + 5000 ng/ml) and high quality control (HQC; EL + 15,000 ng/ml), covering the intended calibration curve range. Stock solution of IS at 10 μg/ml was prepared in 0.1% FA/acetonitrile 1:1 (v/v) and stored in polypropylene vials at 4°C. The IS working solution (30 ng/ml) in 100 mM ABC buffer was freshly prepared prior to sample preparation.
Plasma collection
Plasma samples were collected with institutional review board approval of the C1-INH-HAE investigator sites listed in the ‘Acknowledgements’ section with the blood collections procedures described in previous reports [Citation19]. Each patient was previously confirmed to be C1-INH-HAE and samples were collected between attacks (i.e., basal). Though the information on whether an individual patient had type I or type II was not obtained with these samples, it is known that both forms of C1-INH-HAE are due to a dysregulation of the contact system. Briefly, the SCAT-169–4.5/5 blood collection tube (5.0 ml) was removed from refrigerator. Approximately 5 ml of blood was drawn into the tube using the catheter with multisample luer adapter. Contents of the tube were not allowed to contact the stopper or end of the needle during procedure. Vacuum was exhausted prior to removing the tube from the nonpatient end of the needle. The blood was mixed with the tube additives by slowly inverting the tube eight- to ten-times immediately after blood collection and before centrifugation. The blood sample was centrifuged at 2000 × g for 20 min at room temperature (20–25°C) within 1 h of sample collection. The plasma samples were stored at -70°C or colder until use. Informed consent forms were obtained from each healthy or patient subject.
Sample preparation
Protein pellet digestion
Samples were processed in a high-throughput 96-well plate format, following procedure reported previously by our lab with minor modification [Citation37]. Briefly, aliquots of 25 μl plasma samples (calibration standards, QCs, blanks or study samples) were transferred into a 96-well plate (2 ml capacity). The proteins were denatured by adding 2.5 μl of 10% SDS, followed by vigorous vortexing for 5 min on an Eppendorf Thermomixer (Eppendorf, NY, USA). Protein reduction was carried out by adding 5.0 μl of 500 mM DTT and subsequent incubation for 60 min at 37°C at 700 r.p.m. Plasma proteins were then precipitated by adding 0.7 ml of CH3OH to each well followed by vigorous vortexing for 5 min. After centrifugation at 2000 × g for 10 min, the 0.68 ml of supernatant was removed and discarded by Tomtec liquid handler (Tomtec, CT, USA). The resulting protein pellet was washed with 0.7 ml of methanol and after centrifugation, and 0.68 ml of the supernatant was discarded gain. The plate was placed in a nitrogen blower to remove the remaining methanol. The protein pellets were resuspended in 500 μl of 100 mM ABC digestion buffer by vigorous vortexing for 5 min. In total, 10 μl of 500 mM IAA and 25 μl of IS working solution (30 ng/ml) were added, mixed well and kept in the dark for 30 min at 600 r.p.m. for alkylation. To the protein suspension was added 10 μl of 8.0 mg/ml freshly prepared chymotrypsin solution in 100 mM ABC buffer (1:20 E/S). The proteolytic digestion was carried out at 50°C at 1000 r.p.m. vortexing for 3 h in the dark. The samples were acidified by adding 10 μl of formic acid, followed by vigorous vortexing for 5 min, to quench the digestion.
Peptide enrichment by solid phase extraction
The acidified peptide mixture was centrifuged at 3000 × g for 10 min. The supernatant (460 μl) was transferred to a Waters MCX plate (30 μm, 10 mg, Waters) preconditioned sequentially with 1 ml of CH3CN and 1 ml of 1.0% FA. After sequentially washing with 900 μl of 1.0% FA, 900 μl of water and 900 μl of CH3OH, the peptides were eluted by 600 μl of CH3OH containing 5.0% ammonia hydroxide (v/v) and collected in a clean 96-well collection plate. The eluent was evaporated to dryness under a steady stream of nitrogen at 37°C. The sample was reconstituted in 100 μl of reconstitution solution (CH3OH:water [10:90; v/v] containing 1.0% HFBA [v/v]). Then, each sample (20 μl) was injected into the ion-pair 2D-LC–MS/MS system.
Method validation
Validation followed the FDA guidelines for bioanalytical method validation and white paper [Citation38,Citation39]. The method was validated for linearity, accuracy, precision, matrix effects, pellet digestion efficiency and solid phase extraction (SPE) efficiency. Calibration curves were constructed utilizing peak area ratios of SP to IS with a weighted (1/x2) least-squares linear regression analysis. Accuracy (expressed as percentage error) and precision (expressed as percentage relative standard deviation) were calculated at five levels of QC samples (EL, EL + 200 ng/ml, EL + 600 ng/ml, EL + 5000 ng/ml and EL + 15,000 ng/ml). The intraday accuracy and precision were analyzed using six replicates of each QC level every day. This process was repeated three-times to determine the interday accuracy and precision over 3 days. Mean accuracy and precision statistics for QC samples were calculated using Microsoft Excel. The criteria for intraday or interday accuracy and precision were ≤ ± 25% for LLOQ and ≤ ± 20% for LQC, MQC and HQC. The SPE extraction efficiency was determined using SP-spiked digestion solution of the blank beagle plasma samples (n = 5) at the concentrations of 1.5 ng/ml of SP. SPE extraction efficiency was determined by dividing the peak area of SP spiked before SPE extraction by the peak area of a SP sample spiked after extraction at equal concentration in the same matrix. Matrix effect evaluations were performed by assessing the accuracy of LQC samples utilizing six independent lots of human plasma fortified to the LQC samples. Plasma matrix stability of cHMWK after freeze–thaw (4 cycles) or bench-top stability at 4°C (8 h) as well as processed sample stability in an autosampler (48 h) were also assessed.
Statistics
Student’s t-test for unpaired values and receiver operator curve were utilized to evaluate the statistical significance of the differences between healthy subjects and C1-INH-HAE patients and p-value (<0.05) was considered statistically significant.
Results & discussion
LC–MS strategy & SP selection
HMWK and cHMWK share the same amino acid sequence minus the presence of bradykinin in cHMWK, where the different C-terminal peptides of the heavy chains and the N-terminal peptides of the light chains can differentiate the two proteins (). Plasma kallikrein cleaves HMWK at site Lys380-Arg381 and at site Ser390-Ser644 to generate bradykinin as well as a heterogeneous heavy chain and a 56 kDa light chain. The 56 kDa light chain in cHMWK is further truncated both in vitro and in vivo to form a stable 46 kDa light chain (Lys438-Ser644) () [Citation40]. As the 46 kDa cHMWK is the final cleavage product and is presumably more stable, we measured 46 kDa isoform as a potential biomarker of contact system activation. For this purpose, experimental conditions were optimized in order to measure peptide derived from N-terminus of 46 kDa light chain as a cHMWK surrogate. Trypsin and Lys-C digestion did not generate the specific peptide to differentiate HMWK and cHMWK. In contrast, chymotrypsin, Glu-C and Asp-N digestion all successfully generated unique peptides (). Chymotrypsin was selected for assay development as the resulting peptide KHNLGHGH from the digestion by chymotrypsin possessed the excellent MS responses and the enzyme cost is more manageable compared with Glu-C and Asp-N. Although chymotrypsin is less specific, which is usually not the first option for protein quantitation, we found the digestion at the desired site is highly specific as no other N-terminal peptides derived from the 46 kDa protein were identified. The peptide sequence was further blasted against the protein database to ensure it has no overlap with the hypothetical chymotrypsin peptides derived from other human proteins except HMWK.
Table 1. Signature peptides derived from digestion of 46 kDa light chain with different enzymes.
In previous reports, western blotting has been a method of choice to measure the relative abundance of the total HMWK and cHMWK as a CI-INH-HAE biomarker [Citation12,Citation24]. In contrast, our LC–MS assay measures the absolute quantitation of cHMWK (without normalized to total HMWK) with an aim to explore whether cHMWK level alone is correlated with C1-INH-HAE disease.
Protein pellet digestion
One challenge for LC–MS-based protein quantitation is to enrich target protein biomarkers in the presence of highly abundant endogenous proteins in biological samples. To improve assay sensitivity for target protein, IP in conjunction with LC–MS/MS is widely used [Citation29–31,Citation41]. A variety of commercially available anti-HMWK, antilight chain and antiheavy chain antibodies were assessed as IP reagents but none was able to efficiently pull down the targeted proteins (data not shown). Recently, direct pellet digestion of serum proteins by proteases following CH3OH precipitation has been described in several papers [Citation42–45]. This ‘reagent-free’ approach was capable of simplifying sample manipulation and cleaning up salts, protease inhibitors and other hydrophilic components in serum to improve the digestion. Efficient and reproducible pellet digestion by chymotrypsin is a critical step to ensure the accuracy and precision of the cHMWK assay. We further optimized the pellet digestion procedures previously described [Citation37] by using SDS (0.1%) to completely denature proteins to facilitate the enzyme digestion. DTT as a reducing agent minimized background in the subsequent LC–MS cHMWK assay (data not shown). After CH3OH precipitation, SDS, reducing agent, small MW proteinase inhibitors in SCAT169, phospholipids and other endogenous soluble species were easily removed from the protein pellets.
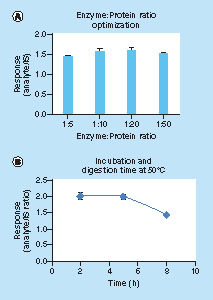
The protein–enzyme ratio and digestion temperature were optimized, aiming to enhance digestion efficiency and/or shorten the digestion time. In our experiments, 3 h digestion at 50°C with a chymotrypsin: protein ratio of 1:20 gave rise to reproducible results with improved S/N ratio (A & B). In contrast, digestion at 37°C overnight would compromise digestion efficiency (data not shown). The SDS and CH3OH denaturation in conjunction with DTT reduction facilitated the dissociation of cHMWK from endogenous binding proteins and promoted the chymotrypsin digestion efficiency of the cHMWK. As justified from the relative peak areas of the SP/IS, the overall cHMWK recovery combining CH3OH crash and digestion steps were 57.5 ± 1.7% at 10 μg/ml (n = 5) in SCAT169 human plasma.
(A) enzyme:protein ratio optimization; (B) response over digestion time at 50°C and IS: 25 ng/ml.
IS: Internal standard.
Chromatographic separation of SP
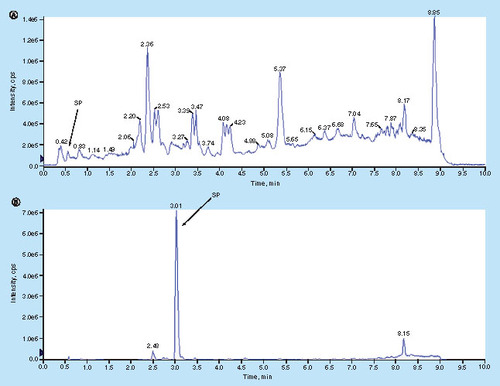
Human plasma contains thousands of proteins with a huge dynamic range of expression. Upon pellet digestion, the proteins were chopped into hundreds of thousands of peptides, many of which would co-elute and interfere with the analyte measurement under the routine chromatographic conditions. In addition, the SP (KHNLGHGH) is a short and very hydrophilic peptide, limiting its retention on the reversed phase C18 column without ion-pair reagent in the mobile phase. As a matter of fact, after peptide sample extracted from the pellet digestion of 100 μg/ml cHMWK spiked in human plasma was injected to a 1D-LC–MS/MS system using the C18 column in mobile phases containing 0.1% of FA (v/v), the signal of the SP was barely detectable due to the poor retention of the peptide and significant ion suppression (A). Different ion paring agents such as 0.1% TFA and 0.1% HFBA were included in the mobile phases aiming to enhance the retention. TFA was able to improve ion signal, but peptide still eluted out of the column too soon. An opposite effect was observed when HFBA was utilized. In order to obtain sufficient analyte retention yet without significant ion suppression, we included 1% (v/v) of HFBA in the injection solution while maintained 0.1% of TFA (v/v) in the mobile phase, the combination leading to much improved chromatographic retention as well as the sensitivity enhancement (B). Obviously high concentration of HFBA in injection solution significantly increased the retention of SP on the first LC column, and by the time SP was eluted from the second column, the HFBA was already washed out, therefore the ion suppression is no longer an issue. To our best knowledge, it was the first time that two ion-pair reagents were utilized in tandem to retain extremely polar peptide on reversed-phase column yet maintain the assay sensitivity.
cHMWK (100 μg/ml) plasma extract was injected to 1D-LC–MS system using selected ion monitoring (SIM: 450.2) and an Agilent Metasil AQ C18 Column (2.0 mm × 100 mm, 5 μm). (A) Without ion-pair reagents: mobile phase A was 0.1% of FA (v/v) in water and mobile phase B was 0.1% of FA (v/v) in CH3OH; injection solution was CH3OH:water (5:95; v/v) containing 0.1% of FA (v/v). (B) With ion-pair reagents: mobile phase A was 0.1% of TFA (v/v) in water and mobile phase B was 0.1% of TFA (v/v) in CH3OH; injection solution was CH3OH:water (10:90; v/v) containing 1.0% of HFBA (v/v). The same graduate elution conditions were utilized in (A) and (B): 0 min/1.0% B, 0.5 min/1.0% B, 5.0 min/50.0% B, 7.0 min/90.0% B, 8.0 min/90.0% B, 8.1 min/1.0% B and 10.0 min/1.0% B at a flow rate of 0.45 ml/min. Injection volume: 20 μl.
FA: Formic acid; HFBA: Heptafluorobutyric acid; TFA: Trifluoroacetic acid.
Although, the 2D-LC–MS/MS has been widely utilized in large scale protein identification and global protein quantification in proteomics field, only recently has it gained attention as a valuable approach to targeted protein quantitation in regulated study [Citation37,Citation46]. The system enriches the target peptide on the column and circumvents the need for immunoaffinity reagents, yet reaches low nanogram/ml sensitivity and maintains high throughput. In contrast to global proteomic identification aiming to identify as many peptides as possible, the targeted quantitation only focuses on the specific SP [Citation37]. The narrow valve switch window (1.3 min) significantly reduced the total number of peptides pouring onto the second column, greatly enriching the targeted peptide and minimizing the interferences by the coeluting peptides [Citation37,Citation47]. The 2D-LC–MS/MS described here offered LLOQ of 200 ng/ml for cHMWK protein, at least fivefold increase in comparison with 1D-LC–MS/MS (LLOQ: 1000 ng/ml; data not shown).
Fit-for-purpose assay validation results
Surrogate matrix & calibration curve
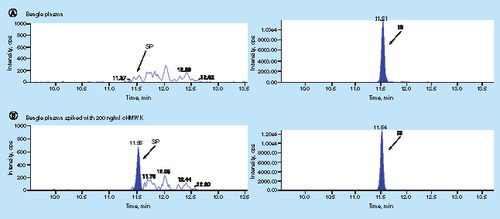
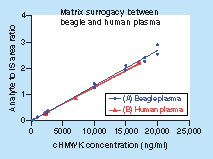
A fit-for-purpose validation was performed for the cHMWK LC–MS assay [Citation30,Citation48]. It is a common challenge for biomarker analysis to find an appropriate matrix for the preparation of standard curve. Ideally, the matrix does not contain the SP sequence or generate irrelevant MRM responses at the retention time window of SP, yet it should be identical or similar to the sample matrix based on digestion efficiency, ionization effects and SPE extraction efficiency. Screening of human serum showed that endogenous cHMWK concentration is highly variable and will interfere with the assay. Therefore, we decided to use plasma from another species as a surrogate. In the preliminary study, cyno, mouse, rat and beagle plasma were evaluated and only beagle plasma gave rise to clean background and insignificant interferences to the SP signal (A, Graph 1). The parallelism of the surrogate matrix (beagle plasma) and sample matrix (human plasma) was demonstrated by comparing the slope difference of the two calibration curves prepared in two matrices. The relative error (RE) of the slope difference is 6.9%, demonstrating a good surrogacy of the beagle plasma because the pellet digestion efficiency, ionization effects and extraction recovery are similar to human plasma (). The good linear response by interday calibration curves was demonstrated over the concentration range of 200–20,000 ng/ml in human plasma (n = 3). The Pearson correlation (R2) ± SD was 0.9955 ± 0.00115. The slope ± SD of calibration curves was 0.000147 ± 0.0000289. RE typically ranged from -13.1 to 14.0% between back-calculated standard concentrations and nominal values. Representative chromatograms obtained from surrogate plasma in the LLOQ standard (200 ng/ml) are shown in B, Graph 1. There were no significant interfering peaks from endogenous compounds observed at the retention time of the SP in blank surrogate matrix.
(A) beagle plasma Y = 0.000135X + 0.000275; R2 = 0.9915 and (B) human plasma Y = 0.000126X + 0.00168; R2 = 0.9927 to demonstrate matrix surrogacy. Each concentration level included duplicate samples.
Validation precision & accuracy
Five QC levels (EL, LLOQ, LQC, MQC and HQC), each in six replicates, were evaluated in three consecutive runs to assess intrarun and inter-run accuracy and precision. As demonstrated in , the intraday and interday precision ranges from 2.1 to 12.5% and 5.8 to 12.7% respectively. The intraday and interday accuracy ranges from -13.2 to 0.6% and -13.8 to -3.5%. All the calibration standards and the QCs passed the acceptance criteria (≤ ± 20.0%) ().
Table 2. The intraday (n = 6) and interday (n = 18) precision (% relative standard deviation) and accuracy (% relative error) of the 2D-LC–MS/MS method.
SPE extraction efficiency, matrix effects & matrix stability
SPE was utilized to enrich and desalt the peptides prior to LC–MS/MS. SPE extraction efficiency was assessed by dividing the peak area of SP spiked before SPE extraction by the peak area of the same amount of SP spiked after extraction. Since reversed-phase cartridge did not retain the SP, strong cation exchange cartridge (Waters MCX) was utilized. The extraction efficiency of SP using the strong cation exchange cartridge was 52.6 ± 2.64% (n = 5). Six independent lots of human plasma were fortified to the LQC samples. Matrix effect evaluations were conducted by assessing the accuracy of the six individual LQC samples. The results demonstrated all of the QC samples met the accuracy criteria (≤ ±20.0%), and there was no significant matrix effects for the cHMWK assay.
Matrix stability of macromolecules, including protein biomarkers, is a critical validation parameter. The analyte cHMWK is an enzymatic product generated from cleavage of the HMWK by endogenous serine proteinase plasma kallikrein, and can be constantly generated in vitro during plasma sample collection, storage and sample preparation if proteolytic activity of the plasma kallikrein is not inhibited [Citation6,Citation25]. SCAT169 reagent contains a variety of different protease inhibitors aiming to halt the kallikrein activity and thus to stabilize HMWK [Citation49]. Freeze–thaw (4 cycles) or bench-top stability at 4°C (8 h) as well as processed sample stability in an autosampler (48 h) were also assessed. As shown in Supplementary Table 1, the RE (%) of the stability test results ranged from -9.0 to -7.1%, which were well within the RE% acceptance criteria. The data in Supplementary Table 1 demonstrated that the cHMWK was stable in plasma treated with SCAT169 inhibitor.
Analysis of cHMWK in healthy individuals & C1-INH-HAE patients
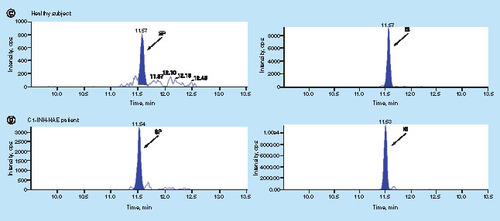
The concentration of 46 kDa cHMWK in plasma from 39 healthy controls and 27 C1-INH-HAE patients were tested utilizing the described method. All SCAT169 plasma samples were collected without any special treatment (dialysis, deproteination and delipidation). C1-INH-HAE patient demographics were shown in Supplementary Table 2. Assay sensitivity was sufficient for measuring 46 kDa cHMWK levels in all subjects (39 healthy controls and 27 C1-INH-HAE patients). Representative chromatograms for monitoring cHMWK level in healthy subjects and C1-INH-HAE patients are shown in (C1 and D1). The concentration of cHMWK in plasma was 415.2 ± 98.2 ng/ml (n = 39) for healthy subjects and 1359 ± 1090 ng/ml (n = 27) for C1-INH-HAE patients, respectively. Hence, plasma levels of 46 kDa cHMWK in the C1-INH-HAE patients were significantly higher than that in healthy subjects (; p < 0.0001). In addition, receiver operator curve in Supplementary Figure 3 supports above claim as well (the area under the curve is 1.0 and p < 0.0001). These results are consistent with the previous reports that patients with C1-INH-HAE exhibit a dysregulated contact system between attacks and thus exhibit elevated cHMWK and bradykinin in plasma [Citation12,Citation14–17,Citation24]. Overall, we have demonstrated that the reagent-free LC–MS assay is able to accurately measure the plasma 46 kDa cHMWK concentration in the presence of a large amount of endogenous HMWK. Consistent with previous observations [Citation12], our data from a limited number of healthy subjects and C1-INH-HAE patients suggest that the concentration of 46 kDa cHMWK alone can differentiate the two groups. However, additional investigations are required to validate the utility of 46 kDa cHMWK as a clinical biomarker for C1-INH-HAE diagnosis and/or therapeutic responses.
![Figure 7. cHMWK plasma level in healthy subjects (415.2 ± 98.2 ng/ml [n = 39]) and C1-INH-HAE patients (1359 ± 1090 ng/ml [n = 27]).](/cms/asset/9e9a1175-00b1-4b03-aa6f-2608fb23aa48/ibio_a_12355087_f0008.jpg)
Conclusion
We presented a novel LC–MS assay for quantitative measurement of the 46 kDa cHMWK in human plasma using a surrogate matrix. The assay is robust, specific and sensitive, enabling quantitation of the analyte in both healthy subjects and C1-INH-HAE patients. Most notably, the described assay does not need the time-consuming production and characterization of specific antibody reagents to distinguish cHMWK and HMWK. Our preliminary data on a limited sample size shows that the 46 kDa cHMWK, the final product of the plasma kallikrein proteolytic digestion, allows differentiation of C1-INH-HAE patients and healthy control group. Further studies are needed to validate its potential use as a C1-INH-HAE biomarker.
Future perspective
The novel LC–MS/MS assay described here takes advantage of the unique peptide sequence resulting from the chymotrypic digestion of the cHMWK in the presence of a high abundance of HMWK. The assay circumvents the use of immunoaffinity reagent to capture the target protein and is truly ‘reagent-free’. This assay can easily be implemented as an alternative to the immunoassay where antibody reagents are not readily available. Further studies should be directed in larger populations to validate the utility of the 46 kDa cHMWK as a potential biomarker for disease diagnosis, progress and therapeutic efficacy for C1-INH-HAE. In addition, a follow-up study can be set up to investigate whether the assay can detect elevated cHMWK in plasma from HAE patients with normal C1-INH, a condition that has not been firmly established to be mediated by a dysregulation in contact system activation. In addition, an isotopically labeled cHMWK protein can be introduced as an IS to minimize the variability in the sample preparation if its clinical usage is further proven in large scale of patient populations.
Background
Although cHMWK has been proposed as a potential biomarker for C1-INH-HAE, it remains challenging to quantify the analyte due to the interference from its high-molecular-weight kininogen precursor.
Experimental
A novel ion-pair 2D-LC–MS/MS assay combined with pellet digestion using chymotrypsin to measure 46 kDa cHMWK in human plasma.
Ion-pair 2D-LC–MS/MS techniques utilizing a narrow valve switch window (1.3 min) significantly reduced the background and interference.
Results
The technique was successfully applied to the determination of cHMWK in healthy subjects and C1-INH-HAE patients. The ion-pair 2D-LC–MS/MS techniques enhanced the sensitivity by at least fivefold in comparison with 1D-LC–MS/MS.
Conclusion
A robust, reagent-free LC–MS assay is developed for the quantitation of 46 kDa cHMWK, allowing differentiation of C1-INH-HAE patients and healthy subjects.
ibio_a_12355087_sm0001.docx
Download MS Word (83.6 KB)Acknowledgements
HAE plasma was collected by the following biomarker study investigators: C Akin (Brigham and Womens Hospital), J Anderson (Clinical Research Center of Alabama, Alabama Allergy & Asthma Center), A Banerji (Massachusetts General Hospital), J Bernstein (University of Cincinnati), E Brooks (UT Health Science Center at San Antonio), P Busse (Mount Sinai School of Medicine), M Cicardi (University of Milan), H Li (Institute for Asthma and Allergy, MD, USA), D McNeil (Columbus, OH, USA), L Schwartz and J Wedner (Washington University School of Medicine).
Supplementary data
To view the supplementary data that accompany this paper, please visit the journal website at: www.tandfonline.com/doi/suppl/10.4155/bio-2017-0105
Financial & competing interests disclosure
All authors are full-time employees of Shire and own stock. Shire funded the study. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Zuraw BL , ChristiansenSC. HAE pathophysiology and underlying mechanisms. Clin. Rev. Allergy Immunol. 51 (2), 216–229 (2016).
- Zeerleder S . C1-inhibitor: more than a serine protease inhibitor. Semin. Thromb. Hemost. 37 (4), 362–374 (2011).
- Davis AE, 3rd, LuF, MejiaP. C1 inhibitor, a multi-functional serine protease inhibitor. Thromb. Haemost. 104 (5), 886–893 (2010).
- Cicardi M , BerettaA, ColomboM, GioffreD, CugnoM, AgostoniA. Relevance of lymphoproliferative disorders and of anti-C1 inhibitor autoantibodies in acquired angio-oedema. Clin. Exp. Immunol. 106 (3), 475–480 (1996).
- Kaplan AP , GreavesMW. Angioedema. J. Am. Acad. Dermatol. 53 (3), 373–388 ; quiz 389–392 (2005).
- Nussberger J , CugnoM, AmstutzC, CicardiM, PellacaniA, AgostoniA. Plasma bradykinin in angio-oedema. Lancet351 (9117), 1693–1697 (1998).
- Agostoni A , CicardiM. Hereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patients. Medicine (Baltimore)71 (4), 206–215 (1992).
- Colman RW , SchmaierAH. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood90 (10), 3819–3843 (1997).
- Berrettini M , LammleB, WhiteTet al. Detection of in vitro and in vivo cleavage of high molecular weight kininogen in human plasma by immunoblotting with monoclonal antibodies. Blood68 (2), 455–462 (1986).
- Cugno M , CicardiM, BottassoBet al. Activation of the coagulation cascade in C1-inhibitor deficiencies. Blood89 (9), 3213–3218 (1997).
- Mori K , NagasawaS. Studies on human high molecular weight (HMW) kininogen. II. Structural change of HMW kininogen by the action of human plasma kallikrein. J. Biochem. 89 (5), 1465–1473 (1981).
- Baroso R , SellierP, DefendiFet al. Kininogen cleavage assay: diagnostic assistance for kinin-mediated angioedema conditions. PLoS ONE11 (9), e0163958 (2016).
- Couture R , BlaesN, GirolamiJP. Kinin receptors in vascular biology and pathology. Curr. Vasc. Pharmacol. 12 (2), 223–248 (2014).
- Gainer JV , MorrowJD, LovelandA, KingDJ, BrownNJ. Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N. Engl. J. Med. 339 (18), 1285–1292 (1998).
- Leeb-Lundberg LM , MarceauF, Muller-EsterlW, PettiboneDJ, ZurawBL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 57 (1), 27–77 (2005).
- Longhurst H , CicardiM. Hereditary angio-oedema. Lancet379 (9814), 474–481 (2012).
- Schmaier AH . Assembly, activation, and physiologic influence of the plasma kallikrein/kinin system. Int. Immunopharmacol. 8 (2), 161–165 (2008).
- Banerji A , BusseP, ShennakMet al. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N. Engl. J. Med. 376 (8), 717–728 (2017).
- Chyung Y , VinceB, IarrobinoRet al. A Phase 1 study investigating DX-2930 in healthy subjects. Ann. Allergy. Asthma. Immunol. 113 (4), 460–466 ; e462 (2014).
- Kenniston JA , FaucetteRR, MartikDet al. Inhibition of plasma kallikrein by a highly specific active site blocking antibody. J. Biol. Chem. 289 (34), 23596–23608 (2014).
- Cugno M , ZanichelliA, BellatorreAG, GriffiniS, CicardiM. Plasma biomarkers of acute attacks in patients with angioedema due to C1-inhibitor deficiency. Allergy64 (2), 254–257 (2009).
- Schapira M , SilverLD, ScottCFet al. Prekallikrein activation and high-molecular-weight kininogen consumption in hereditary angioedema. N. Engl. J. Med. 308 (18), 1050–1053 (1983).
- Kaufman N , PageJD, PixleyRA, ScheinR, SchmaierAH, ColmanRW. Alpha 2-macroglobulin-kallikrein complexes detect contact system activation in hereditary angioedema and human sepsis. Blood77 (12), 2660–2667 (1991).
- Suffritti C , ZanichelliA, MaggioniL, BonanniE, CugnoM, CicardiM. High-molecular-weight kininogen cleavage correlates with disease states in the bradykinin-mediated angioedema due to hereditary C1-inhibitor deficiency. Clin. Exp. Allergy44 (12), 1503–1514 (2014).
- Cugno M , CicardiM, CoppolaR, AgostoniA. Activation of factor XII and cleavage of high molecular weight kininogen during acute attacks in hereditary and acquired C1-inhibitor deficiencies. Immunopharmacology33 (1–3), 361–364 (1996).
- An B , ZhangM, QuJ. Toward sensitive and accurate analysis of antibody biotherapeutics by liquid chromatography coupled with mass spectrometry. Drug Metab. Dispos. 42 (11), 1858–1866 (2014).
- Becker JO , HoofnagleAN. Replacing immunoassays with tryptic digestion-peptide immunoaffinity enrichment and LC–MS/MS. Bioanalysis4 (3), 281–290 (2012).
- Heudi O , BarteauS, ZimmerDet al. Towards absolute quantification of therapeutic monoclonal antibody in serum by LC–MS/MS using isotope-labeled antibody standard and protein cleavage isotope dilution mass spectrometry. Anal. Chem. 80 (11), 4200–4207 (2008).
- Neubert H , FanYY, Fernandez OcanaM. Quantification of protein biomarkers in tissues: new capabilities with pellet digestion peptide immunoaffinity LC–MS/MS. Bioanalysis8 (15), 1551–1555 (2016).
- Neubert H , GaleJ, MuirheadD. Online high-flow peptide immunoaffinity enrichment and nanoflow LC–MS/MS: assay development for total salivary pepsin/pepsinogen. Clin. Chem. 56 (9), 1413–1423 (2010).
- Palandra J , FinelliA, ZhuM, MasferrerJ, NeubertH. Highly specific and sensitive measurements of human and monkey interleukin 21 using sequential protein and tryptic peptide immunoaffinity LC–MS/MS. Anal. Chem. 85 (11), 5522–5529 (2013).
- Qu M , AnB, ShenSet al. Qualitative and quantitative characterization of protein biotherapeutics with liquid chromatography mass spectrometry. Mass Spectrom. Rev. 36 (6), 734–754 (2016).
- Sano S , TagamiS, HashimotoYet al. Absolute quantitation of low abundance plasma APL1beta peptides at sub-fmol/ml Level by SRM/MRM without immunoaffinity enrichment. J. Proteome Res. 13 (2), 1012–1020 (2014).
- Ackermann BL , BernaMJ. Coupling immunoaffinity techniques with MS for quantitative analysis of low-abundance protein biomarkers. Expert Rev. Proteomics4 (2), 175–186 (2007).
- Berna MJ , ZhenY, WatsonDE, HaleJE, AckermannBL. Strategic use of immunoprecipitation and LC/MS/MS for trace-level protein quantification: myosin light chain 1, a biomarker of cardiac necrosis. Anal. Chem. 79 (11), 4199–4205 (2007).
- Patel BB , BarreroCA, BravermanAet al. Assessment of two immunodepletion methods: off-target effects and variations in immunodepletion efficiency may confound plasma proteomics. J. Proteome Res. 11 (12), 5947–5958 (2012).
- Shen Y , ZhangG, YangJet al. Online 2D-LC–MS/MS assay to quantify therapeutic protein in human serum in the presence of pre-existing antidrug antibodies. Anal. Chem. 87 (16), 8555–8563 (2015).
- US FDA. Guidance for Industry: Bioanalytical Method Validation (2001,). www.Fda.Gov/Downloads/Drugs/Guidance/Ucm070107.Pdf.
- Jenkins R , DugganJX, AubryAFet al. Recommendations for validation of LC–MS/MS bioanalytical methods for protein biotherapeutics. AAPS J. 17 (1), 1–16 (2015).
- Zhang JC , ClaffeyK, SakthivelRet al. Two-chain high molecular weight kininogen induces endothelial cell apoptosis and inhibits angiogenesis: partial activity within domain 5. FASEB J. 14 (15), 2589–2600 (2000).
- Fung EN , BryanP, KozhichA. Techniques for quantitative LC–MS/MS analysis of protein therapeutics: advances in enzyme digestion and immunocapture. Bioanalysis8 (8), 847–856 (2016).
- Furlong MT , TitschC, XuW, JiangH, JemalM, ZengJ. An exploratory universal LC–MS/MS assay for bioanalysis of hinge region-stabilized human IgG4 mAbs in clinical studies. Bioanalysis6 (13), 1747–1758 (2014).
- Gong C , ZhengN, ZengJ, AubryAF, ArnoldME. Post-pellet-digestion precipitation and solid phase extraction: a practical and efficient workflow to extract surrogate peptides for ultra-high performance liquid chromatography--tandem mass spectrometry bioanalysis of a therapeutic antibody in the low ng/ml range. J. Chromatogr. A1424, 27–36 (2015).
- Jiang H , ZengJ, TitschCet al. Fully validated LC–MS/MS assay for the simultaneous quantitation of coadministered therapeutic antibodies in cynomolgus monkey serum. Anal. Chem. 85 (20), 9859–9867 (2013).
- Yuan L , ArnoldME, AubryAF, JiQC. Simple and efficient digestion of a monoclonal antibody in serum using pellet digestion: comparison with traditional digestion methods in LC–MS/MS bioanalysis. Bioanalysis4 (24), 2887–2896 (2012).
- Sandra K , MortierK, JorgeLet al. LC–MS/MS quantification of next-generation biotherapeutics: a case study for an IgE binding Nanobody in cynomolgus monkey plasma. Bioanalysis6 (9), 1201–1213 (2014).
- Zhang G , ZhangY, FastDM, LinZ, SteenwykR. Ultra sensitive quantitation of endogenous oxytocin in rat and human plasma using a two-dimensional liquid chromatography-tandem mass spectrometry assay. Anal. Biochem. 416 (1), 45–52 (2011).
- Lee JW , DevanarayanV, BarrettYCet al. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm. Res. 23 (2), 312–328 (2006).
- Pellacani A , BrunnerHR, NussbergerJ. Plasma kinins increase after angiotensin-converting enzyme inhibition in human subjects. Clin. Sci. (Lond.)87 (5), 567–574 (1994).