
Abstract
Aim: In order to evaluate placental transfer of certolizumab pegol (CZP), a more sensitive and selective bioanalytical assay was required to accurately measure low CZP concentrations in infant and umbilical cord blood. Results & methodology: A new electrochemiluminescence immunoassay was developed to measure CZP levels in human plasma. Validation experiments demonstrated improved selectivity (no matrix interference observed) and a detection range of 0.032–5.0 μg/ml. Accuracy and precision met acceptance criteria (mean total error ≤20.8%). Dilution linearity and sample stability were acceptable and sufficient to support the method. Conclusion: The electrochemiluminescence immunoassay was validated for measuring low CZP concentrations in human plasma. The method demonstrated a more than tenfold increase in sensitivity compared with previous assays, and improved selectivity for intact CZP.
Introduction
Certolizumab pegol (CZP) is a PEGylated, humanized fragment antigen-binding monoclonal antibody (mAb) that targets and neutralizes both membrane-bound and soluble TNF-α [Citation1]. CZP has been approved for the treatment of adult patients with rheumatoid arthritis, axial spondyloarthritis, psoriatic arthritis and Crohn’s disease, and is commercially available in more than 60 countries worldwide [Citation2–8].
Women suffering from these chronic inflammatory diseases often require effective therapeutic interventions to control disease activity during pregnancy. Adequate disease control is crucial to ensure the best fetal and maternal health, since high disease activity is associated with an increased risk of adverse pregnancy outcomes [Citation9–13]. Anti-TNF agents provide an effective therapeutic option, however, they are often discontinued during the early stages of pregnancy to limit potential in utero exposure of the fetus and to reduce the risk of congenital malformations during organogenesis [Citation14–16]. There is a need for accurate and robust information to guide therapeutic decision making for these patients.
Active transplacental transport of immunoglobulin G (a maternal antibody) from mother to infant is mediated by the neonatal fragment-crystallizable (Fc) receptor (FcRn), which is expressed in the chorionic villi of the placenta [Citation16,Citation17]. CZP does not contain an Fc region and is therefore not expected to cross the placenta via the FcRn-mediated active transport mechanism [Citation18]. In vitro and ex vivo placental transfer models as well as animal studies have confirmed minimal transfer of CZP through the placental barrier [Citation19–21]. Furthermore, in two investigator initiated studies using ELISA technology, Mahadevan et al. and Förger et al. reported minimal levels of placental transfer of CZP compared with other anti-TNFs [Citation19,Citation20]. However, the ELISA used in these studies was developed and validated for pharmacokinetic (PK) determination in adults treated with CZP, and was therefore designed to detect therapeutic levels in patients, as opposed to the low levels of CZP expected from placental transfer in umbilical cords and infant blood. For this purpose, greater sensitivity and specificity are required, particularly when considering the small and unique molecular structure of CZP compared with other immunoglobulin G-based anti-TNFs.
This study aimed to validate a new immunoassay with enhanced sensitivity and selectivity for the accurate measurement of low CZP concentrations, such as those expected from transfer across the placenta [Citation20].
Materials & methods/experimental
Assay format & reagents
A variety of reagents and platforms were assessed to establish an assay able to measure low levels of CZP in lithium-heparin plasma. The Meso Scale Discovery® (MSD®) electrochemiluminescence immunoassay (Meso Scale Diagnostics, MD, USA) was ultimately selected as the platform of choice due to its robustness, its improved sensitivity over ELISA and its availability in bioanalytical facilities [Citation22].
While the MSD electrochemiluminescence assay utilized several critical reagents similar to those used in the previous ELISA (e.g., TNF-α as a capture reagent) [Citation23], it is important to highlight the key differences between the two assays. The ELISA used a horseradish peroxidase-conjugated anti-human kappa light chain antibody whereas the MSD assay used a biotinylated anti-PEG mAb, revealed with a ruthenium SULFO-TAG™ streptavidin conjugate. This detection antibody was chosen to enhance the specificity of the assay for CZP, since it exclusively targets its PEG moiety, which is absent in other anti-TNFs.
The anti-PEG mAb (clone PEG-B-47; Epitomics/Abcam) was compared with a series of anti-PEG antibodies developed in-house to determine the conditions for optimum binding to recombinant TNF-α-bound CZP. TNF-α (R&D systems) was chosen as the capture reagent over an anti-idiotype antibody for the new MSD method, to ensure consistency with the previous ELISA method [Citation23] and avoid affinity-related discrepancies in the immunocapture of CZP.
This method was developed in UCB Laboratories (Slough, UK), then transferred to and validated by Covance Laboratories, Inc. (VA, USA).
Assay protocol
Recombinant human TNF-α was coated onto the wells of an MSD plate and incubated overnight at 2–8°C, followed by blocking with 1% milk in phosphate-buffered saline and washing with phosphate-buffered saline (Tween-20® was excluded due to the presence of PEG within the formulation). On the day of analysis, calibrators, quality-control samples and unknown samples (diluted to a 1/100 minimum required dilution) were added to the plate and incubated for 1 h, followed by washing. The plate was then incubated for 1 h with biotinylated rabbit anti-PEG antibody and washed again. MSD SULFO-TAG™ streptavidin conjugate was then added (). The plate was then incubated for another hour, followed by a washing step. Finally, MSD read buffer was added to each well and the plate was read on an MSD Sector Imager 6000.
†The SULFO-TAG™ label emits light upon electrochemical stimulation initiated at the electrode surface of the multiarray assay plate.
Fab’: Fragment antigen-binding; PEG: Polyethylene glycol; TNF: Tumor necrosis factor.
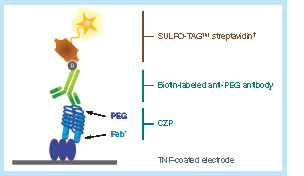
Assay validation
The MSD assay was validated in line with international established standards for bioanalytical methods to support PK determination [Citation24–26]. The following validation characteristics were established: calibration curve performance, accuracy, precision, selectivity, dilution linearity and sample stability. Acceptance criteria for each validation characteristic are detailed in Supplementary Table 1.
As part of a cross-validation assessment, a series of spiked quality control samples were also used to assess consistency of results when compared with the previously established ELISA method. Validation samples were prepared with control lithium-heparin human plasma (Bioreclamation; NY, USA), spiked with a CZP stock solution (UCB Celltech, Slough, UK).
Calibration curve performance
CZP stock solution was diluted in human lithium-heparin plasma to prepare calibration standards at concentrations of 10, 5 (upper limit of quantitation; ULOQ), 4.5, 2, 0.8, 0.32, 0.125, 0.064, 0.032 (lower limit of quantitation; LLOQ), and 0.016 μg/ml, and 0.016 and 10 μg/ml calibrators were used as anchor points and were not subjected to acceptance criteria. Performance characteristics of the calibration standards and a blank sample were monitored over 32 separate experiments. Standard curve performance was evaluated by both the coefficient of variation (CV) for each data point as well as the recovery percentage for all levels between ULOQ and LLOQ. The calibration function was determined using a four-parameter logistic function with (1/y2) weighting, in SoftMax Pro software version 5.0.1.
Accuracy & precision
Accuracy and precision of the method were determined by analyzing validation samples at five different CZP concentrations (0.032 [LLOQ], 0.1, 0.5, 4, and 5 μg/ml [ULOQ]) over six analytical runs. Replicates were measured in the same assay run to evaluate intra-assay accuracy and precision, while interassay precision and accuracy were determined by running the assay standard and controls with different analysts. In both cases and for all CZP concentrations, accuracy was determined as the cumulative mean percentage bias, while precision was determined as the cumulative mean CV. Total error (the overall error caused by both imprecision and inaccuracy of the measurement procedure) was also calculated.
Selectivity
To ensure that the detection of CZP was not compromised by the presence of unrelated compounds (e.g., endogenous proteins), selectivity was assessed using lithium-heparin plasma from treatment-naive patients with rheumatoid arthritis (n = 10), Crohn’s disease (n = 10), psoriasis (n = 10), and from otherwise healthy volunteer donors (n = 10). It was deemed unethical to assess selectivity in newborn infants, but plasma from healthy adult subjects was considered a suitable surrogate. To ensure that no matrix-related interference was observed, CZP levels were assessed in unspiked samples, and in samples spiked at the LLOQ (0.032 μg/ml). The appropriate institutional review board approval was obtained and the principles outlined in the Declaration of Helsinki for all human experimental investigations were followed.
Dilution linearity
Additional experiments were performed to assess the linearity of dilution of an ultra-high concentration. CZP was added to human lithium-heparin plasma to form a 25 μg/ml stock solution. Test samples were created by performing twofold serial dilutions of the stock solution, giving a CZP concentration range of 25–0.0122 μg/ml. Concentrations between 3.125 and 0.049 μg/ml were within the range of the assay and were evaluated against the back-calculated concentration of 25 μg/ml. The back-calculated concentration of the individual samples diluted into the assay range was deemed acceptable if they were within 20% of the expected value (25% for LLOQ and ULOQ). ≥80% of the samples were required to meet these criteria.
Sample stability
Stability of plasma samples at room temperature, 4°C, and between -60 and -80°C was determined for samples spiked with CZP at 0.1 and 4 μg/ml. Freeze/thaw stability was also assessed using 0.1 and 4 μg/ml samples, which were repeatedly frozen and thawed up to 15-times before analysis. Stability assessments were performed using freshly prepared calibration standards.
Cross-validation with ELISA
To evaluate consistency with the ELISA method, six healthy human plasma samples were spiked at 1.0, 8.0, 24.0 and 50 μg/ml with CZP. These samples were tested in triplicate, in two assay runs of both the ELISA and MSD methods. Due to the dissimilar calibration range of the two methods (range of the previous ELISA method: 0.41–1332.0 μg/ml) [Citation23], differing dilutions were required to allow the concentration of the samples to fall within the respective range of the standard curve. The percentage bias, CV and percentage difference between the methods were all required to be ≤20% in order to be considered acceptable.
Results & discussion
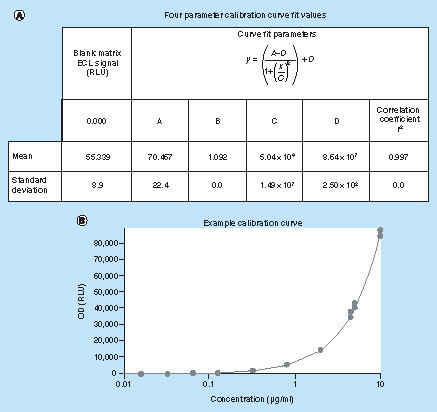
Calibration curve performance
An acceptable calibration curve range was established between 0.032 μg/ml (LLOQ) and 5.0 μg/ml (ULOQ) (). For the 32 reported analytical runs, the assay bias ranged from -4.0 to 5.2%. Interassay variability ranged from 1.9 to 8.1% CV.
(A) Four parameter calibration curve fit values. (B) Example calibration curve.
4 Parameter Fit: y = (A – D)/(1 + (x/C)B) + D, where A = 64.067, B = 1.107, C = 5.42 × 105, D = 7.33 × 106 and R2 = 0.998.
ECL: Electrochemiluminescence; OD: Optical density; RLU: Relative luminescence unit.
Accuracy & precision
Accuracy and precision were established across the analytical range of CZP concentrations (0.032–5.0 μg/ml). Results for the intra-assay and interassay experiments are summarized in . The mean total error ranged between 17.0 and 20.8% for the three quality control samples tested within the calibration curve range, which achieved the targeted acceptance criterion of total error ≤30%. Total error for the LLOQ (23.1%) and ULOQ (18.3%) samples also met the acceptance criterion. Acceptance criteria were met for all runs in both interassay and intra-assay evaluations of precision and accuracy, with the exception of a single run in which a mean intra-assay bias value did not achieve the targeted bias of ≤20%. All other levels were within acceptance criteria on this run.
Table 1. Interassay and intra-assay accuracy and precision.
Selectivity
Of the ten healthy donor plasma samples spiked with CZP at the LLOQ level, nine met acceptance criteria with bias ranging from -16.9 to 21.3% and all CV values were ≤25% (one sample exhibited a bias of -57.9%; A). All unspiked samples returned values below the LLOQ.
Recovery of CZP in lithium heparin plasma samples from (A) healthy volunteers (n = 10), (B) rheumatoid arthritis patients (n = 10), (C) Crohn’s disease patients (n = 10) and (D) psoriasis patients (n = 10). Horizontal dashed lines correspond to the ± 20% bias acceptance criterion.
CZP: Certolizumab pegol.
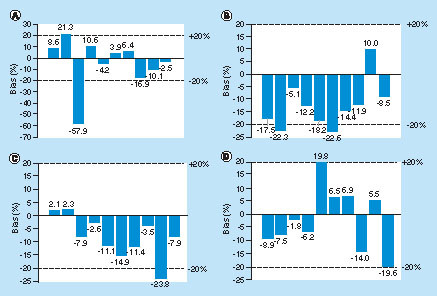
Plasma samples spiked at the LLOQ from rheumatoid arthritis patients demonstrated bias ranging between -22.6 and 10.0%, with all CV values ≤25% (B). Samples spiked at the LLOQ from Crohn’s disease patients demonstrated bias ranging from -23.8 to 2.3%, with all CV values ≤20% (C). Samples spiked at the LLOQ from psoriasis patients exhibited bias ranging from -19.6 to 19.8% (D). All unspiked samples returned values below the LLOQ for both patient groups.
Dilution linearity
Linearity of the method was established for dilutions up to 1/512, which were within the analytical range of 0.032–5.0 μg/ml. Of the seven values measured within the range of the curve, five (71%) were within 20% of the expected back calculated concentration, which did not meet the acceptance criterion (). However, the mean dilutional linearity was acceptable for both bias and CV, indicating that there is sufficient linearity of dilution to support the method. All samples demonstrated CV ≤20% for individual replicate pairs.
Table 2. Dilution linearity.
Stability
Freeze-thaw stability for both 0.1 and 4 μg/ml samples was acceptable for six freeze/thaw cycles (Supplementary Table 2). Stability of both samples at room temperature was deemed acceptable up to 24 h (Supplementary Table 3). Acceptance criteria were met when both samples were stored between 2 h and 8°C for 69 h (Supplementary Table 4). Short-term (29 days) and long-term (31 months + 14 days) stability between -60 and -80°C was considered acceptable (Supplementary Table 5).
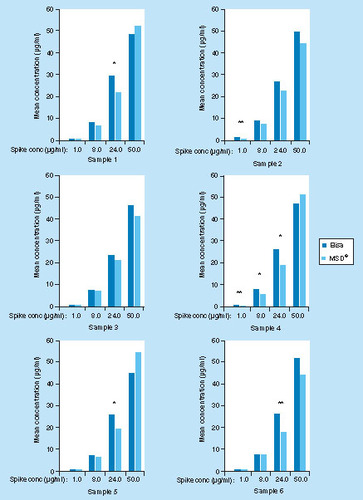
Cross-validation with ELISA
Overall, 7 out of 24 spiked samples returned values with greater than 20% difference between the methods; 3 samples exhibited a difference ≥30% (). None of the unspiked samples returned detectable values above the respective assay LLOQ.
*>20% difference between methods.
**>30% difference between methods.
Conc: CZP concentration; ELISA: Enzyme-linked immunosorbent assay; MSD®: Meso scale discovery® platform.
Discussion
This study aimed to validate a new electrochemiluminescence assay for the quantification of low CZP concentrations such as those expected in the umbilical cords and infants of women treated with CZP during pregnancy [Citation20]. Validation experiments for this CZP assay demonstrated high sensitivity, selectivity, accuracy and precision.
The sensitivity of the electrochemiluminescence assay compares favorably with the previous ELISA developed by UCB Pharma to determine CZP levels in human blood. The UCB Pharma ELISA has an LLOQ of 0.41 μg/ml and was used by Mahadevan et al. to measure CZP plasma concentration in mothers, infants and umbilical cords [Citation19]. In addition to the ELISA provided by UCB Pharma, Förger et al. also used a commercial ELISA with a minimum detection level varying between 0.41 and 0.60 μg/ml [Citation20]. By contrast, the LLOQ of the new electrochemiluminescence assay (0.032 μg/ml) is more than ten-times lower than the previous method, representing a substantial increase in sensitivity. The use of TNF-α as a capture reagent, combined with the use of an anti-PEG antibody as a detection reagent means the new assay is also highly specific for CZP.
ELISAs have been widely used in the past as they provide a relatively simple, high-throughput method. The previous UCB Pharma ELISA was developed to support PK analyses of CZP across a range of indicators and was validated in line with industry guidance available at the time for bioanalytical methods, as provided by the US FDA and the European Medicines Agency [Citation27,Citation28]. Therefore, the previous ELISA was optimized for measuring the therapeutic range of CZP concentrations typically seen in treated adults, but it had not been designed to reliably measure the low CZP plasma levels expected in the umbilical cords and infants of women exposed to CZP during pregnancy () [Citation19–21]. The new MSD electrochemiluminescence assay provides a robust platform for the accurate measurement of low CZP concentrations in human blood.
Table 3. Summary of the meso scale discovery electrochemiluminescence assay characteristics compared with the previous UCB Pharma ELISA used for the measurement of certolizumab pegol concentration in human plasma.
The MSD electrochemiluminescence assay also has limitations, mainly concerning the practicalities of implementing this technology in a global clinical program. In this study, the selectivity of the assay was validated in patients with rheumatoid arthritis, Crohn’s disease and psoriasis, but further testing will be required to confirm assay selectivity in other patient populations. It is also worth noting that access to equipment and expertise, as well as having the capacity to correctly label reagents for electrochemiluminescence detection is needed to ensure that the validity of the assay is not compromised in future studies. The fact that the assay platform is currently supplied by just one vendor (MSD), compared with more widely available ELISA technology, also highlights the need for cross-validation of the method described in this paper.
Conclusion & future perspective
These experiments validated a fit-for-purpose immunoassay designed to measure low concentrations of CZP in human plasma. Crucially, the new assay validated using the MSD system exhibited a more than tenfold increase in sensitivity compared with the previous ELISA [Citation23] with improved selectivity for intact CZP as demonstrated by plasma samples from healthy volunteers and patients with rheumatoid arthritis, Crohn’s disease and psoriasis. Accuracy and precision met acceptance criteria in both interassay and intra-assay evaluations. In addition, dilution linearity and sample stability across a range of conditions were considered acceptable and sufficient to support the method.
Continued changes in patient population and demand for increasingly accurate drug quantification methods, as well as the drive to understand potential sub-population within research programs, has led to the need for ever-evolving bioanalytical strategies. The enhanced sensitivity of the MSD assay has important implications for studies investigating placental transfer of CZP between mothers and infants and has been successfully implemented in clinical trial samples (NCT02019602). The MSD assay may also be used to evaluate the use of CZP in other matrices such as breast milk, and in other patient population where improved sensitivity and selectivity are required.
Aim
To validate a more sensitive and selective certolizumab pegol (CZP) immunoassay required to support clinical trials investigating placental transfer of CZP.
Results & discussion
A new electrochemiluminescence immunoassay method was validated to measure circulating levels of CZP in human plasma. This method is more selective and is more than ten-times more sensitive than previously established ELISA methods.
As opposed to the previous ELISA method, this new immunoassay detects full-length CZP with an anti-PEG antibody.
The assay is suitable for assessing very low levels of CZP in human plasma, such as those expected in the infants of mothers exposed to CZP during pregnancy.
Conclusion
The electrochemiluminescence immunoassay has been validated to measure CZP levels in plasma samples collected from mothers, umbilical cords and infants.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
ibio_a_12355067_sm0001.docx
Download MS Word (19.6 KB)Acknowledgements
The authors thank the patients, the investigators and their teams who took part in this study. The authors also acknowledge C Ecoffet and SE Auteri, UCB Pharma, Brussels, Belgium, for publication coordination.
Supplementary Data
Financial & competing interests disclosure
J Smeraglia, J-P Silva and K Jones are employees of UCB Pharma. UCB sponsored the study and the development of the manuscript/publication and reviewed the text to ensure that from a UCB perspective, the data presented in the publication are scientifically, technically and medically supportable, that they do not contain any information that has the potential to damage the intellectual property of UCB, and that the publication complies with applicable laws, regulations, guidelines and good industry practice. The authors approved the final version to be published after critically revising the manuscript/publication for important intellectual content. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
The authors acknowledge E Thurtle, MChem, R Milho, PhD, and J Bárdos, PhD, from Costello Medical Consulting, Cambridge, UK, for medical writing and editorial assistance in preparing this manuscript for publication, based on the authors’ input and direction.
References
- Melmed G , TarganSR, YasothanU, HanicqD, KirkpatrickP. Certolizumab pegol. Nat. Rev. Drug Discov. 7, 641–642 (2008).
- European Medicines Agency . Annex 1: Summary of product characteristics (Cimzia) (2017). www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001037/WC500069763.pdf.
- European Medicines Agency . CIMZIA European public assessment report (2015). www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/001037/human_med_001294.jsp&mid=WC0b01ac058001d124.
- Mease P , FleischmannR, DeodharAAet al. Effect of certolizumab pegol on signs and symptoms in patients with psoriatic arthritis: 24-week results of a Phase III double-blind randomised placebo-controlled study (RAPID-PsA). Ann. Rheum. Dis. 73, 48–55 (2014).
- van der Heijde D , FleischmannR, WollenhauptJet al. Effect of different imputation approaches on the evaluation of radiographic progression in patients with psoriatic arthritis: results of the RAPID-PsA 24-week Phase III double-blind randomised placebo-controlled study of certolizumab pegol. Ann. Rheum. Dis. 73, 233–237 (2014).
- Reich K , OrtonneJP, GottliebABet al. Successful treatment of moderate to severe plaque psoriasis with the PEGylated Fab′ certolizumab pegol: results of a Phase II randomized, placebo-controlled trial with a re-treatment extension. Br. J. Dermatol. 167, 180–190 (2012).
- Schreiber S . Certolizumab pegol for the treatment of Crohn’s disease. Ther. Adv. Gastroenterol. 4, 375–389 (2011).
- Landewé R , BraunJ, DeodharAAet al. Efficacy of certolizumab pegol on signs and symptoms of axial spondyloarthritis including ankylosing spondylitis: 24 week results of a double-blind randomized placebo-controlled Phase III study. Ann. Rheum. Dis. 73, 39–47 (2014).
- Broms G , GranathF, LinderMet al. Birth outcomes in women with inflammatory bowel disease: effects of disease activity and drug exposure. Inflamm. Bowel Dis. 20, 1091–1098 (2014).
- de Man YA , HazesJM, van der HeideHet al. Association of higher rheumatoid arthritis disease activity during pregnancy with lower birth weight: results of a national prospective study. Arthritis Rheum. 60, 3196–3206 (2009).
- Mahadevan U , SandbornWJ, LiDKet al. Pregnancy outcomes in women with inflammatory bowel disease: a large community-based study from Northern California. Gastroenterology133, 1106–1112 (2007).
- Norgaard M , LarssonH, PedersenLet al. Rheumatoid arthritis and birth outcomes: a Danish and Swedish nationwide prevalence study. J. Intern. Med. 268, 329–337 (2010).
- Jakobsson GL , StephanssonO, AsklingJet al. Pregnancy outcomes in patients with ankylosing spondylitis: a nationwide register study. Ann. Rheum. Dis. 75, 1838–1842 (2016).
- Gisbert JP , ChaparroM. Safety of anti-TNF agents during pregnancy and breastfeeding in women with inflammatory bowel disease. Am. J. Gastroenterol. 108, 1426–1438 (2013).
- Hyrich KL , VerstappenSM. Biologic therapies and pregnancy: the story so far. Rheumatology (Oxford)53, 1377–1385 (2014).
- Hazes JM , CouliePG, GeenenVet al. Rheumatoid arthritis and pregnancy: evolution of disease activity and pathophysiological considerations for drug use. Rheumatology (Oxford)50, 1955–1968 (2011).
- Maidji E , McDonaghS, GenbacevO, TabataT, PereiraL. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal Fc receptor-mediated transcytosis. Am. J. Pathol. 168 (4), 1210–1226 (2006).
- Simister N . Placental transport of Immunoglobulin G. Vaccine21 (24), 3365–3369 (2003).
- Mahadevan U , WolfDC, DubinskyMet al. Placental transfer of anti-tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 11 (3), e286–e224 (2013).
- Förger F , ZbindenA, VilligerPM. Certolizumab treatment during late pregnancy in patients with rheumatic diseases: low drug levels in cord blood but possible risk for maternal infections. A case series of 13 patients. Joint Bone Spine83 (3), 341–343 (2016).
- Porter C , Armstrong-FisherS, KopotshaTet al. Certolizumab pegol does not bind the neonatal Fc receptor (FcRn): consequences for FcRn-mediated in vitro transcytosis and ex vivo human placental transfer. J. Reprod. Immunol. 116, 7–12 (2016).
- Meso Scale Discovery . Singleplex and Multiplex Assay List (2017). www.mesoscale.com/∼/media/files/handouts/assaylist.pdf.
- Wade J , ParkerG, KosuticGet al. Population pharmacokinetic analysis of certolizumab pegol in patients with Crohn’s disease. J. Clin. Pharmacol. 55 (8), 866–874 (2015).
- DeSilva B , SmithW, WeinerRet al. Recommendations for the bioanalytical method validation of ligand binding assays to support pharmacokinetic assessments for macromolecules. Pharm. Res. 20 (11), 1885–1990 (2003).
- Shah VP , MidhaKK, FindlayJWet al. Bioanalytical method validation: a revisit with a decade of progress. Pharm. Res. 17 (17), 1551–1557 (2000).
- Viswanathan CT , BansalS, BoothBet al. Quantitative bioanalytical method and implementation: best practices for chromatographic and ligand binding assays. Pharm. Res. 24 (10), 1962–1973 (2007).
- Guideline on Bioanalytical Method Validation (EMEA/CHMP/EWP/192217/2009 Rev.1 Corr.2) . European Medicines Agency, London, UK (2011). www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf.
- Guidance for industry: Bioanalytical Method Validation . Center for Drug Evaluation and Research, US FDA, MD, USA (2013). www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm368107.pdf.