
Abstract
The 2017 11th Workshop on Recent Issues in Bioanalysis took place in Los Angeles/Universal City, California, on 3–7 April 2017 with participation of close to 750 professionals from pharmaceutical/biopharmaceutical companies, biotechnology companies, contract research organizations and regulatory agencies worldwide. WRIB was once again a 5-day, week-long event – a full immersion week of bioanalysis, biomarkers and immunogenicity. As usual, it was specifically designed to facilitate sharing, reviewing, discussing and agreeing on approaches to address the most current issues of interest including both small- and large-molecule analysis involving LC–MS, hybrid ligand-binding assay (LBA)/LC–MS and LBA approaches. This 2017 White Paper encompasses recommendations emerging from the extensive discussions held during the workshop, and is aimed to provide the bioanalytical community with key information and practical solutions on topics and issues addressed, in an effort to enable advances in scientific excellence, improved quality and better regulatory compliance. Due to its length, the 2017 edition of this comprehensive White Paper has been divided into three parts for editorial reasons. This publication (Part 3) covers the recommendations for large-molecule bioanalysis, biomarkers and immunogenicity using LBA. Part 1 (LC–MS for small molecules, peptides and small molecule biomarkers) and Part 2 (hybrid LBA/LC–MS for biotherapeutics and regulatory agencies’ inputs) are published in volume 9 of Bioanalysis, issues 22 and 23 (2017), respectively.
| Abbreviations | ||
| ACE | = | Acid-capture-elution |
| ADA | = | Antidrug antibody |
| BAV | = | Biomarker assay validation |
| BEST | = | Biomarkers, EndpointS, and other Tools |
| BMV | = | Bioanalytical method validation |
| CDR | = | Complementarity-determining region |
| CDx | = | Companion diagnostics |
| CLIA | = | Clinical Laboratory Improvement Amendments |
| CLSI | = | Clinical and Laboratory Standards Institute |
| Cmax: | = | Maximum concentration |
| CMC | = | Chemistry, Manufacturing, and Controls |
| COU | = | Context of use |
| CPPC | = | Cut point positive control |
| CRO | = | Contract Research Organization |
| CSF | = | Cerebrospinal fluid |
| CV | = | Coefficient of variation |
| FFP | = | Fit for purpose |
| FFPE | = | Formalin-fixed paraffin-embedded |
| GMP | = | Good manufacturing practices |
| IHC | = | Immunohistochemistry |
| IQR | = | Interquartile range |
| IS | = | Internal standard |
| IVD | = | In vitro diagnostic device |
| LBA | = | Ligand-binding assay |
| LCMS | = | Liquid chromatography–mass spectrometry |
| LLOQ | = | Lower limit of quantitation |
| LPC | = | Low positive control |
| LTS | = | Long-term stability |
| MOA | = | Mechanism of action |
| MRD | = | Minimum required dilution |
| NAb | = | Neutralizing antibody |
| PC | = | Positive control |
| PD | = | Pharmacodynamic |
| PK | = | Pharmacokinetic |
| PMA | = | Premarket approval |
| PTM | = | Post-translational modifications |
| QC | = | Quality control sample |
| RNA | = | Ribonucleic acid |
| SPEAD | = | Solid-phase extraction with acid dissociation |
| TE | = | Treatment emergent |
| ULOQ | = | Upper limit of quantitation |
| WRIB | = | Workshop on Recent Issues in Bioanalysis |
Index
Introduction
Discussion topics
Discussions, consensus and conclusions
Recommendations
References
The 11th edition of the Workshop on Recent Issues in Bioanalysis (11th WRIB) was held in Los Angeles/Universal City, California, on 3–7 April 2017 with attendance of over 750 professionals from pharmaceutical/biopharmaceutical companies, biotechnology companies, contract research organizations and regulatory agencies worldwide. The workshop included three sequential core workshop days, two additional advanced specialized sessions and four training courses that together spanned an entire week in order to allow exhaustive and thorough coverage of all major issues in bioanalysis, biomarkers and immunogenicity.
As in previous years, this year’s WRIB gathered a wide diversity of industry international opinion leaders and regulatory authorities working on both small and large molecules to facilitate sharing and discussions focused on improving quality, increasing regulatory compliance and achieving scientific excellence on bioanalytical issues.
The actively contributing chairs included Dr Eric Yang (GlaxoSmithKline), Dr Jan Welink (EMA), Dr An Song (Genentech), Dr Hendrick Neubert (Pfizer), Dr Fabio Garofolo (Angelini Pharma), Dr Shalini Gupta (Amgen), Dr Binodh DeSilva (Bristol-Myers Squibb) and Dr Lakshmi Amaravadi (Sanofi).
The participation of regulatory agency representatives continued to grow at WRIB [Citation1–15], covering topics and discussions in bioanalysis, biomarkers and immunogenicity:
Dr Sean Kassim (US FDA), Dr Sam Haidar (US FDA), Dr Seongeun (Julia) Cho (US FDA), Dr Nilufer Tampal (US FDA), Dr Jan Welink (EU EMA), Dr Olivier Le Blaye (France ANSM), Mr Stephen Vinter (UK MHRA), MS Emma Whale (UK MHRA), Dr Isabella Berger (Austria AGES), Mr Gustavo Mendes Lima Santos (Brazil ANVISA) and Dr Mark Bustard (Health Canada) – Regulated Bioanalysis
Dr Shashi Amur (US FDA), Dr John Kadavil (US FDA) and Dr Yoshiro Saito (Japan MHLW-NIHS) – Biomarkers
Dr Kara Scheibner (US FDA), Dr João Pedras-Vasconcelos (US FDA), Dr Pekka Kurki (Finland Fimea), Dr Isabelle Cludts (UK MHRA-NIBSC), Dr Laurent Cocea (Health Canada) and Dr Akiko Ishii-Watabe (Japan MHLW-NIHS) – Immunogenicity
As usual, the 11th WRIB was designed to cover a wide range of topics in bioanalysis, biomarkers and immunogenicity suggested by members of the community, and included daily working dinners and lectures from both industry experts and regulatory representatives, which culminated in an open-panel discussion among the presenters, regulators and attendees in order to reach consensus on items presented in this White Paper.
At this year’s WRIB, 40 recent issues (‘hot’ topics) were addressed and distilled into a series of relevant recommendations. Presented in the current White Paper are the exchanges, consensus and resulting recommendations on these 40 topics, which are separated into the following areas:
LCMS for small molecules:
Biomarkers and Peptide Bioanalysis (five topics);
Bioanalytical Regulatory Challenges (seven topics).
Hybrid ligand-binding assay (LBA)/LCMS for biotherapeutics:
Pharmacokinetic (PK) Assays and Biotransformations (two topics);
Immunogenicity Assays (one topic);
Biomarker Assays (five topics).
LBA for biotherapeutics:
Immunogenicity Assays (11 topics);
PK Assays (4 topics);
Biomarker Assays (5 topics).
In addition to the recommendations on the aforementioned topics, there is a section in the White Paper that focuses specifically on several key inputs from regulatory agencies.
Due to its length, the 2017 edition of this comprehensive White Paper has been divided into three parts for editorial reasons. This publication (Part 3) covers the recommendations for large molecule bioanalysis, biomarkers and immunogenicity using LBA. Part 1 (LCMS for small molecules, peptides and small molecule biomarkers) and Part 2 (hybrid LBA/LCMS for biotherapeutics and regulatory agencies’ inputs) are published in volume 9 of Bioanalysis, issues 22 and 23 (2017), respectively.
Discussion topics
The topics detailed below were considered as the most relevant ‘hot topics’ based on feedback collected from the 10th WRIB attendees. They were reviewed and consolidated by globally recognized opinion leaders before being submitted for discussion during the 11th WRIB. The discussions, consensus and conclusions are in the next section and a summary of the key recommendations is provided in the final section of this manuscript.
Immunogenicity
2016 US FDA & 2015 EU EMA immunogenicity draft guideline/guidance
Immunogenicity risk assessment
Can we clarify the definition of ‘low-risk’ biotherapeutics that may have varying mechanisms of action? How can consistency be achieved in risk categorization across different modalities? Should we rethink immunogenicity testing strategies for ‘low-risk’ biotherapeutics based on safety and efficacy? If yes, how? How does risk category translate into inclusion of immunogenicity assays in pivotal and prepivotal studies? Can we clarify the relationship between antibody incidence, risk and clinical relevance? Relative immunogenicity: what are suitable criteria for determining comparability between two products (e.g., biosimilars)?
Assay cut points
Change in the statistical approach for deriving the confirmatory cut point using the 99th percentile versus the 99.9th percentile of the negative control population will result in more false positives. Do you think this change could present a challenge in clearly ascertaining any clinical impact of immunogenicity? Do you think that the establishment of ‘in-study’ cut points, if not scientifically warranted, can slow data reporting? Do additional study samples confirm positive if the cut point is changed from 99.9% (considered too stringent) to 99%?
Assay sensitivity
Can the change in the requirement to target ‘100 ng/ml’ of the positive control (PC) be clarified? Should the assay sensitivity evaluation be performed in both individual and pooled samples from treatment and naive subjects? Can the selection approach for minimal required dilution (MRD) of test sample, negative control and low positive control (LPC) be clarified?
Drug tolerance
What is the current best practice to determine drug tolerance? How is the impact of the drug tolerance limit on assay sensitivity and poststudy monitoring evaluated/processed? Why do we need to have highly sensitive and drug-tolerant assays? Consideration No. 1: Assays with greater sensitivity in the presence of drug (i.e., higher drug tolerance) detect more antidrug antibody (ADA). But with these more sensitive assays, there may not be an evident correlation between ADA positivity and loss of efficacy or even clinical relevance, at least in the time frame under investigation. Consideration No. 2: If drug is present at higher concentrations than ADA, the drug will likely still demonstrate some efficacy despite the patient being ADA ‘positive’ in a highly drug-tolerant assay. How is the impact of ADA on the benefit/risk profile of a biologic determined when there is no direct relationship between ADA status and efficacy? What if drug tolerance cannot be improved, resulting in ADA assays with significant drug interference? Can these assays still be accepted if there are no other options?
Positive controls
What is the best approach to improve the reliability of the PC? Is use of a purified ADA as the PC appropriate? And/or the PC used versus extrapolation to actual ADA in the sample? And/or PC versus human immune response? What about adding a medium PC? Are positive samples ‘definitely positive’ or just ‘relatively positive’ due to the choice of PC, assay sensitivity, drug tolerance, assay specificity?
ADA isotyping & ADA specificity
Are isotyping, epitope mapping and cross-reactivity assays always needed? If not, in which cases are they needed? Do we need to detect all isotypes of an ADA response? There is extensive literature indicating that a mature (persistent) antibody response consists of predominantly IgG and reporting persistent response may be better correlated with impact on efficacy, therefore, should we focus on detecting only IgG?
ADA assay reproducibility
How reproducible are positive results from screening assays when sample values are relatively close to the cut point? How can we ensure that study samples that were screened positive initially continue to be positive when assayed a second time with a confirmatory assay? How are conflicting results in screening versus the unspiked sample in the confirmation assay dealt with? How relevant are these low positive samples with variable results in the screening assay? How relevant are low confirmation cut points when they are below or around 20%, similar to the variability of the assay? How relevant is it to determine confirmation cut points by looking at signal inhibition in antibody-negative/naive samples, with signals usually equivalent or very close to the assay background? Should we revisit how to determine confirmation cut point determination and utilize samples with a very low positive response (close to the assay sensitivity)? Would this approach be more representative of the real inhibition in an antibody-positive sample? Should PC be implemented as part of monitoring assay performance (especially over time)? Is the lower end sufficient, or should there be a high end limit? Is there need to include the middle range of the assay as well? Relevant LPC – setting a concentration that will result in assay failure approximately 1% of the time. Is the signal of the LPC relevant to study samples? Is an ADA assay acceptance criteria for precision of 30% acceptable? If yes, why?
Complex cases
Which option is more relevant in the case of healthy-naive samples versus study population: repeat analysis of a small subset of samples (∼50) versus single analysis of a larger number of samples? There can be differences in assay variability between healthy-naive samples and drug-naive, diseased population samples. How can this be taken into account when establishing cut points? When do we need to re-evaluate the cut points? In the case of preexisting ADA, how are cut points determined when there are confirmed titers of preexisting antibodies? How should we consider the incidence of ADA development versus the impact of ADA response on safety and efficacy? How are cut points determined in the case of high ADA incidence with no apparent clinical impact on safety and efficacy? What is the clinical relevance of ‘low’ titer ADA responses?
Outliers, outlier analysis & removal
Regarding outlier determination, is an outlier factor of 1.5 appropriate? Why are we modeling cut point data to remove analytical outliers? Should cut point determination mimic production conditions (e.g., more than two analysts, multiple plate lots, substrate/read buffer lots, different laboratories, etc.)? This increased variability will likely result in higher cut points. Analytical outliers occur during production runs, so why are not we capturing that as part of our cut point determination?
Hemolysis, lipemia & long-term stability for ADA assays
Recently, the FDA has increased the inspections on ADA assays. Should hemolysis and lipemia be tested for ADA assays as they are for PK assays? Should long-term stability (LTS) studies of controls for ADA assays be performed as they are for PK assays?
Monitoring biologically active biomarkers & evaluation of neutralizing antibody via neutralization of this activity
Should a pharmacodynamic (PD) readout be considered to better reflect in vivo neutralization than some artificial in vitro neutralizing antibody (NAb) assays? Can we define and agree on a strategy on cases in which a holistic assessment of PK/PD and ADA can replace dedicated in vitro NAb assays? Is a scientific approach where the decision to deploy a standalone NAb assay, or other/biomarker assay(s) of NAb impact on a case-by-case basis, acceptable? What datasets are appropriate to best inform the presence and impact of NAb? For a low-risk molecule where there is no impact on PK or efficacy, what is the clinical relevance of NAb data?
PK assays
Use of internal standard in LBA to improve precision & accuracy: is it possible? How?
The use of an internal standard (IS) is required for LCMS and ensures accuracy and precision. LBA do not use IS and may show poorer precision compared to LCMS. Is it possible to use IS for LBA? Which steps/tests in LBA may be better controlled by an IS and why (e.g., dilutions, plate treatments/readers, temperature variation, pipetting, liquid handler performance, analyst errors and techniques, reagent addition, adsorption, matrix effects, sample stability)? Is the use of an IS in LBA going to improve accuracy and precision? Can automation and emerging technologies reduce LBA variability? Are automation and emerging technologies able to impact any of the above causes of variability? Can IS be used in combination with automation to further improve accuracy and precision in LBA? Beyond reducing variability, do you think that the use of an IS in LBA can help minimize the impact of analyst mistakes? If yes, why? Can it help in assay troubleshooting and investigations? If yes, why? Can it be used for real-time monitoring of liquid handlers? We have quality controls (QCs) in LBA, do they not monitor these issues? Which operations of the assay process are controlled by QCs and which operations are not? Based on the advantages of IS for LBA, how should the ideal IS (QCs/controls) be designed? How does it differ from traditional QCs? Can pieces of the LBA workflow be controlled independently? Could there be interactions between unit operations? Does it help to control only parts of the entire assay (e.g., only the coating step, only the sample step)? Are IS widely feasible for all or a majority of LBA assays/platforms? QCs have become a regulatory expectation with prescribed performance requirements. Should IS in LBA eventually become a regulatory expectation?
What assay should be developed: free, total or active drug PK assays?
What type of PK assay should be developed in order to measure ‘free’, ‘total’ or ‘active’ drug concentrations? How can the biology and mechanism of action (MOA) help guide the development of the right assay? Use of ‘ligand-free’ matrix is a prerequisite for accurate ‘free’ drug quantification [Citation15–17], but not fully covered in the current guideline, which focuses on ‘total’ drug quantification. What is the current practice by the industry? To what extent is this considered during assay development and validation? What ‘surrogate matrices’ are favored as a ligand-free matrix: buffer, matrix of an alternative species, depleted matrix?
Novel biological therapeutics: understanding their biology & MOA to develop innovative modality-based bioanalytical strategies
How do you operate when there is not enough established experience or guidelines in place for the development and bioanalytical support of new modalities? How are bioanalytical methods, able to quantify what is needed for new modalities, designed? What are the unique scientific and regulatory challenges in developing these novel constructs? What is the industry currently doing to address them? Why should we focus on new modality MOA and biology for bioanalytical method design? Will the industry and regulators accept biological-activity-based assay data as a surrogate for PK measurement? Will the industry view biological-activity-based methods as advantageous or are they impractical to conduct? Is there a future in cell-based PK assays?
Combination therapies with biotherapeutics: what unique additional concerns should be considered in method development & validation?
Considering bioanalytical complexities, will industry and regulators be amenable to developing assays delivering the most reliable data (can be total or free)? What additional information would be required to convert such an analyte into the desired analyte (e.g., free)? How frequently is such information available? If the two therapies have a similar scaffold or belong to the same subclass of immunoglobulins, what is the reagent screening strategy? How can we avoid cross-reactivity? For PK, immunogenicity and NAb assays, how do we address interference in the assays and specificity concerns? Should we switch from the assays used in support of monotherapy to multiplexed assays? If we implement a new multiplexed assay to replace the individual assays, how do we bridge from monotherapy to combination therapy? Can an ADA response directed against one biotherapeutic cross-react to the coadministered biotherapeutic? Can cross-reactivity be identified through ADA testing? If we conduct a human trial with an experimental drug biological in combination with an on-market biological drug in comparison to the on-market biological drug alone, what is the expectation for assessing immunogenicity of the marketed biological drug in such a trial (i.e., either in subjects receiving the marketed biological drug alone or in subjects receiving both the experimental drug and the marketed biological drug)?
Biomarkers
Review of the 2015 & 2016 WRIB White Paper biomarker recommendations
For which types of biomarkers, might the results be considered to be relative quantitative as opposed to absolute quantitative? How does this impact the way the assay is validated? What should the requirements of precision (%CV) and accuracy (%bias) be for biomarker assays? What is the preferred approach to biomarker assay validation (BAV) when there is no official reference standard? What is the preferred approach to BAV when there is no blank matrix?
Biomarker stability: how do the preanalytic factors affect protein biomarker measurements: can we establish some ‘gold standards’?
What are the critical preanalytical considerations and best practices to ensure biomarker stability in collected samples? Can we establish some ‘gold standards’ or are there already ‘gold standards’ used by the industry and agreed upon with the regulators? What factors are important to consider during the sample collection phase at the clinical site? What factors are important to consider during processing at the site? How is material loss during sample processing minimized? What factors are important to consider during shipping to the laboratory? What factors are important to consider during storage until analysis? What are the best approaches to evaluate biomarker sample stability to ensure the quality of biomarker data? Are there some internal QCs for stability that can be applied to ensure quality? What are the best approaches to evaluate sample stability for RNA and miRNA biomarkers? What are the best approaches to evaluate sample stability for metabolites and lipid biomarkers? What are the best approaches to evaluate sample stability for protein biomarkers? What are the main stability challenges with formalin-fixed and paraffin-embedded (FFPE) samples from cancer patients for protein biomarker analysis? What are the current industry standards to determine the minimum percent of tumor required for the assay? What is the role of the bioanalyst versus the pathologist in developing the sampling protocol? How can we ensure that the tumor slides are carefully marked and affixed? How can we ensure that the percent of tumor is thoroughly and correctly calculated as surface area? This information is key for providing reliable biomarker data. What can the industry do to put in place standardized, comprehensive guidelines for correctly handling human specimens to guarantee their integrity? What is the impact of this lack of guidelines or ‘gold standards’ on clinical research?
Challenges & cutting-edge solutions in biomarker assays for mitigating variability & improving reliability
What is the best industry practice to minimize the impact of multiple factors on biomarker assay variability? What are the factors that impact the variability the most (e.g., sample collection, multiple dilutions and dilution volumes, stability, lack of universal reference standard, spiking approach/technique, severe adsorption issues, recovery)? What is the best industry practice for the method development and systematic optimization of biomarker assays, the step-by-step evaluation and reduction/minimization of less rugged steps that significantly impact variability? How can biomarker assay performance be improved by minimizing adsorption at each step for both endogenous biomarkers and spiked samples? What are the recommendations to improve biomarker assay precision based on pipetting techniques and/or automation?
Utility of specific Clinical and Laboratory Standards Institute guidelines for biomarkers in clinical drug development: building on the 2016 recommendations on global harmonization of BAV
What are the factors taken into consideration when assessing the robustness and reliability of a biomarker assay? What are the main scientific and regulatory expectations for biomarker assay performance? Where can we practically use Clinical and Laboratory Standards Institute (CLSI) guidelines for guidance on BAV? How are Clinical Laboratory Improvement Amendments (CLIA) assays different from biomarkers applied during drug development? What aspects of CLSI guidance are applicable to biomarkers applied during drug development? What is the current application and understanding of the definition of context of use (COU) for biomarkers in drug development across the whole industry? What is the relationship of COU between Center for Devices and Radiological Health (CDRH; premarket approval [PMA]/510k), Center for Drug Evaluation and Research (CDER; BAV) and CLSI (CLIA) requirements? Are there universal criteria that can be set prospectively for biomarker assays? What more can we do as a bioanalytical industry to design an industry/regulator harmonized global BAV guidance? What are the latest updates on ongoing US FDA supported activities for BAV? What is the status of the framework development for a US FDA BAV guidance? What resulted from the Crystal City VI discussion on BAV [Citation18,Citation19]? Will BAV guidance still be included in the revision of the 2013 FDA bioanalytical method validation (BMV) draft guidance [Citation20]? What is the EU EMA perspective on BAV and ongoing FDA activities (C-Path Initiative)? Are there any EMA/FDA interactions on BAV guidance/guideline? Are there any additional recommendations building on the 2016 White Paper recommendations on global harmonization of BAV [Citation15]?
Clinical biomarker fit-for-purpose validation in support of biotherapeutics: how to overcome scientific & regulatory challenges
How can we ensure that fit-for-purpose (FFP) BAV fully supports the intended purpose of the data in a clinical setting? What are the main lessons learned from FFP BAV in support of relevant clinical studies and label claims? What was the regulators’ feedback on submitted studies using FFP BAV? For a PD or target biomarker in a clinical trial, what BAV elements should be considered to make it FFP? What experiments should be considered/designed to ensure that the free target assay is more reliable? What is the industry standard for handling relative quantitative results in highly regulated clinical settings? What is the regulators’ point of view? What can be done for ensuring enough assay sensitivity and specificity to guarantee clinical performance? What is current industry best practice for optimizing assays toward increased specificity, sensitivity and robustness? How is biological variability considered? For the novel immuno-enhanced hybrid LBA/LCMS assays, have we used this technology to support any clinical studies? Compared with the traditional ELISA assay, under what circumstances can we decide to use a hybrid LBA/LCMS assay to support clinical trials?
Discussions, consensus & conclusion
Immunogenicity
2016 US FDA & 2015 EU EMA immunogenicity draft guideline/guidance
Biologics are a class of drugs of growing importance in the treatment of human diseases. The cost to the biotechnology industry of developing a biologic from early stage all the way to licensing is estimated to exceed US$2 billion [Citation21,Citation22], and many factors can impinge the approval. One potential obstacle impacting product development is the occurrence of immune responses against the biologic drug in treated patients. These immune responses can impact efficacy and safety of the product and their development is influenced by factors that can be patient/disease specific, product specific or related to route of delivery. The goal of immunogenicity studies of biotechnology-derived protein drugs is to detect immunogenicity, evaluate its clinical significance and, if necessary and possible, to mitigate it. These efforts will continue over the life cycle of the product. Evaluation of immunogenicity is a multidisciplinary exercise that proceeds from the risk assessment of immunogenicity, development of valid assays, integration of immunogenicity testing into the clinical program, evaluation of the impact of immunogenicity on pharmacokinetics, safety and efficacy, and risk management of immunogenicity.
Despite the progress of bioanalytical methods, subsequent steps in the investigation of immunogenicity can be invalidated by suboptimal immunogenicity assays. The development of valid assays is critical as it is the foundation upon which all subsequent assessments are built. Ideally, the same assay(s) should be used both in the pre- and post-licensing immunogenicity studies. The aim is to establish correlations between induced ADA and other clinical outputs such as pharmacokinetics, safety and efficacy. If such correlations are found, possibilities for post-approval immunogenicity monitoring should be explored. The EMA released the first guideline on immunogenicity testing for recombinant therapeutic proteins in 2006 [Citation23], and a revised draft in 2015 [Citation24]. The FDA released their first draft guidance in 2009 and a new draft in 2016 [Citation25]. Both the revised draft EMA guideline and the draft FDA guidance were still undergoing revisions at the time of the meeting, however, the EMA guideline was subsequently adopted in May 2017 [Citation26]. Numerous discussions have been ongoing within industry and regulators at WRIB about the content and ramifications of these documents on immunogenicity assay development, validation and use in clinical testing of recombinant therapeutic protein drugs.
Immunogenicity risk assessment
The EU general guidance on immunogenicity of therapeutic proteins has been revised based on experience from regulatory submissions and new information in the literature [Citation26]. The revised guideline incorporates a risk-based approach for assessment enabling better alignment with the guidance on immunogenicity assessment of monoclonal antibodies [Citation27] along with an integrated analysis of clinical data (pharmacokinetics, pharmacodynamics, efficacy and safety) and immunogenicity data to understand the clinical consequences of induced ADAs. The risk-based approach should be justified in an integrated summary of immunogenicity at the time of licensing. The goal of this new element is to facilitate regulatory review and to promote a multidisciplinary systematic approach to immunogenicity. Since ADA assays are critical for a rigorous assessment of immunogenicity, the guideline emphasizes the need for developing and validating assays suitable for their intended purpose along with implementation of a strategy, which allows clinical correlation of any induced drug-specific ADA.
One discussion point was the potential to achieve consistency in the risk categorization for different biotherapeutic modalities. For example, the guidance does not define or provide recommendations for ‘low-risk’ biotherapeutics that may have varying mechanisms of action. Instead, the risk is evaluated for each product by considering all product-, disease- and patient-related risk factors. The risk categorization of a biotherapeutic in development drives when an assay needs to be in place and how the data are interrogated. For higher-risk category molecules, an earlier and faster pace of assay development should take place as well as more frequent sampling and interim analysis. Harmonizing the industry approach to the different risk levels could be beneficial in order to achieve consistency in risk categorization. While the White Papers and guidance documents define risk factors, a consistent way of risk categorization has not been achieved [Citation25,Citation26,Citation28,Citation29]. It was concluded that there is not a risk score classification based on therapeutic modality. The MOA and other factors (e.g., patient population) need to be considered. In general, novel biotherapeutic molecules are perceived as higher risk than known types of biotherapeutics where there is clinical experience. In addition to new structures, novel production/process systems may include factors that could enhance their immunogenicity risk (e.g., impurities). Sponsors should perform an immunogenicity risk assessment early during drug development for each unique biotherapeutic, taking into consideration the probability and potential consequences of immune responses and including information regarding Chemistry, Manufacturing, and Controls (CMC), clinical and bioanalytical assay development.
Sponsors are expected to be transparent with regulators on immunogenicity strategies early in drug development, presenting bioanalytical plans for early stages based on the information available at the time of the assessment (e.g., for a low-risk molecule, samples can be banked and tested based on clinical data). Immunogenicity risk assessments should be updated based on emerging clinical data and sample testing strategies modified as appropriate. The risk assessment should be considered an ongoing process that evolves as more information becomes available and could lead to reclassification of the immunogenicity risk. In fact, early clinical data might be critical in providing information on the effect of ADA on PK and safety. The risk evaluation translates into the immunogenicity assessment strategy in pivotal and prepivotal studies. Although regulators may ask for NAb data in the more closely scrutinized pivotal studies, it is recognized that other datasets (PK, PD) may inform neutralizing impact of ADA and all datasets should be integrated to form a complete picture. Data from early clinical studies can be incorporated into the integrated summary of immunogenicity, which includes, but is not limited to, nonclinical and clinical assessments (if available) for ADA, cytokines, complement activation and other immunological parameters.
A consensus was reached that there is not always a clear relationship between ADA incidence and risk. Risk is an assessment (prospective initially) that encompasses the likelihood of developing ADA and the anticipated clinical consequences of an immune response to the drug based on known factors. The ADA incidence is the proportion of patients that seroconvert or boost their preexisting ADA during the study period as a result of exposure to the drug [Citation30]. There can be high incidence of ADA with no clinical impact or low incidence with severe clinical sequelae.
The approach for comparative evaluation of immunogenicity of biosimilar drugs is also included in the guidance. Relative immunogenicity has come into the spotlight along with biosimilars. Most bioanalysts use an ADA assay using the biosimilar active substance as the assay reagent. This is acceptable with regulators, but the bioanalyst should have a plan to address a possible difference in the immunogenicity between the biosimilar and its reference product. Suitable criteria for determining comparability between two products were discussed. A parallel arm study with a same dose scheme in a head-to-head comparison is the standard requirement. An in-depth analytical characterization (including stability data) is more relevant than similar production or manufacturing period products. For ADA sample analysis, a single assay with the biosimilar active substance for initial evaluation is recommended but ADA specificity to both the biosimilar and the reference product should be assessed in the confirmatory assay. Differences in ADA incidence and, most importantly, effect on clinical outcomes, are assessed case-by-case. It was recommended that sponsors communicate early on with regulators regarding study design and immunogenicity studies (especially for studies with a small number of patients).
Assay cut points
The assay cut point is the level of response of the assay that defines whether a sample response is positive or negative. The expectations outlined in the EMA guideline and the FDA draft guidance [Citation25,Citation26] are to set a 95% assay cut point, resulting in 5% false positives for screening assays and a 99% cut point for confirmatory and neutralizing assays, resulting in a 1% false-positive rate. The validation cut point should be confirmed in pretreatment clinical study samples or representative disease subjects. If the in-study cut point is significantly different from the validation cut point, this new cut point may be used especially when it results in a more sensitive ADA detection. If the new cut point makes the assay less sensitive, the validation cut point may still be used. The impact of the new cut point versus the validation cut point should be fully assessed before making the replacement.
With the new draft guidance released in 2016, the FDA is currently considering using a screening cut point with a 90% confidence interval for the 95th percentile. However, changing the statistical approach will result in more false positives and could present a challenge in clearly ascertaining any clinical impact of immunogenicity. Furthermore, it is likely that additional study samples will confirm positive if determination of the confirmatory cut point is changed from 99.9% (considered too stringent) to 99%. The regulators are presently evaluating these concerns. Industry should be encouraged to bring examples forward that can help with this evaluation.
It was discussed if the establishment of an ‘in-study’ cut point can slow data reporting. The consensus was that the evaluation of in-study cut points is always scientifically sound since cut points determined with the study/disease population should provide a more accurate representation of the variability within the given population. Therefore, this information is crucial to determine whether the validation cut point, which was typically set using commercial samples, is suitable for the study population and an accurate assessment of the disease population variability and the incidence rate of immunogenicity. For pivotal studies, a sufficiently large subset of samples can be used to confirm validation cut points or re-establish study- or population-specific cut points for screening and confirmatory assays.
Assay sensitivity
The lowest concentration of ADA that is equal to or above the assay cut point has been set to 100 ng/ml, according to the current FDA draft guidance [Citation25]. As stated in the document, the FDA has traditionally recommended sensitivity of at least 250–500 ng/ml, but the new target recommendation is based on data from two publications [Citation31,Citation32] and the fact that most current assay formats are more sensitive than legacy methodologies. The relevance of these publications and the recommendation is still under consideration by the FDA and Industry/Regulators’ consensus was not reached during the discussion. Regulators have reported that they consistently see assay sensitivity <20 ng/ml during data review. It is recommended to have sponsors work together with the FDA if sensitivity at or below 100 ng/ml cannot be achieved (e.g., PC characteristics, molecule-specific factors). It should be considered that sensitivity is based on a PC which may or may not be representative of actual responses, and pushing sensitivity lower and lower will increase detection of very low responses, which can increase detection of clinically meaningless positive samples. Given the nature of polyclonal ADA response, reference control ADA from an animal species may have different affinities and isotypes. All types of surrogate ADAs have limitations that should be taken into account in the interpretation of the immunogenicity results in clinical trials. For example, a high-affinity surrogate ADA may show good sensitivity, but such ADA control can tolerate harsh assay conditions that could limit detection of low-affinity ADA if the assay is not properly controlled.
Data-driven assessment of assay sensitivity is needed, especially when surrogate ADA cannot be successfully generated (e.g., affinity purification challenges for polyclonal PC ADA due to drug characteristics). If a PC ADA cannot be successfully generated or purified, the desired sensitivity may not be achieved.
The selection approach for MRD of test samples, negative control and LPC is under internal discussion by the FDA. The current recommendation is to provide the procedure to establish MRD as part of the summary of assay development work (e.g., validation report that includes summary of development data). The LPC may need to be adjusted during clinical development since acceptance criteria may differ. Screening assays typically have ranges of signal ratios and confirmation assays are % inhibition. Hence, the concentration of the LPC may be different for screening and confirmation assays. LPC (as a PC) should be consistently above the assay cut point; when it is below (even with ∼1% assay failure rate), theoretically, it should not be confirmed. However, the confirmatory assessment (such as drug competitive inhibition) may not work when absolute signal is low and therefore the sample cannot achieve the level of % inhibition to be determined positive.
Drug tolerance
The ADA assay sensitivity and drug tolerance strongly depend on affinity/binding characteristics of the selected PC (antibody) and are not fully representative for a broad variety of ADA in study samples. Inadequate drug tolerance continues to be the most common reason for regulatory objections to ADA assays. The EMA guideline expects that the drug tolerance level is higher than the drug level in patient samples to allow evaluation of the true immunogenicity of the drug. It is expected that all means to reach a satisfactory drug tolerance are explored, including changing the initial assay format if necessary. Another important issue is if an assay has poor drug tolerance, small differences in trough concentrations of drug may actually be indicative of impact on exposure, but it may be difficult to tell if these effects are ADA related or not. It is best to examine drug tolerance early in assay development. These data will inform the regulators’ assessment of impact on assay sensitivity when reviewing immunogenicity data. The current best practice to determine drug tolerance and to control the impact of the drug tolerance limit on assay sensitivity and post-study monitoring is to assess two or more levels of ADA (target sensitivity level included) and several levels of drug (including expected trough level). Study samples should be collected at trough levels of drug or after an appropriate washout period. Integration of ADA results with other clinical data by relating PK drug levels to ADA results for specific sampling time points should be considered to help interpret ADA data.
Highly sensitive and drug-tolerant assays have been encouraged by regulators, but have resulted in some interpretation challenges. Assays with greater sensitivity and/or higher drug tolerance detect more ADA, but the detected ADA may not correlate with loss of efficacy and/or clinical impact if analyzed by simply comparing the ADA-positive and -negative populations. In these more sensitive assays, there is a risk of missing correlations between ADA positivity and clinical impact. However, assays with poor drug tolerance may be unable to detect ADAs in a significant proportion of samples. In this situation, clinical correlations may also be missed. The correlation of ADAs to efficacy depends on the antibody titer and neutralization capacity, which may change over time.
Furthermore, the presence of ADAs may sterically hinder detection of drug in the PK assay and therefore it may seem that there is an impact on exposure when there is not. However, it should also be noted that if ADA affects the drug’s detection in the PK assay, then it likely affects its ability to bind target. Hence, the impact on exposure ADAs may cause a bias in the drug-concentration assay, which reflects an impact on exposure to active drug. Moreover, if drug is present at higher concentrations than ADA, the drug may still demonstrate efficacy despite the patient being ADA ‘positive’ in a highly drug-tolerant assay. However, the impact of other ADA-related factors may still need to be considered, such as drug clearance or sensitivity responses. Longer-term exposure may be required to better understand these relationships, which may not be evident during the timeframe of a clinical study. Despite these issues, there is an expectation to detect ADA and interpret ADA positivity in relation to clinical outcomes. Characteristics of ADA responses (e.g., persistent vs transient responses, titer levels) may be more relevant to assess correlations with efficacy, safety or pharmacokinetics in comparison to ADA incidence alone.
There was a discussion on how to evaluate the impact of ADA on the benefit–risk profile of a biotherapeutic in the absence of a clear relationship between ADA positivity and impact on efficacy. This issue can be further investigated by looking at the correlation of efficacy to the titer of ADAs and to the neutralizing capacity as well as by sufficiently long follow-up. Beyond the case where there is ADA detected but no impact on efficacy, the below cases should be also taken into consideration:
When no ADA is detected but there is an impact on efficacy, then impact on exposure should be evaluated;
When ADAs are detected but there is no impact on efficacy, then it may be that these low-level ADAs are not clinically meaningful;
When there is loss of efficacy but no ADA detected, and there is a drop in exposure, then there is the possibility that the assay failed to detect ADA that are present;
When there is loss of efficacy, but no impact on PK and no detection of ADA, then the loss of efficacy is unlikely to be due to ADA.
Finally, conclusions should also take into consideration whether or not the drug was expected to be low risk for immunogenicity incidence and impact. It could be that the ADA is detected in only a few patients, which indicates the need for a review of the postmarketing risk assessment. Correlations may be linear or nonlinear in nature.
It is recommended in cases where drug tolerance is poor resulting in ADA assays with significant drug interference, the sponsor should provide the totality of the data and demonstrate a good faith effort to develop a drug-tolerant assay by considering exploring acid dissociation, acid-capture-elution (ACE), solid-phase extraction with acid dissociation (SPEAD), precipitation and acid method (PANDA) [Citation33] or an alternative assay format [Citation34,Citation35]. However, if acid dissociation is used, there is a risk of denaturing ADA due to pH treatment, or the potential to release the soluble target from the therapeutic:target complex [Citation36]. It has been clearly stated by the regulators that whatever the chosen strategy, it must aim at achieving reliable detection of ADA. For this reason, adequate assay development is critical.
Concerns have been expressed by some regulators that the reported incidence of ADA is becoming less clinically relevant. In the 2016 draft guidance from the FDA [Citation25], they state that “…the impact of ADA on safety and efficacy may correlate with ADA titer and persistence rather than incidence”. This apparent reduction in correlation between ADA incidence and clinical efficacy end points is to a great extent the result of improved ADA assay methodologies, which have significantly improved in the areas of assay sensitivity and drug tolerance. It was agreed that the current industry practice is to report treatment-emergent (TE) ADA incidence on the labels of therapeutic proteins. However, it was also discussed that there is a growing number of cases where a better correlation with reduced efficacy was found when ADA responses were stratified into persistent versus transient responses, with only persistent responses leading to loss of efficacy.
Positive controls
PCs are often monoclonal antibodies or affinity-purified polyclonal antibodies from hyperimmunized animals. The ways to improve the reliability of the PC were discussed. It was clear that extrapolating to the actual ADA level in the sample by using a PC from an animal was not acceptable. Regardless of the type of PC chosen, a sample that tests positive is considered positive, as positivity is based on the cut point that is determined using negative samples. The best approach to improve the reliability of the PC was also discussed in depth, and purified ADA from hyperimmunized animals was confirmed as a good source for PC. With respect to ADA control levels, a medium control may be explored in validation but was not considered a requirement for routine testing. The FDA recommends low, medium and high PCs. Affinity purification procedures of polyclonal PC ADA should be carefully examined. If such procedures only generate a very small quantity of high-affinity ADA, such preparations may not be suitable as ADA controls. Positive ADA serum may be used as it includes a complex mixture of ADA with various affinities and epitope specificities.
ADA isotyping & ADA specificity
Isotyping, epitope mapping and cross-reactivity assays are often not needed. A cross-reactivity assay is considered to be crucial if there is an endogenous counterpart to the drug. If a strong safety signal is detected that calls for further characterization of the nature of ADA, isotyping may be required. ADA domain specificity for multidomain therapeutics should be assessed in confirmatory assays [Citation37]. Detailed, precise epitope mapping or differentiation between linear versus conformational epitopes are rarely requested.
Available literature indicates that, while the early ADA response is often of the IgM class, a mature (persistent) antibody response is predominately IgG and reporting persistent response may correlate better with any impact on efficacy end points. It is therefore important to understand antibody response kinetics. Although typical bridging assays are designed to detect all isotypes, it is understood that mature immune responses to chronic drug administration are expected to be predominantly IgG.
Setting ADA assay cut points
ADA assay reproducibility
While there is an increasing expectation for both highly sensitive and drug-tolerant ADA assays by both the FDA and EMA, there is also an evolving concern that the current approach in the reporting of ADA-incidence data may not be as relevant to clinical outcomes (PK, efficacy or safety). Assays with greater sensitivity usually generate higher positivity rates because they can detect low-level ADA-positive samples. Higher titer ADA responses (quartile analysis) have been shown to correlate to a higher degree with loss of PK or efficacy [Citation32]. These higher titer responses in the assay are more likely to have an impact on drug clearance, and might, in principle, be detected in less sensitive assays with lower drug tolerance. However, higher titer responses are not known without titration with a sensitive assay. In addition, a threshold for clinically significant ADA levels is not always known at the time of the study and could evolve throughout the course of clinical development. Acceptance of an insensitive assay is problematic since ADA detection may be further compromised by other factors that are independent of true ADA levels (e.g., variation in drug levels in samples). In general, ADAs are assessed based on a tiered approach: screening, confirmatory and titration, followed by additional characterization assays (e.g., a NAb assay). A statistically based quartile analysis of titers based on study data may be informative relative to PK and efficacy end points.
A critical factor in determining ADA assay sensitivity and performance is the cut point; setting this threshold correctly allows an assay to be less susceptible to detecting responses that are not clinically relevant. In the screening assay, the cut point is used to characterize whether a study sample is putatively positive and should move to the confirmatory assay, and the confirmatory cut point is used to determine whether the ADA is specific for the study drug. As such, the data and statistical methodology used to determine these cut points are critical to ensuring an accurate assessment of ADA-positive samples in the study population. Several important aspects of cut point assessment were discussed. These include outlier removal (outlier factors, analytical and biological outliers), the use of small sample sizes (n < 50) as a representation of the disease population, and the appropriateness of modeling these data. When sample signals are relatively close to the cut point, screening positive results can be variable and may not be reproducible. In order to ensure that study samples that screen positive initially are positive again when assayed with the confirmatory assay (without the drug), development of an assay that addresses biological variability is preferred. Ensuring the assay is well controlled with a clearly defined procedure (e.g., shaker speed, controlled incubator temperature, etc.) is important. Use of % CV for sample screen and confirmatory raw data signal for assay pass/fail may not be suitable. If variability is observed (e.g., >30% signal difference), a repeat test strategy could be implemented.
It was agreed that, unfortunately, conflicting results in the screening assay versus the unspiked sample in the confirmation assay do occur. Most companies consider only the % inhibition in the confirmation assay, not the signal from the unspiked sample. It is recommended to take multiple time points (sampling) to truly assess the ADA status of the patient and to provide information on the kinetics of the ADA response. The data should be reported as is and the interpretation discussed with regulators/sponsors. Consideration should be given to the concept that the confirmation cut point should not be lower than the empirically determined variability in the assay, for example, confirmation assays where percent inhibitions less than 20% do not make sense when assay variability can be 20%. The option of determining confirmation cut points by utilizing samples with a very low positive response (close to the assay sensitivity) was discussed, as well as if this approach would be more representative of the real inhibition in an antibody-positive sample. It was concluded that this approach (conducted by testing naive individuals spiked with PC at the assay sensitivity level) could be considered in development to assess the appropriateness of the confirmation cut point determined with unspiked naive samples.
It is recommended to include a minimum of a low- and high-positive and -negative control in all runs to monitor assay performance. Monitoring of assay performance should involve raw signals (not only S/N). Assessing trends over time (e.g., through Levy–Jennings graphs) should be considered. When setting a concentration that will result in assay failure approximately 1% of the time (minimum requirement), either LPC (or CPPC) set at 1% assay failure level or approximately two- to three-fold S/N level approaches can be considered. If setting LPC at 1% assay failure rate, the intent is to have the LPC fall below the cut point 1% of the time. When LPC is set at approximately two- to three-fold S/N level, assay failure should be based on statistical bounds (e.g., lower 99%) such that the LPC fails the acceptance criteria 1% of the time. This is to ensure that the LPC is not too high so as to be meaningless. Assays may fail more than 1%. For example, PC ranges can be set based on mean ± 2SD, which should result in 5% fail. Attendees reported that typically more plates fail due to analyst error than due to the 1% LPC being below the cut point. However, regulators noted that they do look at failure rate of the LPC. It was recommended to consider using LPC at approximately two- to three-fold S/N level for validation (as long as the resulting levels are not several folds higher than the majority of positive study samples) and assess the 1% failure level on validation data. For ADA assays, the acceptance criteria of 30% for assay precision was generally considered too high by panelists, higher than recommended in regulatory guidance documents. However, this depends on the assay and controls and should be assessed empirically. Certain assays (e.g., cell-based NAbs) may have precision levels more than 20%, but what would be acceptable for exploratory studies may not be appropriate for registration studies. It was acknowledged that it is unusual that precision requirements for ADA assays are more stringent than for PK assays.
Complex cases
Several complex cases for determining ADA assay cut points were discussed. In the case when the biological variability, and therefore putative cut point, are different between validation and in-study populations, the relevance of two options was debated: 1) repeat analysis of a small subset of samples (∼50 or fewer); or 2) single analysis of a larger number of samples. It was concluded that the cut point determined in validation using 50 samples is provisional. Different populations may require a reassessment of the cut point determined during validation, and possibly implementation of an in-study cut point may be necessary. If more than 50 baseline samples are available, it is also acceptable to use a greater number of samples analyzed fewer times. The use of a greater number of samples may, in fact, provide a more accurate representation of the variability within the study population and lead to a more appropriate and relevant cut point.
The case of preexisting antibodies was also discussed. Low-level biological reactivity that is reflective of the population should be included in cut point assessments; otherwise the cut points could be artifactually low, resulting in a higher incidence of in-study false positives. It was recommended to remove actual preexisting positives that have higher signals from cut point calculations. The focus should be on TE positivity, which includes treatment-induced and treatment-boosted responses. Indeed, for some therapeutics (e.g., pegylated proteins), preexisting anti-PEG antibodies do occur [Citation38] and in these cases treatment-boosted positivity may be relevant. It is recommended to report treatment-induced and treatment-boosted ADA separately. Individuals that are positive at baseline but do not show treatment-boosted ADA are not to be included in the overall ADA incidence. The final case examined was the determination of cut points in case of high ADA incidence with no apparent clinical impact on safety and efficacy. The conclusion was that cut point determination is not affected by high TE ADA incidence. Clinical response assessments during the timeframe of a clinical study may not reveal an impact of ADA on efficacy. A change in PK is a measurable end point that is potentially a cleaner gauge of ADA clinical impact.
Outliers, outlier analysis & removal
Analytical outliers do occasionally occur, and can greatly impact cut point determination. It was argued that cut point determination should mimic routine testing conditions. Increased variability due to, for example, more than two analysts, multiple plate lots, substrate/read buffer lots, different plate readers, different laboratories and so on, will likely result in higher cut points and so should be captured as part of cut point determination. However, the consensus was that production run analytical outliers, changing laboratories or substrates and so on, generally have a relatively minor impact on cut point determination versus the biological variability with different individual samples (consider multiple vendors for serum samples) or a larger number of samples. It was noted that day-to-day variability should also be included in cut point determination. There was a discussion on the appropriate outlier factor to employ when there is a need to remove genuine outliers. It was agreed that a 1.5-fold outlier factor is not always appropriate, and often removes a large number of samples that are not true outliers and reflect biological variability. Such noise – representative of real study samples – should not be removed. Otherwise cut points might be low and only related to instrument noise and analytical variability without capturing biological variability. Other statistical approaches to outlier removal may be acceptable (e.g., 3.0 outlier factor, non-interquartile range [IQR] methods). If the outlier removal approach results in the removal of large numbers of outliers (e.g., >10%), there should be an investigation with respect to the assay, study population and statistical methods used, as it may indicate that the criteria for outlier removal may not be appropriate, especially if using samples from the study/disease population.
Hemolysis, lipemia & LTS for ADA assays
The necessity to test for the impact of hemolysis and lipemia for ADA assays was discussed. Based on the current industry experience, interference from hemolysis and lipemia does not seem to impact LBA ADA assays and these tests should not be required for ADA immunoassays even if they are used, as per EMA guidelines, for LBA PK assays [Citation39]. The discussion of whether LTS studies of controls for ADA assays are needed continued following initial conclusions reported in the 2016 White Paper [Citation15]. At that time, it was recommended that up to 3 months stability could be acceptable. If the surrogate ADA stability has been established, the same ADA may not need to be reassessed in a different animal species or human. This year’s discussion also affirmed that PC stability is not relevant to stability of samples. IgG stored under suitable controlled conditions (-20°C or -80°C) has been demonstrated in the literature to be stable.
Monitoring biologically active biomarkers & evaluation of NAb via neutralization of this activity
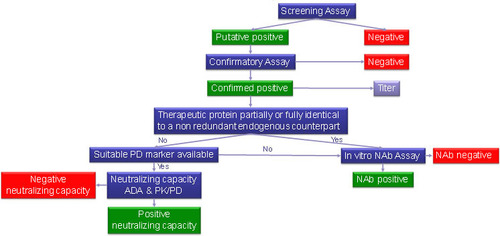
Although there has been much discussion within the biopharmaceutical industry to consider an integrated assessment of ADAs and PK/PD as a means to understand presence and impact of NAbs, regulatory agencies are still requiring the standard three-tiered approach (including in vitro NAb assays for characterization) [Citation23]. In general, ADAs can lead to an impact on efficacy or on safety. There was agreement that NAb assays are essential for high-risk molecules, such as replacement therapies. However, for low-risk molecules with an antagonistic MOA, there is interest in using an integrated data approach. NAbs are thought to influence clinical effects; however, only some clinical consequences are mediated by neutralizing antibodies (i.e., reduced or diminished efficacy and loss of function of the endogenous counterpart). For low-risk molecules where NAb impact would result in lower exposure and thereby potentially reduced efficacy, the impact can frequently be seen more readily by integrating PK, PD and ADA datasets. This topic has been widely discussed in the 2015 and 2016 White Papers [Citation12,Citation15] and during those discussions the regulators asked the industry to provide sound data and a clear strategy to reach a consensus on the way forward. Hence, this year’s discussion started with the consideration that cell-based NAb assays measure downstream functional changes (e.g., increase or decrease in an analyte downstream of a signaling pathway), which is a direct measure of a biomarker of NAb impact in the context of the assay. Some real case studies were considered (fully humanized IgG1 anti-BDCA2 mAb, IFN-β and anti-TNF monoclonals) and an in-depth evaluation was done on the practical decision tree proposed by the European Immunogenicity Platform (EIP) to choose which circumstances might allow the use of PK/PD and ADA data to assess the neutralizing capacity of ADAs.
Overall, an integrated assessment of PK/PD and ADA may be able to predict the NAb status, but it cannot discriminate if an observed increased clearance/decreased PD is due to Abs that can form immune complexes and accelerate drug clearance only or a combination of such immune complexes and neutralizing antibodies, unless we can discriminate between total and active drug by appropriately characterized PK assays. Thus, NAb assay and PK/PD measurements provide complementary information. Increased clearance/decreased PD is expected to impair the efficacy of a therapeutic protein (irrespective of the presence of NAbs). However, in the presence of NAbs, a deficiency syndrome might occur (in addition to impaired efficacy) if the therapeutic protein is partially or fully identical to a nonredundant endogenous counterpart. In these cases, NAbs represent a potential safety issue [Citation40]. For these therapeutic proteins, dedicated NAb assays are warranted, and it is important to deploy a dedicated NAb assay from the beginning of clinical development. For therapeutic proteins which are neither partially, nor fully, identical to nonredundant endogenous counterparts, it may not be important if an observed decreased efficacy is due to complex formation and/or neutralizing antibodies [Citation41]. For these therapeutic proteins, it is deemed appropriate to use an integrated assessment of ADA and PK/PD to assess the neutralizing capacity of ADAs. However, this is not accepted by either the EMA or the FDA at this stage – an NAb assay is still expected by both agencies for most cases.
The decision to deploy a standalone NAb assay or other biomarker assay of NAb impact should occur on a case-by-case basis, preferably in consultation with regulators. The totality of data from different assays (PK, ADA, PD, NAb) that best reflect the in vivo situation should be used. PK, ADA, PD (without NAb) may be sufficient for regulatory submissions in some situations, but these assays will not necessarily replace a NAb assay. It is suggested to use the decision tree in as a guide. Sponsors should discuss alternative testing plans with regulatory agencies if differing from recommended tiered approaches.
PK assays
Use of IS in LBA to improve precision & accuracy: is it possible?
The use of an IS is required for LCMS bioanalytical assays and ensures accuracy and precision. Moreover, in many chemical and biochemical assays, an IS can also be used to ensure proper dilution of the sample and to monitor assay performance. Quantitative LBAs that support PK evaluations typically utilize a standard curve and QCs to monitor the performance of these assays. However, assays that support potency or those that do not have a standard curve may benefit from an IS to control the assay performance. A case study was discussed where the IS concept was applied to monitor sample dilution bias in a relative potency ELISA under GMP [Citation42]. A fluorescent dye was spiked into test articles and reference standards at the first assay step and carried downstream through all subsequent sample dilution steps. After the sample and reference curves were added to the plate, the IS was used to quantitate any dilution bias between the samples and standards. The measured dilution bias was used to adjust the final ELISA test results rather than calculating the reportable results using only the nominal sample dilutions. This internal dilution standard approach has resulted in improved assay precision for this type of assay. It was discussed whether a similar approach could be applied to LBA in bioanalysis. This proposal was very interesting and it generated a healthy debate. However, it was concluded that the use of IS is best suited for LBA-based potency assays under GMP. It was discussed that while there is no perceived benefit, the use of an IS could only be done with two different capture antibodies in order to prevent the IS from competing with the main analyte, which then removes the advantage of normalizing the results as the capture and binding of a second molecule adds variability.
What assay should be developed: free, total or active drug PK assays?
Elucidation of the PK properties of new drug candidates and understanding of the PK/PD relationship is a crucial part of drug development. The power of an established PK/PD model is highly dependent on the data provided for PK modeling and thus requires clearly defined high quality bioanalytical data. It is important to define what is meant by free and total drug assays as the assay can be ligand-free or ligand-bound to the drug (total). Alternatively, a free assay can be drug unbound to ADA (free) or bound to ADA (total). The new generation of highly potent (multidomain) biologics is administered at low doses. A significant fraction could be present in a sample in a complexed, and thus neutralized form, bound to a soluble target and/or ADA. Depending on the required information, a bioanalytical strategy which clearly differentiates between the different drug forms might be required. On the one hand, since free/active drug exposure is considered to be the most relevant information allowing for a meaningful understanding and interpretation of PK and PD data, active drug assays become increasingly important. On the other hand, knowledge of true total drug data might be required to gain an understanding of the drug clearance. In addition, depending on the mode of action of multidomain biologics, the bioanalytical strategy might even provide information on the specific activities of the different domains of the drug.
It was agreed that the selection of the bioanalytical strategy requires a clear understanding of what form of the drug (e.g., free/active, total) needs to be monitored under careful consideration of the drug format, the disease biology and its mode of action. Building on this, appropriate bioanalytical methods need to be developed taking into consideration the bioanalytical challenges. For accurate free drug quantification and preparation of appropriate QC samples, the potential erroneous bias of the result due to the presence of soluble ligand in the matrix has to be considered. As described in references [Citation15–17], critical steps are calibration and sample dilution. Use of soluble ligand-containing matrix for the calibration of a free drug assay might result in a systematic shift of the calibration curve and its use for sample dilution might result in the formation of new drug-ligand complexes since additional ligand is added to the equilibrium at every dilution step. The relevance of the effects on the accurate free drug quantification requires a case specific evaluation, taking into consideration, among others, soluble ligand concentrations in the matrix as well as the assay sensitivity. The proposed use of ‘ligand-free’ matrix to circumvent any soluble target-related bias of the free drug assay result [Citation16,Citation17] was considered as a solution. However, the current practice in industry is still fragmented. The discussions from 2016 on this topic has clearly raised the bar on the importance of this topic and stimulated the interaction between industry and regulators. Relative effects of the surrogate matrix and the fact that the surrogate matrix cannot be used for QCs need to be further discussed with new case studies by multiple companies at next year’s meeting.
Novel biological therapeutics: understanding their biology & MOA to develop innovative modality-based bioanalytical strategies
Viral gene therapy has been tried for several years now, targeting a number of genetic and acquired conditions including hemophilia, cancer, cardiovascular diseases and muscular dystrophy. Viral vector delivery is a very promising technique in delivering the gene of interest to the target cells or tissues due to its relative ease of production, capacity, ability to infect a variety of different cell types and efficiency of transfection in the targeted cells. Numerous viruses have been tested as delivery vehicles including lentiviruses, adeno-associated viruses and adenovirus. During drug discovery and development, a number of significant challenges have been discovered and addressed. However, numerous questions and concerns remain open as further development of the viral delivery-based gene therapy is advancing. One of the significant challenges related to development of the viral gene therapy modality is potential impact of pre- and post-administration immunogenicity. A suite of immunogenicity assays should be considered in support of a gene therapy product, including pre- and post-administration immune responses against viral capsid, transgene product and transfected cells (only if using allogeneic cells but not if autologous cells are used) [Citation43–45].
Other novel modalities discussed were drug conjugates, bispecifics, prodrugs, oligonucleotides and cell therapies. It was unanimously reconfirmed that methods need to be FFP or what is technically feasible, and it needs to be scientifically justifiable. It was clearly pointed out that some of the novel therapeutics do not require PK assays, but may only require PD assays. For instance, PK assays may not be relevant in gene therapy using hematopoietic stem cells because the transduced stem cells migrate to the bone marrow, engraft and begin to repopulate peripheral blood. Thus, the ‘API’ is no longer in circulation. A surrogate assay (e.g., quantitative PCR) can be developed to detect vector sequences in peripheral blood, as an indicator of successful engraftment of transduced cells in bone marrow. In addition to serving as potential surrogate PK markers, vector persistence can also serve as safety biomarkers, especially for integrating vector-based delivery systems. However, they come with the price of being cumbersome and low throughput. Any assay type should be confirmed for ability to reflect on the biotherapeutic mode of action and activity. Then, the LBA could be a surrogate to the cell-based assay if you can prove that a downstream activity could be correlated to a binding event.
Combination therapies with biotherapeutics: what unique additional concerns should be considered in method development & validation?
Combination therapies with two (or more) biotherapeutics are an exciting new area of development [Citation12]. Regardless of serial, concomitant or fixed dose combinations, both biotherapeutics are likely to be present in circulation at the same time. This is particularly true when both therapies fall in the same modality; for example, two monoclonal antibody therapeutics are likely to have similar half-lives and therefore likely to be present in samples at the same time. Guidance exists for clinical trials using combination therapies [Citation46,Citation47], however, none mention the bioanalytical or immunogenicity considerations, which include issues such as common scaffolds between molecules, cross-reactivity of ADA and bridging data from earlier studies.
Consensus was reached on the fact that, typically, the same approaches to monotherapy should be applied to combination therapy assays unless there are specific MOAs that are at the interplay (e.g., synergistic effects). The assays selected should be specific to the relevant molecules. It was agreed that if the two therapies have a similar scaffold or belong to the same subclass of immunoglobulins, current practice is to use two separate assays and check each for cross-reactivity. However, if long-term use of the combination is anticipated, a multiplex assay may be considered, driven by practical, ethical and regulatory issues (e.g., amount of sample volume required for multiple assays, how many drugs are present in combination). For a PK assay, testing for interference at the maximum concentration (Cmax) and at the lowest expected concentrations should be performed. If the molecules have identical scaffolds and ADAs are not directed against the complementarity-determining region (CDR), then cross-reactivity should be identified through ADA testing. When a marketed biologic is being tested in combination with an experimental biologic (not previously approved), the immunogenicity testing approach applied during the clinical development of the marketed biologic may continue to be used. This will allow continuity and enable a certain degree of comparison of immunogenicity across the monotherapy and combination therapy.
Biomarkers
Review of the 2015 & 2016 WRIB White Paper biomarker recommendations
Discussion of topics regarding BAV began with the 2012 White Paper [Citation5] and have continued every year thereafter. Many recommendations were proposed, especially in the 2015 and 2016 documents [Citation12,Citation15]. These recommendations were reviewed this year to verify how many companies were applying them after another year of experience and to ensure they were still relevant and scientifically sound.
Accuracy (relative)
When it comes to protein biomarker assays, the term ‘relative accuracy’ is more appropriate than ‘accuracy’. The challenge is primarily related to the fact that in most cases a true reference standard is not available that reflects the exact nature of the endogenous biomarker. Therefore, protein biomarker assays generally yield relative quantitative rather than absolute quantitative results. However, some small molecule assays can demonstrate absolute quantitation due to the availability of a true reference standard. It is important for a biomarker/bioanalytical scientist to understand that simply applying PK assay type criteria may not be applicable to every aspect of a given biomarker assay, especially if the assay primarily demonstrates relative quantitation. There were no additional recommendations following the discussion other than underscoring the importance of understanding the COU, the biomarker biology/assay performance and interpreting the results accordingly.
Precision
Discussion regarding precision of biomarker assays concluded that PK assays and their associated precision criteria may not be the best fit for some biomarker assays. The consensus was that there is no ‘one size fits all’ recommendation. Acceptable precision depends on the biology of the biomarker and the expected effect size in relation to the clinical COU. A CV of more than 20% may be acceptable for biomarkers when expecting a large magnitude of change; however, more importantly, small changes in biomarker response may not be detected if the precision bias is too large.
Reference standards
There were no new recommendations regarding reference standards used for biomarker assays. The recommendation to obtain a reference standard as close as possible to the endogenous molecule and to demonstrate parallelism and specificity is still the best option.
Matrix
There were no new recommendations regarding matrix selection for biomarker assays. The expectation is that a similar matrix for the reference standard as the samples would be the best approach whenever possible. However, a protein-based buffer can also serve as a good substitute, if parallelism and selectivity have been demonstrated. It was concluded that the recommendations in prior white papers [Citation12,Citation15] and the outputs of Crystal City VI conference reports [Citation18,Citation19] are still valid for accuracy, precision, reference standards and matrix.
Biomarker stability: how do the preanalytic factors affect protein biomarker measurements – can we establish some ‘gold standards’?
It is widely recognized that biomarkers can add immense value during drug discovery and the development continuum all the way up to clinical practice. While the discovery and development of biomarkers holds huge promise, it is clear that in order to draw meaningful conclusions, the analysis needs to be as close as possible to the time of collection of samples. This seemingly simple task is ridden with multiple challenges including preanalytic factors (e.g., collection, storage, shipping and handling), which can have a huge impact on the biomarker’s stability, thereby affecting data reliability [Citation48,Citation49]. Since the impact on sample analysis can be quite substantial, it is important to evaluate sample collection and handling procedures in the assay development phase and subsequently provide the clinical sites with detailed instructions on controlling preanalytical variables [Citation50] in a procedure manual (or lab manual), including specifics such as collection kits, collection tubes, exact processing methods with detailed steps, timing and temperature of each step, rate of freezing, aliquot size, etc. It is also important to balance details with the feasibility of executing specialized instructions at clinical sites. Keep the instructions and collection materials as simple as possible to ensure compliance. Phase I sites may be able to handle more complex procedures, while for global Phase II/III sites, complex procedures may not be feasible. Thus, as a drug program advances, staged efforts should be made to simplify sample collection and handling or even consider alternative biomarker approaches to collect the key scientific data. It may be worth investing efforts in auditing clinical sites and training site personnel either in-person or remotely via video. Also consider implementing continuous monitoring at the clinical site(s) after personnel training to ensure that the procedure(s) are followed consistently by all personnel throughout the trial.
Efforts in evaluating preanalytic factors during assay development should reflect the complexity and level of decision-making required for the biomarker being studied. Importantly, one should understand the scientific and operational limitations of conducting biomarker analyses in a clinical setting and thereby inform the overall biomarker strategy for a drug-development program. Fundamentally, one must understand the biomarker and its properties as it relates to the collection and processing of samples. For example, specialized equipment or materials may be required to stabilize labile analytes at the time of collection, such as protease or phosphatase inhibitors. Cerebrospinal fluid (CSF) or urine sample collection may require antiadsorptive agents and/or low-binding tubes. Once samples are collected, consider how to minimize material loss or degradation during sample processing, aliquoting and storage with special attention to timing, aliquot volumes, storage tubes, stabilizers, temperature, vortexing, centrifugation, appropriate lighting and so on. The site must have the required equipment to appropriately handle samples, such as refrigerated centrifuges and -80°C freezers. Following sample collection and initial processing, one must consider all the elements that affect sample storage at the site and during shipping. It is equally important to consider the level of tracking required to ensure sample integrity during the journey from the clinical site to the bioanalysis lab, temperature monitors may be appropriate to identify any deficiency in sample handling. Sample chain of custody must be reconstructable, so that one can establish in what piece of equipment and at what temperature samples have been stored. Storage should occur at a previously validated temperature, and it must be controlled and monitored for as long as samples remain in the unit. Many compounds are stable for a longer time at -80°C than at -20°C; however, these freezers are not always available at all clinical sites. If only -20°C freezers are available, one should understand the analyte’s stability during assay validation at this temperature for specific storage durations. More frequent shipments to the bioanalytical lab may be needed to ensure timely sample analysis or maintain long-term storage within the established stability window. Best practices are to discuss and implement a shipping plan and schedule early on to ensure sample integrity during shipment and timely receipt by bioanalytical lab personnel. Therefore, due to potential lack of LTS at -20°C, the samples shipped to the central lab or bioanalytical lab are then kept stored at -80°C. In such cases, the stability assessment of the biomarker during assay validation must mimic the storage time and temperature conditions at the clinical site and central lab or bioanalytical lab. Furthermore, it should be planned that samples shipped to the lab are received by trained personnel to ensure appropriate handling and storage. Best practices are to discuss and implement a shipping schedule early on. A reliable shipping vendor should be chosen with experience navigating through customs processes to minimize delays, offering service to refill dry ice if needed. It is recommended to use a three-container system filled with dry ice and equipped with a temperature monitor.
Ultimately, the quality of biomarker data is often largely dependent on understanding the stability variables of the biomarker as much possible during assay development. If the biomarker is fragile yet critical, one may need to identify the necessary conditions to preserve the stability. Short-term and LTS testing should be performed using incurred (unspiked) samples to assess the stability of the endogenous analyte in matrix. To evaluate sample stability for biomarkers, the ideal practice is to obtain freshly prepared samples through an internal donor program or by other means early on in during the assay development phase. If that is not feasible, attempt to get the freshest samples possible and assign time ‘0’. In practice, often the earliest time point that can be measured is after one freeze-thaw and stability of one freeze–thaw should be assumed, but be aware that this approach has the potential to lead to significant bias. It may be appropriate to include internal QCs for sample stability for some analytes, but practices may vary from biomarker discovery to clinical biomarker development. In the case of sample stability for RNA biomarkers, it is recommended to use housekeeping genes for gene expression (e.g., 6–7 gene panel), but controls for miRNA are still evolving. However, it is important to ensure that these housekeeping genes or controls do not change in the disease population in the study. Research to develop such IS to assess sample quality is in progress in various laboratories.
While blood is the preferred source for biomarker analysis since it can be collected in a relatively noninvasive manner in most situations, oncology biomarkers are often measured directly in biopsies or surgically resected tumor tissues. These tissue samples are either frozen or FFPE. FFPE-thin sections are routinely generated by pathologists for the analysis of tumor pathology through hematoxylin and eosin (H&E) staining and microscopic analysis. These sections are also used for immunohistochemistry (IHC) for the detection of specific proteins. According to experts, there are more than a billion FFPE tissue samples archived in hospitals and tissue banks around the world due to their long-term storage at ambient temperature and minimal storage space requirements. However, the preanalytical processing required for these preparations presents challenges introducing variation impacting quality and reliability of assay results, whether using IHC or MS end points. For example, fixation time and the quality of the fixation buffer can have an impact on stability since molecular changes may occur during the process; the most sensitive is phosphorylation state of the protein. Given that no pathologists were present, it was not possible to comprehensively discuss the specific challenges around FFPE stability. Instead, there was consensus that bioanalytical scientists should work closely with pathologists to ensure appropriate collection and handling of FFPE samples required with respect to the expected biomarker analyses. To date, IHC is widely used for biomarker measurement and represents a useful tool for tumor diagnosis and patient selection. However, this technique is at best semiquantitative and provides limited possibilities for multiplexing. In contrast, developments in MS have shown that this technology is quantitative and reproducible despite the extent of formalin cross-links. Furthermore, MS provides high multiplexing capabilities measuring up to several hundred proteins in one assay. However, both MS and the noncell-based LBAs can only analyze tissue homogenates and cannot address the variability caused by tissue heterogeneity, or provide the spatial information of biomarker expression. When incorporating an internal control for tissue quality, ß-actin is often used. Tissue slices can be treated for IHC similar to an immunoassay where stability is assessed over time. It was concluded that assessing post-translational modifications (PTMs) is more challenging and is often influenced by preanalytical factors compared with total protein. Additionally, fixation time and the quality of the fixation buffer can have an impact on stability.
Challenges & cutting-edge solutions in biomarker assays for mitigating variability & improving reliability
Given the increased interest and importance surrounding biomarker applications during drug development, the panel discussed how to mitigate variability and improve the consistency of biomarker assay performance, despite the complex issues associated with them. The panel also discussed that precision and reproducibility are at least as critical for a biomarker assay as for a PK method, and the criteria for acceptance depend on understanding the biological and analytical variability and the COU for the biomarker data [Citation51–53]. In order to attempt to counter some of the many factors that contribute to biomarker assay variability, it is recommended to characterize critical reagents, standardize all steps and use automation as much as possible. There was consensus that factors that can affect biomarker assay variability are no different than the factors that can impact any other LBA method and include changes in critical reagents, pipette technique, uniform timing, plate coating and equilibrium binding. In order to attempt to counter some of these issues, it was agreed that consistent pipetting is critical and similarly automation may be desirable approach. Adsorption of both endogenous biomarkers and spiked samples can contribute to imprecision of measurements, and may be minimized by using low binding tubes or detergents. Importantly, best practice is to incorporate endogenous QCs in assay validation and sample testing to assess assay precision and monitor assay drift. Caution should be used when adopting a commercial kit assay. It is important to evaluate specificity and parallelism as different manufacturers’ kits may measure different forms of the same analyte, or even not the intended analyte. Also, lot-to-lot differences should be evaluated in assay development or validation. Overall, it is important to verify the information provided by the vendor and assess kit performance with the endogenous analyte in appropriate matrix. It was agreed that practices to reduce assay variability are similar between biomarker assays and PK or immunogenicity assays, and include analyst training and qualification. There is no simple, ‘one size fits all’ solution. Every analyte is different and each critical step in the method should be appropriately evaluated during assay development.
Utility of specific CLSI guidelines for biomarkers in clinical drug development: building on the 2016 recommendations on global harmonization of BAV
Building on 2016 recommendations on global harmonization of BAV [Citation15], the applicability of CLSI guidelines to BAV and utilization was discussed. The FDA database of recognized consensus standards includes over 100 CLSI consensus standards and guidelines. These are directed toward the CLIA-regulated laboratory setting post-FDA clearance and approval, but can also serve as valuable information sources for bioanalytical methods pertinent to biomarker testing in the drug development arena. As the intended use of these documents is not BMV for drug development, it is important to understand where their guidance can be appropriate in that context to global developers. However, both FDA bioanalytical guidance and CLSI biomarker guidelines have similar detailed reporting, training and recordkeeping requirements, as well as a quality system dedicated to ensuring traceable and quality results, and so are very similar in the spirit of the content. The main scientific and regulatory expectations for biomarker assay performance minimally include determination of (relative) accuracy, precision, analytical sensitivity, analytical specificity, reportable range (LLOQ, ULOQ) and robustness. Many factors should be taken into account when assessing the robustness and reliability of a biomarker assay. Robustness of an assay is the property that indicates insensitivity against changes of known operational (critical) parameters on the results of the method and hence its suitability for its defined purpose. Reliability is not a defined term, but ruggedness of an assay is the property that indicates insensitivity against inadvertent changes of known critical operational parameters plus any variations not discovered in intralaboratory experiments which may be revealed in interlaboratory experiments. Parameters to assess may include analyte sensitivity, analyte amount, blank matrix, known interferences and cross interferences, and known influences of known factors (e.g., pH and stirring).
It was discussed that since the CLSI guidelines cover many relevant aspects related to biomarker method validation that many concepts could be drawn from them. The use of these guidelines for assistance on BAV is most appropriate for those biomarkers that will have data submitted to regulatory agencies, rather than for exploratory biomarkers used only for internal decision making. Although many aspects of the CLSI guidelines may be instructive references for biomarkers during drug development, none are required. Commonly used biomarkers in drug development that are pathway related or simply exploratory to help understand modulation of disease biology are not required to be qualified, nor do they require the use of these guidelines. The stage and extensiveness of validation will dictate which CLSI guidelines, if any, may have utility. Bioanalysts must define what is necessary and what is not. CLSI guidelines are generally more rigorous than BAV and are required only for in vitro diagnostic devices (IVDs) and for companion diagnostics (CDx) in a commercial diagnostic laboratory setting. Existing literature outlining the FFP validation requirements [Citation51,Citation54] should be used for exploratory biomarkers in drug development, and layers of validation from CLSI documents may be added as appropriate to the COU. It is a continuum.
It is recommended that the definitions of the biomarker-related terms in Biomarkers, Endpoints, and other Tools (BEST) [Citation55], which are accepted by the FDA, and consistent with CLSI guidelines, should be the solitary definitions used for those terms. The current preferred application and understanding of the definition of COU for biomarkers in drug development is provided in BEST [Citation55], and it does not have a direct relationship to Center for Devices and Radiological Health (CDRH; PMA/510k), Center for Drug Evaluation and Research (CDER; BAV) and CLSI (CLIA) requirements. The intended use in a PMA or 510(k) is very specific and, if deviated from, a revalidation for the COU is required. Though CLIA applies to testing of human specimens and requires establishing certain performance characteristics relating to the analytical validity, it does not directly regulate the test itself nor specify exact performance requirements for the use of that test. Therefore, CLIA requirements are not applicable (NA). Bioanalytical validation is different than clinical validation and COU is more descriptive than FFP. Refer to the below for a comparison of some of the parameters to be considered for different levels of bioanalytical validation goals.
Table 1. Comparison of validation conditions for different regulatory requirements.
Although BAV is addressed in some form across many documents, there is not yet a harmonized global BAV Guidance. The latest guidance from the FDA to address this topic, the 2013 draft BMV guidance [Citation20], is still undergoing review following the comment period; many comments suggested separation of BAV from BMV. However, the two meeting reports available following the Crystal City VI meeting [Citation18,Citation19] and recommendations in the 2016 White Paper [Citation15] are still valid. A public workshop (FDA/Critical Path/industry) was held on 14–15 June 2017 to obtain feedback on analytical validation considerations in biomarker qualification [Citation56]. Comments will also be solicited from global regulatory bodies. This will be used by the FDA as scientific input into analytical validation for biomarker qualifications. In an effort to harmonize their efforts and for continued scientific exchange of ideas, quarterly meetings between the FDA and the EMA continue to be held.
Clinical biomarker FFP validation in support of biotherapeutics: how to overcome scientific & regulatory challenges
Development and application of biomarkers begins during the earliest stages of a project and continues through the clinical phase. For clinical biomarker research, one of the goals is establishing scientifically meaningful and reproducible assays to generate reliable data that are suitable for strategic decision-making for a drug development program. Development and validation of biomarker assays are generally more complicated than those used for most pharmacokinetic drug measurements, due to the endogenous nature of the molecules and the wide variety of modalities that serve as biomarkers. In order to ensure that FFP BAV fully supports the intended purpose of the data in a clinical setting, it is crucial to understand and define the biology, the reasons for investigating that biomarker and what the hypothesis is for its utilization, that is, the COU (refer to BEST definitions [Citation55]). Following this definition, the validation and required evaluations should scientifically support the assay. The recommended industry best practice for optimizing assays toward increased specificity, sensitivity, a degree of accuracy and robustness is to implement design input requirements. A study sample analysis should be based on clear communications and understanding with a clinical team to determine the COU including necessary specificity, sensitivity, accuracy and robustness. This allows the bioanalytical scientist to choose the appropriate platform and optimize accordingly. Biological variability has already been defined [Citation57] for inter- and intrapatient variability. The COU will determine if sex, diurnal variations, fasting, age, smoking, etc. need to be considered. Depending on the needs of the assay – time constraints, throughput, cost, testing facility complexity, LBA reagent availability – the assay platform will be selected. Generally, bioanalysts start with LBA; however, if appropriate reagents are not available or do not fit the need, other avenues like hybrid LBA/LCMS assays are pursued and have been used to support clinical studies.
Recommendations
Below is a summary of the recommendations made during the 11th WRIB.
Immunogenicity
2016 US FDA & 2015 EU EMA immunogenicity draft guideline/guidance
Immunogenicity risk assessment
Immunogenicity risk score classification is not based on therapeutic modality. Instead, the MOA and other factors (e.g., patient population) need to be considered for appropriate risk categorization. In general, novel therapeutic molecules are perceived to carry higher risk potential than known types of therapeutics where there is clinical experience. In addition to new structures, novel production systems may also include factors (e.g., impurities) that could alter a product’s immunogenicity risk. When the risk category for the product is higher, well-developed assays will be required earlier in drug development. More frequent sample collection and interim analysis should be considered for high-risk molecules.
Sponsors should typically perform a risk assessment early during drug development based on the prospective evaluation of probability and consequences of immune responses. It should be a multidisciplinary approach that includes information from the drug modality, MOA, CMC and patient population.
Data from early clinical studies can be incorporated into the integrated summary of immunogenicity. The risk assessment may be redefined with data from early clinical studies if in a relevant patient population. Early clinical data may provide insights on the effect of ADA on PK, efficacy and safety. For pivotal studies, sponsors should plan for evaluation of NAb activity, which may require a standalone NAb assay or in some cases may be understood by integration of PK, ADA and PD data.
There may not always be a clear relationship between ADA incidence and clinical risk. The risk is an assessment of the probability and clinical consequences of an immune response to the drug. The ADA incidence is the proportion of subjects that seroconvert or boost their ADA titers as a result of exposure to the drug [Citation30] and is only one factor in determining the risk/probability of an observed immune-mediated adverse effect.
For determining comparability of two biosimilar products, the recommendation is to perform a parallel arm study with a similar dose scheme in a head-to-head comparison. If possible, the lots of the two products should have similar age. For ADA sample analysis, a single assay with the biosimilar reagents for screening is recommended but the ADA specificity should be characterized with both in the confirmatory assay. Communicating early on with regulators regarding study design is recommended, for example, an adaptive design is recommended for studies with a small number of patients.
The evaluation of an in-study cut point must be scientifically sound. The information provided is useful to determine whether the validation cut point using commercial samples is confirmed or not. The establishment of an in-study cut point provides a better representation of the variability of the study/disease population, leading to more appropriate cut point values and, consequently, a more accurate assessment of the true incidence of ADA. For pivotal studies, the evaluation of a subset of samples can be used to confirm or replace validation cut points for screening and confirmatory assays, as appropriate.
Assay sensitivity
There is an expectation to provide the procedure to establish MRD as a summary of development work (e.g., within the integrated immunogenicity summary, or as a link to validation report that includes summary of development data). The LPC may need to be adjusted in clinical development and can have different acceptance ranges for screening and confirmatory assays.
Drug tolerance
The current best practice to determine drug tolerance and to control the impact on assay sensitivity and poststudy monitoring is to assess two or more levels of ADA (target sensitivity level included) and several levels of drug (includes expected trough level). Varying levels of drug concentrations should be tested to determine the drug tolerance threshold.
If assay drug tolerance cannot be improved, resulting in ADA assays with significant drug interference, the sponsor should provide a summary of the totality of the assay development data and demonstrate a good faith effort has been made to develop a drug-tolerant assay.
Regardless of the type of PC chosen, a sample that tests positive is considered positive. Using a PC is the preferred method over extrapolation to actual ADA in the sample or using clinical ADA-positive samples that are in limited supply even when available. A medium-level PC was recommended for assay validation but is not required for production.
Isotyping, epitope mapping and cross-reactivity assays are not always needed. A cross-reactivity assay is crucial if there is an endogenous counterpart to the therapeutic molecule. If a strong safety signal is detected, then subsequent isotyping may be needed. ADA domain specificity for novel multidomain therapeutics is expected in confirmatory assays. Epitope mapping is rarely requested (e.g., to determine if detected ADAs are linear or conformational epitopes) but may be useful to the sponsor in directing future development efforts.
ADA results should be integrated with clinical data by relating PK drug levels to ADA results for specific sampling time points. Interpretations of ADA positivity should be integrated with clinical efficacy, PD and safety outcomes. The categorization of persistent versus transient responses as well the magnitude of the response may provide a better correlation with PK, efficacy or safety.
Setting ADA assay cut points
ADA assay reproducibility
Reporting persistent responses and evaluation of ADA relative to titer quartiles may provide better correlation with impact on PK and efficacy than overall incidence of ADA.
In order to ensure that study samples that screen positive initially are positive again when assayed in the confirmatory assay, an assay that captures biological variability should be developed. Ensuring the assay is well controlled with a clearly defined procedure (e.g., shaker speed, controlled incubator temperature) is a good practice. The use of %CV (n = 2) for assay pass/fail may not be practical.
A common approach for confirmatory assays is to consider the percent inhibition in the assay signal of the drug-spiked sample by comparison within the unspiked sample included in the assay run. It is recommended to take multiple time points to truly assess the ADA status of the individual (as distinct from the sample). The data should be reported as is and the interpretation discussed with regulators/sponsors. Consideration should be given to the concept that the confirmation cut point should not be lower than the empirically determined variability in the assay.
It is recommended to include a minimum of a low- and high-positive and -negative control in all runs to monitor assay performance. The signal should be monitored, as well as S/N. Furthermore, Levy-Jennings graphs should be considered to plot assay signal and assess trends for information purposes.
Consider using LPC at approximately two- to three-fold S/N level for validation (as long as the resulting levels are not several folds higher than the majority of positive study samples) and efforts should be made to assess the 1% failure level on validation data.
Complex cases
Cut point determined in validation using 50 samples is provisional. The appropriateness of the cut point with study samples should be reassessed. If more than 50 baseline samples are available, it is acceptable to use a greater number of samples analyzed fewer times. The use of a larger number of samples may provide a better representation of the variability in the study population.
Actual preexisting positives that have high signals should be removed from cut point calculations. The focus should be on TE positivity, including treatment-induced and treatment-boosted responses [Citation30]. It is recommended to report separately treatment induced from baseline negative samples versus preexisting boosted responses. Individuals that are positive at baseline but do not show boosting of ADA response upon treatment should not be included in the overall ADA incidence calculation.
Cut point determination is not affected by high TE ADA incidence. Clinical response assessments may not reveal an impact of ADA. Clinical findings should be correlated with ADA titers and persistency. A change in PK is a measurable end point that is potentially a cleaner gauge of ADA clinical impact.
Outliers, outlier analysis & removal
Analytical outliers should be removed during cut point determination. It is important to capture biological variability with different individual samples (multiple vendors for serum samples should be considered). Day-to-day variability should be included in cut point determination.
A 1.5-fold outlier factor may not always be the most appropriate. Other statistical approaches to outlier removal may be acceptable if properly justified (e.g., 3.0 outlier factor, non-IQR methods). Large numbers of outliers (e.g., >10%) should be investigated with respect to the assay, study population and statistical methods used. The removal of a large number of outliers may be an indication that the criteria for outlier removal are inappropriate.
Evidence of hemolysis and lipemia impacting ADA assays appears insufficient at the current time. PC stability is not relevant to stability of samples. IgG stored under suitable conditions (-20°C or -80°C) has been demonstrated in the literature to be stable.
The decision to deploy a standalone NAb assay or other/biomarker assay of NAb impact should occur on a case-by-case basis following the decision tree in . The totality of data from different assays (PK, ADA, PD, NAb) that best reflect the in vivo situation should be used. However, it should be noted that regulatory agencies still expect NAb assays and deviation from that situation requires scientific justification.
PK assays
IS may not be readily applicable to bioanalytical PK LBAs at this time because IS can add complexity to an already complex method without adding additional value to the results.
The selection of the bioanalytical strategy for PK assays requires a clear understanding of what form of the drug (e.g., free, active, total) needs to be monitored under careful consideration of the drug format, the disease biology and its mode of action. The proposed use of ‘ligand-free’ matrix to circumvent any soluble target-related bias of the free drug assay result was considered as a possible solution.
Although some novel biotherapeutics may not require true PK assays, an exposure confirming tests are expected, including PD assays. As long as these alternatives are scientifically sound and justified and properly validated, they can be accepted as a surrogate to PK assays.
The same approaches to monotherapy should be applied to combination therapy assays unless there are specific MOAs that are at the interplay (synergistic effects, etc.). The assays selected should be specific to the relevant molecules. If the two therapies have a similar scaffold or belong to the same subclass of immunoglobulins, current practice is to use two separate assays and check each for cross-reactivity. However, if long-term use of the combination is anticipated, a multiplex assay may be considered.
Biomarkers
It was concluded that the recommendations in prior White Papers [Citation12,Citation15] and the outputs of Crystal City VI conference reports [Citation18,Citation19] are still valid for accuracy (relative), precision, reference standards and matrix.
Biomarker stability – key considerations
It is imperative to provide clinical sites with very detailed instructions in a procedure/laboratory manual.
Invest efforts in auditing clinical sites and training site personnel either in-person or remotely via video.
Treat the collection process similar to the development of an assay protocol, in other words, with equal care. It should be kept simple, with detailed instructions. Clinical sites should have the necessary equipment and site staff should be well trained and monitored. Collection time and temperature should be recorded. A continuous monitoring program must be implemented to ensure that the trained personnel of the clinical sites continue to perform appropriately.
The site must have the equipment it needs to collect and preserve the samples appropriately such as refrigerated centrifuges and -80°C freezers.
Samples containing low amount of protein (i.e., CSF or urine) may require antiadsorptive agents and/or low-binding tubes.
Sample processing must follow a proper aliquoting plan with special attention to timing, temperature, vortexing, centrifugation and so on. Labile analytes may need stabilizers, protease or phosphatase inhibitors.
Sample chain of custody must be reconstructable.
Storage should occur at a previously validated temperature, and it must be controlled and monitored for as long as samples remain in the unit.
If samples must remain frozen, it is recommended to use a three-container system containing dry ice and a temperature monitor.
During assay validation, perform short-term and LTS testing using unspiked samples to assess the endogenous analyte in matrix.
In the case of evaluating sample stability for RNA and miRNA biomarkers, it is recommended to use housekeeping genes for gene expression (e.g., six–seven gene panel) for RNA but controls for miRNA are still evolving. However, it is important to ensure that these housekeeping genes or controls do not change in the disease population in the study.
To evaluate sample stability for protein biomarkers, the ideal practice is to obtain freshly prepared samples through an internal donor program or by other means. If that is not feasible, attempt to get the freshest samples possible and assign time ‘0’. However, most often, the earliest time point that can be measured is after one freeze-thaw.
When incorporating an internal control for tissue quality, β-Actin can be used. Tissue slices can be treated for IHC similar to an immunoassay where stability is assessed over time.
It is important to work closely with pathologists to develop a detailed protocol for FFPE sample collection and appropriately train clinical sites.
Automation should be used when possible to achieve consistent pipetting in order to reduce biomarker assay variability. Adsorption of both endogenous biomarkers and spiked samples can be minimized by using low-binding tubes or detergents (e.g., Tween-20 in CSF for Aβ42). Incorporate utilization of endogenous QCs to assess interassay precision and identify assay drift. If using a commercial kit for a biomarker assay, verify the information provided by the vendor and assess kit performance in the appropriate matrix instead of a buffer.
Utility of specific CLSI guidelines for biomarkers in clinical drug development
The main scientific and regulatory expectations for biomarker assay performance minimally include determination of (relative) accuracy, precision, analytical sensitivity, analytical specificity, reportable range (LLOQ, ULOQ) and robustness.
The use of CLSI guidelines for assistance on BAV is appropriate for those biomarkers which will have data submitted to regulatory agencies, rather than for exploratory biomarkers used only for internal decision-making. CLSI guidelines are intended for clinical laboratories dealing primarily with diagnostic biomarkers. Although many aspects of the CLSI guidelines may be instructive references for biomarkers during drug development, none are required.
Definitions on biomarkers are in BEST [Citation55] and are consistent with CLSI guidelines and accepted by the FDA. The stage and extensiveness of validation will dictate which CLSI guidelines may have utility. Bioanalysts must define what is necessary and what is not. CLSI guidelines are generally more rigorous than BAV and are required only for IVDs and for CDx.
Existing literature outlining the FFP validation requirements [Citation49,Citation56] should be used, and layers of validation from CLSI documents added as appropriate to the COU.
In order to ensure that FFP BAV fully supports the intended use of the data in a clinical setting, it is crucial to understand and define the biology, the reasons for investigating that biomarker, the hypothesis is for its use (i.e., the COU), and then design the validation and required evaluations to achieve the most optimal assay for the intended purpose. The recommended industry best practice for optimizing biomarker assays toward increased specificity, sensitivity, accuracy and robustness is to implement design input requirements. When the study has a clear plan on the COU including necessary specificity, sensitivity, accuracy and robustness, the responsibilities of the scientist is then to choose the appropriate platform and optimize the assays accordingly.
Immunogenicity: The ability of a substance, including biotherapeutic substances, to induce an immune response. For the purposes of this article, antidrug antibody (ADA) assays are equivalent to immunogenicity assays that are designed to measure and characterize ADA.
Biomarker: A defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes or responses to an exposure or intervention, including therapeutic interventions. Molecular, histologic, radiographic or physiologic characteristics are types of biomarkers. Categories of biomarkers include: susceptibility/risk biomarker, diagnostic biomarker, monitoring biomarker, prognostic biomarker, predictive biomarker, pharmacodynamic/response biomarker and safety biomarker [Citation57].
Cut point: An established assay-specific limit of the ADA assay that defines the sample response as positive or negative for the presence of ADA.
ADA incidence: The proportion of the study population found to have seroconverted or boosted their preexisting ADA during the study period [Citation30]. Synonymous with ‘treatment-emergent ADA’.
Acknowledgements
The authors acknowledge the US FDA, Europe EMA, UK MHRA, Austria AGES, France ANSM, Brazil ANVISA, Health Canada, Japan MHLW and Finland Fimea for supporting this workshop. S Gupta (Amgen), B DeSilva (Bristol-Myers Squibb), L Amaravadi (Sanofi), E Yang (GlaxoSmthKline), J Welink (EU EMA), A Song (Genentech), H Neubert (Pfizer), F Garofolo (Angelini Pharma) for chairing the workshop and/or the White Paper discussions. All the workshop attendees and members of the bioanalytical community who have sent comments and suggestions to complete this White Paper, and Future Science Group as a trusted partner are also acknowledged.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Savoie N , BoothBP, BradleyTet al. 2008 White Paper: the 2nd Calibration and Validation Group Workshop on recent issues in good laboratory practice bioanalysis. Bioanalysis1 (1), 19–30 (2009).
- Savoie N , GarofoloF, van AmsterdamPet al. 2009 White Paper on recent issues in regulated bioanalysis from the 3rd Calibration and Validation Group Workshop. Bioanalysis2 (1), 53–68 (2010).
- Savoie N , GarofoloF, van AmsterdamPet al. 2010 White Paper on recent issues in regulated bioanalysis and global harmonization of bioanalytical guidance. Bioanalysis2 (12), 1945–1960 (2010).
- Garofolo F , RocciM, DumontIet al. 2011 White Paper on recent issues in bioanalysis and regulatory findings from audits and inspections. Bioanalysis3 (18), 2081–2096 (2011).
- DeSilva B , GarofoloF, RocciMet al. 2012 White Paper on recent issues in bioanalysis and alignment of multiple guidelines. Bioanalysis4 (18), 2213–2226 (2012).
- Stevenson L , RocciM, GarofoloFet al. 2013 White Paper on recent issues in bioanalysis: “Hybrid” - the best of LBA & LC/MS. Bioanalysis5 (23), 2903–2918 (2013).
- Fluhler E , HayesR, GarofoloFet al. 2014 White Paper on recent issues in bioanalysis: a full immersion in bioanalysis (Part 1 – small molecules by LCMS). Bioanalysis6 (22), 3039–3049 (2014).
- Dufield D , NeubertH, GarofoloFet al. 2014 White Paper on recent issues in bioanalysis: a full immersion in bioanalysis (Part 2 – hybrid LBA/LCMS, ELN & regulatory agencies’ input). Bioanalysis6 (23), 3237–3249 (2014).
- Stevenson L , AmaravadiL, MylerHet al. 2014 White Paper on recent issues in bioanalysis: a full immersion in bioanalysis (Part 3 – LBA and immunogenicity). Bioanalysis6 (24), 3355–3368 (2014).
- Welink J , FluhlerE, HughesNet al. 2015 White Paper on recent issues in bioanalysis: focus on new technologies and biomarkers (Part 1 – small molecules by LCMS). Bioanalysis7 (22), 2913–2925 (2015).
- Ackermann B , NeubertH, HughesNet al. 2015 White Paper on recent issues in bioanalysis: focus on new technologies and biomarkers (Part 2 – hybrid LBA/LCMS and input from regulatory agencies). Bioanalysis7 (23), 3019–3034 (2015).
- Amaravadi L , SongA, MylerHet al. 2015 White Paper on recent issues in bioanalysis: focus on new technologies and biomarkers (Part 3 – LBA, biomarkers and immunogenicity). Bioanalysis7 (24), 3107–3124 (2015).
- Yang E , WelinkJ, CapeSet al. 2016 White Paper on recent issues in bioanalysis: focus on biomarker assay validation (BAV) (Part 1 – small molecules, peptides and small molecule biomarkers by LCMS). Bioanalysis8 (22), 2363–2378 (2016).
- Song A , LeeA, GarofoloFet al. 2016 White Paper on recent issues in bioanalysis: focus on biomarker assay validation (BAV): (Part 2 – Hybrid LBA/LCMS and input from regulatory agencies). Bioanalysis8 (23), 2457–2474 (2016).
- Richards S , AmaravadiL, PillutlaRet al. 2016 White Paper on recent issues in bioanalysis: focus on biomarker assay validation (BAV): (Part 3 – LBA, biomarkers and immunogenicity). Bioanalysis8 (23), 2475–2496 (2016).
- Staack RF , JordanG, HeinrichJ. Mathematical simulations for bioanalytical assay development: the (un-)necessity and (im-)possibility of free drug quantification. Bioanalysis4 (4), 381–395 (2012).
- Schick E , StaackRF, HaakMet al. Validation of a ligand-binding assay for active protein drug quantification following the ‘free analyte QC concept’. Bioanalysis8 (24), 2537–2549 (2016).
- Lowes S , AckermannBL. Conference report - AAPS and US FDA Crystal City VI workshop on bioanalytical method validation for biomarkers. Bioanalysis8 (3), 163–167 (2016).
- Arnold ME , BoothB, KingL, RayC. Workshop report: Crystal City VI-bioanalytical method validation for biomarkers. AAPS J. 18 (6), 1366–1372 (2016).
- US Department of Health and Human Services, US FDA, Center for Drug Evaluation and Research, Center for Veterinary Medicine . Draft guidance for industry, bioanalytical method validation. Rockville, MD, USA (2013). http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm368107.pdf.
- Pharmaceutical Research and Manufacturers of America . 2016 Biopharmaceutical research industry profile. PhRMA, Washington DC, USA (April 2016).
- DiMasi JA , GrabowskiHG, HansenRW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J. Health Econ. 47, 20–33 (2016).
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) . Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins. London, UK (2007). EMEA/CHMP/BMWP/14327/2006. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003946.pdf.
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) . Draft guideline on Immunogenicity assessment of biotechnology-derived therapeutic proteins. London, UK (2015). EMEA/CHMP/BMWP/14327/2006 Rev. 1. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/10/WC500194507.pdf.
- US Department of Health and Human Services, US FDA, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research, Center for Devices and Radiological Health (CDRH) . Draft guidance for industry, assay development and validation for immunogenicity testing of therapeutic protein products. Rockville, MD, USA (2016). http://www.fda.gov/downloads/Drugs/…/Guidances/UCM192750.pdf.
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) . Guideline on Immunogenicity Assessment of Therapeutic Proteins. London, UK, EMEA/CHMP/BMWP/14327/2006 Rev. 1 (2017). http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/06/WC500228861.pdf.
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) . Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. London, UK (2012). http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128688.pdf.
- Koren E , SmithHW, ShoresEet al. Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products. J. Immuno. Met. 333, 1–9 (2008).
- Wu B , ChungS, JiangXRet al. Strategies to determine assay format for the assessment of neutralizing antibody responses to biotherapeutics. AAPS J. doi: 10.1208/s12248-016-9954-6 (2016) ( Epub ahead of print).
- Shankar G , ArkinS, CoceaLet al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides—harmonized terminology and tactical recommendations. AAPS J. 16 (4), 658–673 (2014).
- Plotkin SA . Correlates of protection induced by vaccination. Clin. Vaccine Immunol. 17 (7), 1055–65 (2010).
- Zhou L , HoofringSA, WuYet al. Stratification of antibody-positive subjects by antibody level reveals an impact of immunogenicity on pharmacokinetics. AAPS J. 15 (1), 30–40 (2013).
- Zoghbi J , XuY, GrabertRet al. A breakthrough novel method to resolve the drug and target interference problem in immunogenicity assays. J. Immuno. Meth. 426, 62–69 (2015).
- Wessels U , SchickE, RitterMet al. Novel drug and soluble target tolerant antidrug antibody assay for therapeutic antibodies bearing the P329G mutation. Bioanalysis9 (11), 849–859 (2017).
- Stubenrauch K , WesselsU, EssigUet al. Evaluation of a generic immunoassay with drug tolerance to detect immune complexes in serum samples from cynomolgus monkeys after administration of human antibodies. J. Pharm. Biomed. Anal. 52 (2), 249–254 (2010).
- Dai S , SchantzA, Clements-EganAet al. Development of a method that eliminates false-positive results due to nerve growth factor interference in the assessment of fulranumab immunogenicity. AAPS J. 16, 464–477 (2014).
- Gorovits B , WakshullE, PillutlaRet al. Recommendations for the characterization of immunogenicity response to multiple domain biotherapeutics. J. Immunol. Methods408, 1–12 (2014).
- Myler H , HruskaMW, SrinivasanSet al. Anti-PEG antibody bioanalysis: a clinical case study with PEG-IFN-λ-1a and PEG-IFN-α2a in naive patients. Bioanalysis7 (9), 1093–1106 (2015).
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) . Guideline on bioanalytical method validation. EMEA/CHMP/EWP/192217/2009. London, UK (2011). www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf.
- Bendtzen K , GeborekP, SvensonMet al. Individualized monitoring of drug bioavailability and immunogenicity in rheumatoid arthritis patients treated with the tumor necrosis factor alpha inhibitor infliximab. Arthritis Rheum. 54 (12), 3782–3789 (2006).
- Jensen PEH , SellebjergF, SondergaardHBet al. Correlation between anti-interferon-b binding and neutralizing antibodies in interferon-b-treated multiple sclerosis patients. Eur. J. Neurol. 19, 1311–1317 (2012).
- Verch T , RoselleC, Shank-RetzlaffM. Reduction of dilution error in ELISAs using an internal standard. Bioanalysis8 (14), 1451–1464 (2016).
- Mingozzi F , MausMV, HuiDJet al. CD8+ T-cell responses to adeno associated virus capsid in humans. Nat. Med. 13 (4), 419–422 (2007).
- Mendell JR , CampbellK, Rodino-KlapacLet al. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl J. Med. 363 (15), 1429–1437 (2010).
- Mingozzi F , HighKA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood122 (1), 23–36 (2013).
- US Department of Health and Human Services, US FDA, Center for Drug Evaluation and Research . Guidance for industry, nonclinical safety evaluation of drug or biologic combinations. Rockville, MD, USA (2006). https://www.fda.gov/OHRMS/DOCKETS/98fr/05d-0004-gdl0002.pdf.
- US Department of Health and Human Services, US FDA, Center for Drug Evaluation and Research . Guidance for industry, codevelopment of two or more new investigational drugs for use in combination. Rockville, MD, USA (2013). https://www.fda.gov/downloads/drugs/guidances/ucm236669.pdf.
- Mobley A , LinderSK, BraeuerRet al. A survey on data reproducibility in cancer research provides insights into our limited ability to translate findings from the laboratory to the clinic. PLoS ONE8 (5), e63221 (2013).
- Dakappagari N , ZhangH, StephenLet al. Recommendations for clinical biomarker specimen preservation and stability assessments. Bioanalysis9 (8), 643–653 (2017).
- Ellervik C , VaughtJ. Preanalytical variables affecting the integrity of human biospecimans in biobanking. Clin. Chem. 61 (7), 914–934 (2015).
- Lee JW , DevanarayanV, BarrettYCet al. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm. Res. 23 (2), 312–328 (2006).
- Cummings J , WardTH, DiveC. Fit-for-purpose biomarker method validation in anticancer drug development. Drug Discovery Today15 (19–20), 816–825 (2010).
- Cowan K , GaoX, ParabVet al. Fit-for-purpose biomarker immunoassay qualification and validation: three case studies. Bioanalysis8 (22), 2329–2340 (2016).
- Hougton R , GoutyD, AllinsonJet al. Recommendations on biomarker bioanalytical method validation by GCC. Bioanalysis4 (20), 2439–2446 (2012).
- FDA-NIH Biomarker Working Group . BEST (Biomarkers, EndpointS, and other Tools) Resource. US Food and Drug Administration, Silver Spring, MD, USA ; US National Institutes of Health, Bethesda, MD, USA (2016).
- Piccoli SP , SauerJM, AmurSet al. Points to consider document: scientific and regulatory considerations for the analytical validation of assays used in the qualification of biomarkers in biological matrices. Biomarker Assay Collaborative Evidentiary Considerations Writing Group. https://healthpolicy.duke.edu/sites/default/files/atoms/files/white_paper_6_14_17_0.pdf.
- Ricós C , AlvarezV, CavaF. Current databases on biological variation: pros, cons and progress. Scand. J. Clin. Lab. Invest. 59 (7), 491–500 (1999).