
Abstract
The 2022 16th Workshop on Recent Issues in Bioanalysis (WRIB) took place in Atlanta, GA, USA on September 26–30, 2022. Over 1000 professionals representing pharma/biotech companies, CROs, and multiple regulatory agencies convened to actively discuss the most current topics of interest in bioanalysis. The 16th WRIB included 3 Main Workshops and 7 Specialized Workshops that together spanned 1 week in order to allow exhaustive and thorough coverage of all major issues in bioanalysis, biomarkers, immunogenicity, gene therapy, cell therapy and vaccines. Moreover, in-depth workshops on ICH M10 BMV final guideline (focused on this guideline training, interpretation, adoption and transition); mass spectrometry innovation (focused on novel technologies, novel modalities, and novel challenges); and flow cytometry bioanalysis (rising of the 3rd most common/important technology in bioanalytical labs) were the special features of the 16th edition.
As in previous years, WRIB continued to gather a wide diversity of international, industry opinion leaders and regulatory authority experts working on both small and large molecules as well as gene, cell therapies and vaccines to facilitate sharing and discussions focused on improving quality, increasing regulatory compliance, and achieving scientific excellence on bioanalytical issues. This 2022 White Paper encompasses recommendations emerging from the extensive discussions held during the workshop and is aimed to provide the bioanalytical community with key information and practical solutions on topics and issues addressed, in an effort to enable advances in scientific excellence, improved quality and better regulatory compliance. Due to its length, the 2022 edition of this comprehensive White Paper has been divided into three parts for editorial reasons. This publication (Part 3) covers the recommendations on Gene Therapy, Cell therapy, Vaccines and Biotherapeutics Immunogenicity. Part 1 (Mass Spectrometry and ICH M10) and Part 2 (LBA, Biomarkers/CDx and Cytometry) are published in volume 15 of Bioanalysis, issues 16 and 15 (2023), respectively.
| Abbreviations and Definitions | ||
| AAV: | = | Adeno-associated virus |
| Ab: | = | Antibody |
| ACE: | = | Affinity capture elution |
| ADA: | = | Anti-drug antibody |
| Anti-id: | = | Anti-idiotypic |
| BAV: | = | Biomarker assay validation |
| BCR: | = | B cell receptor |
| BLA: | = | Biologics license application |
| BMV: | = | Bioanalytical method validation |
| BsAb: | = | Bispecific antibody |
| BTM: | = | Blood transcription modules |
| CAR-T: | = | Chimeric antigen receptor T Cell |
| CDC: | = | Complement dependent cytotoxicity |
| CDx: | = | Companion diagnostic |
| cGMP: | = | Current good manufacturing practices |
| CHO: | = | Chinese hamster ovary |
| CIC: | = | Circulating Immune Complexes |
| CK: | = | Cellular kinetics |
| CLIA: | = | Clinical Laboratory Improvement Amendments |
| Companion diagnostics: | = | A companion diagnostic device can be an in vitro diagnostic device, testing kit or imaging tool that provides information that is essential for the safe and effective use of a corresponding therapeutic product. |
| COU: | = | Context of Use |
| CP: | = | Cut point |
| CRISPR: | = | Clustered regularly interspaced short palindromic repeats |
| CRISPR-Cas9: | = | It stands for clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9. By delivering the Cas9 nuclease complexed with a synthetic guide RNA (gRNA) into a cell, the cell's genome can be cut at a desired location, allowing existing genes to be edited, removed and/or new ones added. |
| CRO: | = | Contract Research Organization |
| CTD: | = | Common technical document |
| CV: | = | Coefficient of variation |
| DCA: | = | Domain competition assays |
| DDA: | = | Domain detection assays |
| dPCR: | = | Digital polymerase chain reaction |
| ddPCR: | = | Droplet digital polymerase chain reaction |
| DNA: | = | Deoxyribonucleic acid |
| DoE: | = | Design of experiments |
| dsDNA: | = | Double stranded DNA |
| DT: | = | Drug tolerance |
| ECLA: | = | Electrochemiluminescence assay |
| ECD: | = | Extra cellular domain |
| ELISA: | = | Enzyme-linked immunosorbent assay |
| ELISpot: | = | Enzyme-linked immune absorbent spot |
| FFP: | = | Fit for purpose |
| FIH: | = | First in human |
| FIX: | = | Factor IX |
| FPR: | = | False positive rates |
| FSC/SSC: | = | Forward scatter/Side scatter |
| GCP: | = | Good Clinical Practices |
| gDNA: | = | Genomic DNA |
| GTx: | = | Gene therapy |
| HAMA: | = | Human anti-mouse antibodies |
| hFIX: | = | Human Factor IX |
| HLA: | = | Human leukocyte antigen |
| HMW: | = | High molecular weight |
| hFVIII: | = | Human FVIII |
| IC: | = | Immune complex |
| ICS: | = | Intracellular cytokine staining |
| IDE: | = | Investigational device exemption |
| IFN-γ: | = | Type II interferon |
| IMPD: | = | Investigational Medicinal Product Dossier |
| IND: | = | Investigational new drug |
| ISI: | = | Integrated Summary of Immunogenicity |
| ISR: | = | Incurred sample reanalysis |
| IVD: | = | In vitro device |
| KOL: | = | Key opinion leader |
| LBA: | = | Ligand binding assay |
| LCM: | = | Life cycle management |
| LCMS: | = | Liquid chromatography mass spectrometry |
| LDT: | = | Laboratory developed test |
| LIMS: | = | Laboratory Information Management System |
| LLOQ: | = | Lower limit of quantitation |
| LOD: | = | Limit of detection |
| MAA: | = | Marketing authorization application |
| mAb: | = | Monoclonal antibody |
| MHC: | = | Major histocompatibility complex |
| MIQE: | = | It stands for the minimum information for publication of quantitative real-time PCR experiments. MIQE guidelines describe the minimum information necessary for evaluating qPCR experiments and target the reliability of results to help ensure the integrity of the scientific literature, promote consistency between laboratories, and increase experimental transparency. |
| MOA: | = | Mechanism of action |
| mRNA: | = | Messenger RNA |
| MSR: | = | Minimum significant ratio |
| NAb: | = | Neutralizing antibody |
| NGS: | = | Next generation sequencing |
| NHP: | = | Non-human primate |
| NTC: | = | No template control |
| pAb: | = | Polyclonal antibody |
| PAI: | = | Pre-approval inspection |
| PBMC: | = | Peripheral blood mononuclear cell |
| PC: | = | Positive control |
| PCR: | = | Polymerase chain reaction |
| PD: | = | Pharmacodynamic |
| PHA: | = | Phytohemagglutinine |
| PK: | = | Pharmacokinetics |
| PMC: | = | Postmarketing commitment |
| PPV: | = | Positive predictive value |
| PRNT: | = | Plaque reduction neutralization test |
| QC: | = | Quality control |
| qPCR: | = | Quantitative polymerase chain reaction |
| RCV: | = | Replication competent virus |
| RG: | = | Reference genes |
| RIN: | = | RNA integrity number |
| RNA: | = | Ribonucleic acid |
| RNP: | = | Ribonucleoprotein |
| ROA: | = | Route of administration |
| RT: | = | Reverse transcription |
| rVLP: | = | Recombinant virus-like particles |
| SAP: | = | Statistical analysis plan |
| scFv: | = | Single-chain variable fragment |
| SEC: | = | Size exclusion chromatography |
| sgRNA: | = | Single guide RNA |
| SNR: | = | Signal to noise ratio |
| TAb: | = | Total antibody |
| TCR: | = | T cell receptor |
| TE: | = | Target engagement |
| Tfh: | = | T follicular helper |
| TI: | = | Transduction inhibition |
| Transduction inhibition: | = | The inhibition of the transduction of cells by serum or other matrices in a cell-based assay. The inhibition may be caused by antibodies, low molecular weight drugs, or proteins present in the sample. |
| TLF: | = | Tables, listing and figures |
| VCN: | = | Vector copy number |
| Vector shedding: | = | the dissemination of viral vector released outside the treated subject via excreta (e.g., urine and feces), and secreta (e.g. saliva, semen, sweat). |
| VRNT: | = | Virus reduction neutralization test |
| WRIB: | = | Workshop on Recent Issues in Bioanalysis |
Index Part 3
SECTION 1 – Gene Therapy, Cell Therapy and Vaccines
Hot Topics & Consolidated Questions Collected from the Global Bioanalytical Community
Discussions, Consensus & Conclusions
SECTION 2 – Immunogenicity of Biotherapeutics
Hot Topics & Consolidated Questions Collected from the Global Bioanalytical Community
Introduction
The 2022 16th Workshop on Recent Issues in Bioanalysis (WRIB) took place in Atlanta, GA, USA on September 26–30, 2022. Over 1000 professionals representing pharma/biotech companies, CROs, and multiple regulatory agencies convened to actively discuss the most current topics of interest in bioanalysis. The 16th WRIB included 3 Main Workshops and 7 Specialized Workshops that together spanned 1 week to allow an exhaustive and thorough coverage of all major issues in bioanalysis of biomarkers, immunogenicity, gene therapy, cell therapy and vaccines.
Moreover, in-depth workshops on ICH M10 BMV final guideline (focused on this guideline training, interpretation, adoption and transition); special features of the 16th edition included mass spectrometry innovation (focused on novel technologies, novel modalities, and novel challenges); and flow cytometry bioanalysis (rising of the 3rd most common/important technology in bioanalytical labs).
As in previous years, WRIB continued to gather a wide diversity of international, industry opinion leaders and regulatory authority experts working on both small and large molecules as well as gene, cell therapies and vaccines to facilitate sharing and discussions focused on improving quality, increasing regulatory compliance, and achieving scientific excellence on bioanalytical issues.
The active contributing chairs included:
Dr. Chris Beaver (Syneos), Dr. Arindam Dasgupta (US FDA), Dr. Fabio Garofolo (BRI Frontage), Ms. Dina Goykhman (Merck), Dr. James Huleatt (Sanofi), Dr. Akiko Ishii-Watabe (Japan MHLW / ICH M10 EWG), Mr. Gregor Jordan (Roche), Dr. John Kamerud (Pfizer), Dr. Steve Keller (AbbVie), Dr. Lina Loo (Vertex), Mr. Fred McCush (Pfizer), Mr. Luis Mendez (Merck), Ms. Dulcyane Neiva Mendes Fernandes (Brazil ANVISA / ICH M10 EWG), Dr. Luying Pan (Takeda), Mr. Noah Post (Ionis), Dr. Mohsen Rajabi Abhari (US FDA), Dr. Yoshiro Saito (Japan MHLW / ICH M10 EWG), Dr. Daniel Spellman (Merck), Dr. Giane Sumner (Regeneron), Dr. Matthew Szapacs (Abbvie), Dr. Albert Torri (Regeneron), Dr. Montserrat Carrasco-Triguero (Sangamo), Dr. Elizabeth Verburg (Lilly), Dr. LaKenya Williams (BMS), Dr. Karl Walravens (GSK), Dr. Yongjun Xue (BMS)
The participation of major and influential regulatory agencies continued to grow at the 16th WRIB during its traditional Interactive Regulators' sessions including presentations and panel discussions on:
Regulated Bioanalysis and BMV Guidance/Guidelines: Dr. Chris Burns (UK MHRA), Dr. Seongeun Julia Cho (US FDA), Dr. Arindam Dasgupta (US FDA), Dr. Xiulian Du (US FDA), Dr. Akiko Ishii-Watabe (Japan MHLW / ICH M10 EWG), Dr. Elham Kossary (WHO), Dr. Yang Lu (US FDA), Ms. Dulcyane Neiva Mendes Fernandes (Brazil ANVISA / ICH M10 EWG), Dr. Mohsen Rajabi Abhari (US FDA), Dr. Yoshiro Saito (Japan MHLW / ICH M10 EWG), Mr. Stephen Vinter (UK MHRA / ICH M10 EWG), Dr. Yow-Ming Wang (US FDA), Dr. Li Yang (US FDA), Dr. Jinhui Zhang (US FDA)
Biotherapeutic Immunogenicity, Gene Therapy, Cell Therapy and Vaccines: Dr. Nirjal Bhattarai (US FDA), Dr. Eric Brodsky (US FDA), Dr. Isabelle Cludts (UK MHRA), Dr. Heba Degheidy (US FDA), Dr. Shirley Hopper (UK MHRA), Dr. Chad Irwin (Health Canada), Dr. Akiko Ishii-Watabe (Japan MHLW), Dr. Julie Joseph (Health Canada), Dr. Susan Kirshner (US FDA), Dr. Mohanraj Manangeeswaran (US FDA), Dr. Kimberly Maxfield (US FDA), Dr. Joao Pedras-Vasconcelos (US FDA), Dr. Mohsen Rajabi Abhari (US FDA), Dr. Zuben Sauna (US FDA), Dr. Vijaya Simhadri (US FDA), Dr. Therese Solstad (EU EMA/Norway NoMA), Dr. Seth Thacker (US FDA), Dr. Omar Tounekti (Health Canada), Dr. Daniela Verthelyi (US FDA), Dr. Meenu Wadhwa (UK MHRA), Ms. Leslie Wagner (US FDA), Dr. Joshua Xu (US FDA), Dr. Takenori Yamamoto (Japan MHLW), Dr. Lucia Zhang (Health Canada), Dr. Lin Zhou (US FDA).
Biomarkers/CDx and BAV Guidance/Guidelines: Mr. Abbas Bandukwala (US FDA), Dr. Shirley Hopper (UK MHRA), Dr. Kevin Maher (US FDA), Dr. Yoshiro Saito (Japan MHLW), Dr. Yow-Ming Wang (US FDA), Dr. Joshua Xu (US FDA)
The 16th WRIB included the traditional evening roundtables, which were attended by both industry key opinion leaders (KOL) and regulatory representatives. The extensive and fruitful discussions from these roundtables together with the lectures and open panel discussions amongst the presenters, regulators and attendees culminated in consensus and recommendations on items presented in this White Paper.
A total of 63 recent issues (‘hot’ topics) were addressed and distilled into a series of relevant recommendations. Presented in the current White Paper is the background on each issue, exchanges, discussions, consensus and resulting recommendations.
Due to its length, the 2022 edition of this comprehensive White Paper has been divided into three parts for editorial reasons. This publication covers Part 3 recommendations.
Part 1 – Volume 15 Issue 16 Month August 2023
Mass Spectrometry, Chromatography & Sample Preparation (4 Topics)
Hybrid Assays - Replacing Conventional Technologies
Hybrid Assays - New Applications/Approaches
Regulatory Challenges in Mass Spectrometry Bioanalysis
Innovation in Mass Spectrometry & Novel Challenges & Solutions
Mass Spectrometry Novel Technologies, Novel Modalities, & Novel Challenges (4 Topics)
Novel Applications & Novel Technologies in Bioanalysis
Oligonucleotides: Novel Modalities & Novel Method Development
ADC: Novel Modalities & Novel Method Development
Problem Solving for Non-Liquid and Rare Matrices
ICH M10 BMV Guideline & Global Harmonization (12 Topics)
Impact of Global Harmonization on Regulated Bioanalysis
Harmonization of Cross Validation in Regulated Bioanalysis
Patient Centric Sampling in Regulated Bioanalysis
Harmonization of Reference Standard Materials
Common Mass Spectrometry & Ligand-binding Assays Issues
Impact the 3Rs in Regulated Bioanalysis
Regulated Bioanalysis of Tissues & Secondary Matrices
Stability Issues in Regulated Bioanalysis
Harmonization of Endogenous Molecules Validation – Making the most of BMV & BAV Similarities
Novel/Alternative Technologies in Regulated Bioanalysis
LBA Unique Challenges
LBA Single Well Analysis (Singlicate) in Regulated Bioanalysis
Change of the Critical Reagents: “KISS - Keep It Simple & Straightforward”
LBA Carryover Assessment in Regulated Bioanalysis
Commercial, RUO & Diagnostic LBA Kits in Regulated Bioanalysis
Input from Regulatory Agencies on Regulated Bioanalysis/BMV & Biomarkers/CDx/BAV
ICH M10
1: Introduction, 2: General principles, 4: Ligand Binding Assay
3: Chromatography, 5: Incurred Sample Reanalysis (ISR), 6: Partial and Cross Validation, 8: Documentation
7: Additional Considerations
Adoption by ANVISA
US FDA
Bioanalytical Considerations for Antibody-Drug Conjugates (ADC)
Recent Review Experience with Biosimilar Bioanalysis using LBA
Deficiency in Method Validation for Endogenous Analytes
General Considerations in Pharmacokinetic Bioequivalence Studies of Endogenous Compounds in ANDA Submission
Reflections on FDA Remote Evaluation Activities over the Past 2 Years
Regulatory Findings from Recent Inspections
Biomarkers for Biosimilars: US FDA perspective
CDRH CLIA Categorization Processes
Biomarker Qualification and Analytical Guidance
Next-Generation Sequencing (NGS) Panels for Precision Oncology Biomarkers
UK MHRA
Bioanalytical Observations, Findings and Data Integrity Issues
International Reference Standard Materials (RSM) for Biotherapeutics and Advanced Therapies
Japan MHLW
Recent Developments of Biomarker Assay Validation (BAV) in Japan for qPCR Assays
WHO
Inspection & Review of CROs' computerized systems validation
Input from Regulatory Agencies on Immunogenicity, Gene Therapy, Cell Therapy & Vaccines
Immunogenicity
US FDA
Immunogenicity Information in the U.S. Prescribing Information
Assay Signal-to-Noise Ratio (S/N) as A Potential Alternative to Titer for An ADA Response
Preclinical tools for assessing the risk of innate immune response modulating impurities applied to biosimilars
Updates of the US FDA OCP Efforts on Evaluating Clinical Impact of Immunogenicity
Health Canada
Immunogenicity Labelling for Biologics in Health Canada Drug Submissions
UK MHRA
Development of reference material as positive controls for ADA assays
Gene & Cell Therapy & Vaccines
US FDA
Unique Scientific Challenges in the Immunogenicity Assessment of Novel Modalities
Understanding, Assessing and Managing Immune Responses to CAS-proteins
Perspective on Emerging Landscape of Gene Therapies
Application of Flow Cytometry in Cell Therapy; Current Perspective
Serology Assay Validation
EU EMA/ Norway NoMA
Regulatory Perspective on Vaccine Serological Assays- Validation as Clinical Endpoints
UK MHRA
Importance of immunobridging data for vaccine approval: recent experience with COVID-19 vaccines
Health Canada
Cell and Gene Therapies: Regulatory Challenges and Considerations
Authorization of new COVID-19 vaccines: The utility of immunobridging studies
Use of Functional Assays in the Development of Vaccines
Japan MHLW
Anti-SARS-CoV-2 Neutralizing Antibody Titer as a Clinical Endpoint of Vaccine Clinical Study in Japan
Two-Dimensional Droplet Digital PCR as a Tool for Titration and Integrity Evaluation of Recombinant Adeno-Associated Viral Vectors
Part 2 – Volume 15 Issue 15 Month August 2023
Biomarkers & CDx Development & Validation (8 Topics)
BAV for Primary/Secondary End Points in Clinical Studies
Method Development and BAV Strategies for Biomarker & CDx
Fit for Purpose Validation for Endogenous Analytes: BMV vs BAV for Mass Spectrometry and comparison with other Biomarker Assays
BAV for Vaccine Study Endpoints
Difficult Method Development and BAV: Tissues, Complex Matrices and ROS
Extracellular Vesicles Bioanalysis: Latest Developments and Next Steps
A decade of Free/Total Assays Discussions for Biomarker & PK Assays
Challenges with Multiplex Immunoassays for Biomarkers
Cytometry Validation & Innovation (6 Topics)
Vaccine Functional Assays
Cytometry in Tissue Bioanalysis
Innovation in Cytometry
Current Challenges with Cytometry Validation
Biomarkers, RO, Macrophage Polarization and Phagocytosis Measurements
Cytometry Conventional/Novel Technologies and Main Applications -
LBA, Enzyme Assays & Critical Reagents (5 Topics)
Novel Technologies & Automation in LBA
Novel Modalities, Novel Method Development/Validation Challenges
Rare Matrices
Problem Solving for Complex NAb Assays
Critical Reagents Deep Characterization
Part 3 – Volume 15 Issue 14 Month July 2023
Gene Therapy, Cell Therapy & Vaccines (14 Topics)
Immunogenicity
LNP Immunogenicity
Cell Therapy Immunogenicity Risk Assessment
Viral Vectors Immunogenicity
Bridging LBA to assess ADA response to CAR-T
Immunogenicity Assessment for Oligonucleotide-based Therapeutics
Lesson Learned on Cell & Gene Therapy Bioanalytical Strategy
Vaccine Immunogenicity Strategies
Vaccine Clinical Study Endpoints
Technologies
Guidance for Fit-for-Purpose NGS Assay Selection and Validation
NanoString Technology in Gene Expression
Novel Platform for Infectivity Assays
Bioanalytical PK Evaluation for siRNA using stem-loop RT-qPCR
qPCR and ddPCR Method Development and Validation
bDNA for for CRISPR-Cas9 Analysis of sgRNA
Immunogenicity of Biotherapeutics (10 Topics)
New FDA Draft Guidance on Immunogenicity Information in Prescription Drug Labeling
Immunogenicity & Bioanalysis for Drugs that have a Prolongation Effect in vivo
Affinity of ADA in Clinical Samples
Risk-based Approaches, Prediction and Mitigation
Characterization of “high” Incidence Clinical ADA beyond ADA and NAb Assay Testing
T-cell Engager (BiTE) Immunogenicity & Associated Cytokine Release
Target Interference on Screening Assays Cut Point & Importance of Risk Assessment for pH Sensitive Multi-domain Biotherapeutic (MDB)
Preclinical & Clinical Harmonization and Enhanced Tiered & Cut Point Approaches
NAb Assays Integrated Approach
ADA Assay Comparison & Monitoring
SECTION 1 – Gene Therapy, Cell Therapy & Vaccines
Luying Pan1, Johanna Mora13, Karl Walravens3, Leslie Wagner19, Shirley Hopper9, Lina Loo2, David Bettoun4, Sarah Bond5, Francis Dessy3, Sean Downing1, Fabio Garofolo6, Soumi Gupta7, Neil Henderson8, Chad Irwin10, Akiko Ishii-Watabe11, Sumit Kar12, Vibha Jawa13, Julie Joseph10, Ludovic Malvaux3, Jean-Claude Marshall14, Jessica McDevitt13, Susovan Mohapatra15, Jessica Seitzer16, Justin Smith17, Therese Solstad18, Hiroshi Sugimoto1, Omar Tounekti10, Bonnie Wu20, Yuling Wu21, Yuanxin Xu16, Joshua Xu22, Takenori Yamamoto11 & Lin Yang23
Authors are presented in alphabetical order of their last name, with the exception of the first 6 authors who were session chairs, working dinner facilitators and/or major contributors.
The affiliations can be found at the beginning of the article.
HOT TOPICS & CONSOLIDATED QUESTIONS COLLECTED FROM THE GLOBAL BIOANALYTICAL COMMUNITY
The topics detailed below were considered as the most relevant “hot topics” based on feedback collected from the 15th WRIB attendees. They were reviewed and consolidated by globally recognized opinion leaders before being submitted for discussion during the 16th WRIB. The background on each issue, discussions, consensus and conclusions are in the next section and a summary of the key recommendations is provided in the final section of this manuscript.
LNP Immunogenicity
Are lipid nanoparticles (LNP) used for RNAi, mRNA, and mRNA vaccines & gene therapy immunogenic?
Cell Therapy Immunogenicity Risk Assessment
What should be considered when developing the immunogenicity strategy for drug products? What are the expectations for Immunogenicity Risk Assessment for gene or cell therapy programs? Is the traditional tiered approach used for biotherapeutics suitable for gene therapy?
Viral Vectors Immunogenicity
When selecting suitable negative control (NC) for anti-AAV assays, it is challenging to prepare a matrix pool for NC due to the high prevalence of pre-existing anti-AAV Abs. What are the pros and cons of using different types of NC, which can result in dramatic differences in the measured anti-AAV Ab incidence? Without knowing the sensitivity before cut point determination, how could we know the LOD? Should we change to assay signal or normalized value (NV) estimated from other ADA assays? Due to the presence of pre-existing Abs, it may be difficult to identify true outliers. How many rounds of outlier removal should be performed: one round or multiple rounds until no outlier can be identified? Are assays supporting clinical trial inclusion/exclusion criteria and or safety/efficacy monitoring considered as IVD/CDx or investigational use? If Conformite Europeenne (CE) mark assays are modified and validated to meet current US FDA and ICH BMV, are these assays considered outside of CE mark use, where risk justification and regulatory approval for ongoing and new clinical trials are needed according to EU IVDR effective on 26May2022?
Bridging LBA to assess ADA response to CAR-T
Is the bridging assay format a superior platform to assess CAR specific ADA response? Are there any other results from sponsors showing that the flow cytometry assay generated a high assay background? Which assay (bridging assay vs flow cytometry assay) would generate results that better correlate with the clinical outcomes?
Immunogenicity Assessment for Oligonucleotide-based Therapeutics
Regarding ADA testing recommended in the Draft Guidance Clinical Pharmacology Considerations for the Development of Oligonucleotide Therapeutics issued in 2022, how is “low risk” defined? Is it a platform-based approach? Timing of ADA testing in non-clinical studies is not clear if Phase 1/2 study involves multiple dosing in patients. What are the recommendations for testing for dsDNA Abs, NAbs and ADA isotyping for oligonucleotide therapeutics during development? What is a suitable PC for anti-oligonucleotide if an affinity-purified Ab cannot be obtained?
Lesson Learned on Cell & Gene Therapy Bioanalytical Strategy
What is really needed for a CAR-T bioanalytical strategy? What methods are really needed to measure humoral immunity? What peptide design and strategy for assessments of cytotoxic T lymphocyte (CTL or CD8+ T cells) vs effector T cell (CD4+ T cells) responses are recommended? The field of CAR-T needs more studies to assess correlation between anti-CAR humoral and cellular responses with safety and efficacy for both autologous and allogeneic cells. Are these going to be different for NK cells, iPSC derived, gamma/delta T cells? Does immunogenicity differ in complex CAR-T cells with genome editing modifications and added features?
Vaccine Immunogenicity Strategies
In vaccine clinical trials, for vaccine immunogenicity assessment endpoints, measurement below “cut-off” (LLOQ, technical limit, …) are often imputed at half the cut off. Is it correct to similarly impute such measurements when verifying the immunogenicity assay precision during validation? Among the different sensitivity limits (LOB, LOD, LLOQ) assessed during vaccine immunogenicity assay qualification and validation, what is the real added value of the LOD? Could LOD be removed from the list of critical assay characteristics for vaccine immunogenicity assays? How should validation acceptance criteria consider both the intended use of the assay and the qualification data? e.g., if intended use of the assay can allow an intermediate precision CV = 50%, and qualification data suggest CV∼15%, how should we weigh these two elements to define the validation acceptance criteria?
Vaccine Clinical Study Endpoints
What are the current bioanalytical strategies for COVID-19 vaccines clinical trials? What is the status of antibody assays used for regulatory filings in vaccines?
Guidance for Fit-for-Purpose NGS Assay Selection & Validation
What are validation guidelines for NGS assays that include software pipeline for genetic alteration calls? These are often home-grown with no control of versions over a trial, which can affect data.
What are important factors to consider in developing NGS technologies in support of gene and cell therapy products? What are current best practices in NGS applications for discovery vs clinical monitoring, particularly for gene editing risk assessment?
NanoString Technology in Gene Expression
How could gene expression data as a surrogate endpoint of efficacy be used in the context of a drug approval for gene and cell therapy and vaccines?
Novel Platform for Infectivity Assays
Validation of infectivity assays: What is the level of cross comparison required for confidence in novel assay platforms? How do you validate a new infectivity platform?
Bioanalytical PK Evaluation for siRNA using stem-loop RT-qPCR
What are the guidelines for evaluating the performance of PCR methods designed to evaluate copy number in support of gene therapies? What are the advantages and disadvantages of available technologies for PK analysis of nucleic acid therapies? What are the advantages in using stem-loop qPCR to quantify siRNA? Are 2020 and 2021 White Paper in Bioanalysis qPCR recommendations applied for assay method development and FFP Validation criteria? Can qPCR be used for regulated BA? What are the challenges and limitations?
qPCR & ddPCR Method Development & Validation
What validation parameters/ approaches and data reporting units are unique for gene therapy? Can a conversion factor be used for the volume-based unit to assess biodistribution? Can (copy/μg nucleic acid) be used? What is considered negative in qPCR for pathogens in vaccine trials used to support efficacy endpoint? Defining an appropriate positivity cut-off may be challenging as real-time PCR is able to detect extremely low amount of genetic material in a sample. In many situations, this clinically relevant cut off is not available. Therefore, would the LOD be a better cut-off than zero? Is the “standard” 50 cp/μg LOD for VCN qPCR assays an optimal measure of sensitivity and what is the rationale? The cp/μg LOD cutoff commonly used in PCR-based VCN assays is dependent on the primer/probe performance (i.e., the least copies that can be reliably distinguished from background), and the amount of input DNA used in the reaction. Dosing with AAV can lead to a detectable background qRT-PCR (i.e., the DNA template as well as in-vivo expressed gRNA). What are the recommendations of the community/regulators for best practices when presenting/reporting these types of data by the bioanalyst? When does the AAV “background” contribution become meaningful?
Technical considerations for sample preparation: what is the panel's experience with matrix selection for qPCR methods? What are some of the pros and cons of using plasma vs. PAXgene tube? How do you assess instrument reproducibility and validation for ddPCR and its impact on your assay and results? How do you know the sample is partitioned the same every time? How is the homogeneity of the droplet size controlled and ensured, etc.?
bDNA for for CRISPR-Cas9 Analysis of sgRNA
How to determine exclusion criteria based on pre-existing antibodies against Cas9 protein when selecting patients for in vivo use of CRISPR Cas9?
DISCUSSIONS, CONSENSUS & CONCLUSIONS
LNP Immunogenicity
Lipid nanoparticles (LNP) are used as delivery vehicles for RNAi, mRNA, mRNA vaccines & gene therapies. Active RNAs are formulated in LNP with multiple lipid components including but not limited to polyethylene glycol (PEG). The 2021 White Paper in Bioanalysis [Citation30] discussed the use of LNP for gene based therapeutics (GTx) and the bioanalytical and immunogenicity challenges associated with gene editing components like LNP apart from transgene [Citation30].
The 2022 white paper working group discussed a case study where LNP were used to deliver CRISPR/Cas9 intracellularly for in vivo gene editing. This therapeutic had immunogenicity potential due to a PEG lipid composition of LNP following systemic exposure (IV). A risk-based approach was applied for this drug modality that is using LNP targeting specific genes in the liver to reduce disease-causing proteins. Overall immunogenicity risk was considered low because of the single dose, IV route of administration, and liver targeting with rapid drug clearance in blood. Immunogenicity assessment was focused on anti-drug antibody (ADA) responses to the LNP. Data from non-human primate (NHP) studies showed overall lack of treatment emergent ADAs specific to PEG2000, the most immunogenic component of the LNP.
Based on the above case study and other shared experiences, the expert panel discussed whether LNPs used for RNAi, mRNA, and mRNA vaccines & gene therapy require clinical immunogenicity risk assessments. There was a consensus that immunogenicity related to such LNP may be product specific, and in some cases desired since LNPs could act as adjuvants and enhance vaccine induced immune responses. A proactive collaboration with CMC colleagues to understand DS (drug substance) and DP (drug product) related variation as it pertains to LNP will be critical to conduct a sound immunogenicity risk assessment. Platform based approaches may be applied.
Immunogenicity assessments are not limited to antibody responses to the therapeutic protein and any resulting protein or peptide produced, but also include factors such as cytokine release and complement activation. Risk assessment for unwanted immunogenicity, depends on product context of use. For example, risk assessment for desired immunogenicity in the mRNA vaccine category is different from the risk assessment of therapeutic proteins. Validation of methods for assessing immunogenicity of LNPs could follow an approach similar to that recommended by regulatory agencies for assessment of ADA against therapeutic proteins. However, currently there is no expectation to quantify responses to mRNA antigen vs LNPs separately in the clinic for vaccines.
There is a higher likelihood of anti-PEG Abs in patients who have received PEGylated biologics. The immunogenicity risk can be reduced by use of low MW PEG in LNP [Citation32]. However, there is limited information on anti-PEG Ab titers before and after administration of COVID-19 vaccine (where mRNA is delivered using LNPs) and no correlation of anti-PEG Ab presence and SAE has been observed. Hence, the immunogenicity risk assessment should inform if humoral immunogenicity (specifically anti-PEG) needs to be included in clinical studies, and in some cases non-clinical assessments may be useful. Therefore, the consensus was to perform risk assessments and monitor in a relevant animal model to see if needed to assess in humans. However, it may be difficult to evaluate these responses in clinical trials if the events are too rare to pick up. In some cases, SAEs can only be picked up in post-marketing studies once a large number of people have already been immunized.
Samples can be banked at early-stage clinical studies and may be analyzed if there is a clinical safety signal related to immunogenicity depending on product risk. Further discussions on LNP and associated risk of immunogenicity and its impact are anticipated at the WRIB 2023 meeting.
Cell Therapy Immunogenicity Risk Assessment
Cell therapies are being considered as “next generation” or “novel biologics” contributing to a growing percentage across development pipelines. A recent search in clinicaltrials.gov with the term “CART” and “TCR” yielded 230 and 248 active/recruiting studies respectively. With such a rapid growth, novel bioanalytical assays had to be developed and validated with only a draft guidance available as reference [Citation33]. In the meantime, industry experts, have shared best practices around cellular kinetics and immunogenicity assessments to help bridge the gap [Citation34,Citation35].
In the chemistry, manufacturing, and controls (CMC) space, where cells themselves are considered as drugs, current challenges require additional discussion across functional areas. Immunogenicity risk assessments around residuals can be leveraged to set specifications for residual levels in the cell product. The additional risk assessment should consider the innate and adaptive immune responses due to these residuals and not only the intrinsic sequence-based risks due to extracellular domain of the CAR or engineered T-cell receptor (eTCR) [Citation30,Citation36].
A case study was discussed covering the validation/qualification strategy for an in vitro human PBMC cytokine release assay used to assess immunogenicity risk from cell therapy product residuals (viral proteins (AAV) and other residual non-human origin proteins, e.g., Cas9). The PBMC assay qualification included analysis of multiplexed innate and adaptive cytokine panels using relevant positive and negative controls [Citation36,Citation37] and pertinent levels of the residuals. A priory criterion was utilized to define what would be considered an induced immune response. Evaluation of the assay's performance included LOD, LLOQ and reproducibility. The results from the PBMC assay and the literature review were used as justification to perform analytical characterization of the identified residuals and exclude them from the release criteria since their immunogenicity risk was low. The same data justified exclusion of immunogenicity assessment for these residuals in the clinical bioanalytical strategy. This case study demonstrated the benefits of having analytical characterization data from process demonstration runs that covered levels of residuals including on the cell surface of the DP, understanding analytical parameters of the PBMC assay, establishing a priori criteria for defining an induce response, and engaging early with health authorities to get feedback on the strategy.
A second case study was discussed assessing immunogenicity in non-clinical and clinical studies for in vivo CRISPR/Cas9 genome editing therapies. The immunogenicity assessment presented focused on anti-Cas9 protein antibody responses. Data from non-human primate studies showed that antibodies to Cas9 protein (transgene product, and not the drug component Cas9 mRNA) were detected in most of the drug treated animals, even when pre-dose samples were negative. The antibody response was however transient; it peaked at approximately month 2 and had no impact on PK, PD, or safety.
Based on these case studies, consensus was reached on how to draft a bioanalytical strategy for potentially immunogenic residuals in drug products with gene modifications. It was recommended to perform a risk assessment and design assays based on the specific product. Specifically, ex vivo cell therapy and in vivo gene therapy may be associated with different risks. From a regulatory point of view, CMC should be expected to demonstrate that manufacturing is under control and the Cas9 protein does not cross over a certain limit. The CMC cutoff specification is set by experience (literature driven evidence and in house studies). Anti-Cas9 protein response monitoring may not be needed if target antigen is not present in the drug product above the cutoff. Currently, there is no prior evidence where antibody responses to Cas9 protein or other residuals have been implicated in development of safety signals in clinical studies.
A one size fit approach cannot be used for immunogenicity risk assessments for gene or cell therapy programs. Risk assessment needs to be tailored for the specific product and disease indication. Regulatory agencies may ask for an immunogenicity risk assessment as part of the IND submission. For cell therapy, since the genetic modifications are performed ex vivo, in vitro assessments could be used to assess impact of cell therapy process development related residuals like Cas9 protein and AAV. Such assessments should be performed in conjunction with CMC to guide the manufacturing process and determine much needed characterization/specifications.
Another discussion topic was whether the traditional tiered approach used for biotherapeutics is also suitable and relevant for gene therapy. Questions remain on whether long term anti-AAV antibody testing is needed, due to consistently observed high seroconversion rate and high titer, except in cases of redosing where testing may be warranted [Citation37,Citation38].
Viral Vectors Immunogenicity
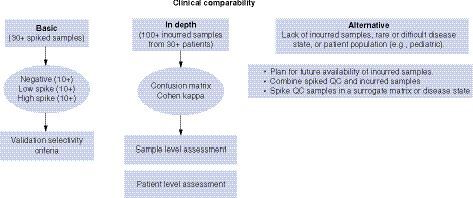
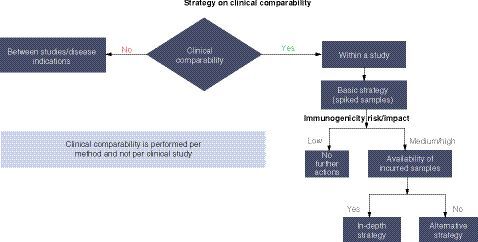
Adeno-associated viruses (AAVs) have been identified as the most important delivery vector in the development of gene therapies. However, anti-AAV antibodies can be found in humans as a result of exposure to the wild-type virus. Depending on the route of administration, these pre-existing antibodies to AAVs may hamper transduction efficiency and potentially reduce therapeutic efficacy of AAV-based gene therapy. Therefore, before vector administration, sensitive and robust assays that measure pre-existing anti-AAV antibodies may become critical to interpretation of clinical outcomes and in some cases, sponsors may decide to use them to determine treatment eligibility for patients.
Pre-existing anti-AAV Total antibody (TAb) and neutralizing antibody (NAb) assays were extensively compared in the 2021 White Paper for Bioanalysis [Citation30]. There was agreement that TAb assays are most often used for ease of use, sensitivity, high throughput and association of TAbs with immunotoxicities. The need for a NAb assay against the viral vector and transgene protein is evaluated on a case-by-case basis. It was previously recommended that sponsors engage with the Regulatory Agencies as soon as possible to determine whether a CDx is required for inclusion/exclusion of patients based on pre-existing antibody level.
The ongoing experience with TAb and NAb assays was further discussed in 2022. Cell based transduction inhibition (CBTI) assays and bridging electrochemiluminescence (ECL) assays are two platforms which are often used to detect the presence of pre-existing anti-AAV NAb and TAb, respectively.
In a case study, the correlation between NAb and TAb results was assessed by testing 210 human samples in both anti-AAV8 NAb and TAb assays. Seventy-two samples were screened negative in both assays and 99 screened positive in both assays with overall concordance of 81%, suggesting a good correlation between two assays.
To determine the best and most suitable platform in this case study, two NAb and two TAb assays were developed and validated at different labs. The performance of NAb and TAb assays within and between different labs was assessed. NAb titers obtained from different labs are rarely comparable due to different assay conditions. Even though the same cell line, reporter gene, and positive antibody were used, the NAb assay sensitivity was ∼10-fold different between the two labs. The overall agreement of positive/negative NAb results between two labs was 72%. Compared to NAb assays, TAb assays showed much less variability with comparable or better sensitivity. The overall agreement of positive/negative TAb results between two labs was 94%. TAb assays have shorter turn-around times for sample testing, are easier to standardize, and amenable to cross-validation in multiple labs. These data demonstrated the advantages of the TAb over NAb assays and supported the choice of the ECL based TAb assay platform for development of both immunogenicity and companion diagnostic assays.
Criteria for selecting samples for cut point assessment was discussed. Due to the presence of pre-existing Abs, it may be difficult to identify true outliers. Typically, 1 to 2 rounds of outlier exclusion are used. Many rounds of outliers are discouraged unless justification is provided. A TAb assay is suggested due to many advantages over a cell based NAb assay. Due to presence of pre-existing anti-AAV Abs, it is impossible to randomly select individual samples for cut point assessment; prescreening individual samples by TAb assay may help. However, for cut point sample selection for NAb assays, the identified ADA negative samples should be further confirmed for NAb negativity in NAb assays.
When using TAb or NAb to determine patients' clinical trial (pivotal study) eligibility, implementation of CDx and PMA (pre-market approval by FDA CDRH) are required by regulatory agencies. The sponsors need to justify the choice of assay and patient's inclusion/exclusion criteria based on the study data.
The final topic was whether assays supporting clinical trial inclusion/exclusion criteria and or safety/efficacy monitoring are considered as IVD/CDx or investigational use. According to US FDA, CDx is used for identifying patients who are most likely to benefit or likely to be at increased risk for serious side effects due to treatment with a particular product, or implemented to monitor response to treatment and adjust as needed to achieve improved safety or efficacy. Generally, CDx is not used for PK assessment. Guidance on GCP and BA method validation need to be followed. For UK MHRA, if the biomarker is used for inclusion/exclusion or for subject management, then an IVD for performance is required, but in all cases, sponsors need to validate the assay for intended use.
Bridging LBA to assess ADA response to CAR-T
CAR-T is an engineered T cell carrying a chimeric antigen receptor (CAR) that redirects killing activity against tumor cells expressing the CAR target. Autologous CAR-T cells are engineered using a patient's own T cells. Immunogenicity determinants on the cell surface may lead to a stronger ADA response when compared with the soluble form of protein, e.g., mAb biotherapeutics. Immune response can prevent CAR-T expansion and eliminate CAR-T from the circulation, resulting in disease relapse or failure of second infusion. The clinical outcomes can be a compromise in treatment efficacy and patient safety, off tumor toxicity, and cytokine release syndrome.
A case study was discussed demonstrating the bioanalytical strategy and the technology platforms for the assessment of ADA response to autologous CAR-T therapy. The results from the comparative analysis of the bridging assay and flow cytometry assay formats were discussed. In one CAR-T program. The original flow cytometry-based ADA method generated a higher background in screening assay due to nonspecific binding of serum antibodies to cell surface consistent with previous data [Citation35]. So, a confirmatory assay was implemented to eliminate the false positive ADA response. When compared with the improved flow cytometry method, the bridging assay format generated similar sensitivity and same ADA classification for clinical samples. Therefore, the bridging assay was the selected format for the autologous CAR-T ADA assessment, which offers a higher operational efficiency than the more tedious 2-day flow cytometry assay. The results indicated that the bridging assay format is a viable platform that would provide a sensitive and reliable method to assess ADA response to the autologous CAR-T therapy [Citation35].
This case study led to a discussion on whether the bridging assay format is a superior platform to assess CAR specific ADA response and which format best correlates with clinical outcome. There was consensus that for ADA assessment, the bridging assay format is common but other assays, e.g., flow cytometry, could also be used. It is sponsor's responsibility to select the suitable assay based on feasibility and context/intended use. For autologous CAR-Ts, bridging format is generally fine, but may not be applicable for allogenic CAR-Ts or other novel platforms. For feasibility reasons, sponsors may first consider evaluating bridging assay format and then move to another assay format if the feasibility of the bridging format is not demonstrated. Regulatory Agencies mentioned that they are flexible to accept suitable assay formats if justified by quality data. For these new treatment modalities, when more data becomes available, the expectations may evolve.
Additional areas which need further delineation includes, i) which format best correlates with clinical outcome ii) suitability for multi-domain CARs which may be difficult to express as soluble extracellular domains required for the bridging assay format. A feasibility assessment on the suitability of assay formats would be needed to address the above-mentioned gaps. The Regulatory Agencies were open to evaluating the feasibility data on a case-by-case basis to decide on the final and most suitable assay format.
Overall, there was agreement that flow cytometry is more variable when compared to immunoassays. Hence, “reasonable” assay precision criteria are difficult to determine for this platform. In this case, the regulatory agencies invite industry sponsors to provide data to support and justify criteria, and if they are reasonable, the agencies may take them into consideration.
Immunogenicity Assessment for Oligonucleotide-based Therapeutics
Oligonucleotide therapeutics (ONTs) are generally short, chemically synthesized DNA or RNA molecules with molecular weight between 6 – 13 kDa that have a significant potential to address genetic diseases that have been challenging to treat with small molecules and biologics. Chemically modified ONTs like antisense oligonucleotides (ASOs) and short interfering ribonucleic acids (siRNAs) have made tremendous progress in last two decades with multiple approvals. ADA assessment against these modalities is routinely conducted as part of non-clinical and clinical studies to determine potential impact of ADA on safety and pharmacokinetics/pharmacodynamics (PK/PD). However, the potential risk from immunogenicity of ASOs and siRNAs is low based on available ADA data for approved ONTs. Given the rapidly evolving field of ONTs, and the use of variety of chemistries that are utilized to develop these therapies, the approaches to assessing immunogenicity should be carefully considered.
The challenges for development of ADA assays for ONTs have been underappreciated. The generation of ADA positive control by immunization of animals is typical for assay development, however, this is not a trivial process for ONTs due to their poor immunogenic properties and issues with affinity-purification of antibodies. This among other factors can lead to poor assay sensitivity and overall assay performance that does not meet the regulatory requirements.
A recently published FDA draft guidance provides recommendations for clinical immunogenicity assessments for ONTs in addition to other available guidance [Citation38–41]. Recent publications also elaborate on unique considerations for immunogenicity assessment for ONT [Citation42–44]. Overall, a product specific risk-based approach should be followed. The need for immunogenicity testing in non-clinical studies is molecule and program specific. For a 2′-O-methoxyethyl modified antisense oligonucleotide (MOE ASO), it was demonstrated that onset, incidence, and titers of ADA responses were comparable to the ADA observations in clinic [Citation45]. Thus, immunogenicity assessment in non-human primates (NHPs) may provide useful information to guide risk assessment and clinical monitoring strategy for ONTs. If non-clinical testing is planned, collecting, and analyzing ADA samples from chronic toxicity studies to evaluate ADA risk may be appropriate due to generally low immunogenicity of the molecules and late onset. The draft clinical pharmacology guidance (FDA Clinical pharmacology considerations for the development of oligonucleotide therapeutics 2022) recommends that, as determined by the immunogenicity risk assessment, it may be adequate to bank samples in early development (e.g., Phase 1/ First-in-human [FIH] studies) for later testing if there is new evidence of altered PK/PD, or immune-mediated adverse events. In certain circumstances, Regulatory Agencies could recommend the assessment for nucleotide sequence-specific antibodies and/or bioactivity (e.g., neutralization, enhancement). Any recommendations for assays will be informed by clinical concerns, such as ONT sequence cross-reactivity, novel structures, or modifications and should be discussed with the Agencies on a case-by-case basis.
Discussions at WRIB aimed to provide additional clarity on what is considered low-risk and extent of characterization needed (NAbs, isotyping etc.). The recommendation was to use literature to inform risk assessment given that ADA assays may not be available early in development, collect and bank samples from nonclinical and clinical studies for later use, and leverage data from FIH study samples to assess the risk and plan clinical monitoring for late phase clinical studies. Risks are product and disease specific, and a platform approach (i.e available ADA data from other ONTs using the same chemistries) can be used to inform risk assessment. Development of an ADA assay for ONT (total drug in case of conjugates) is necessary for high-risk products.
The recommendations for testing for dsDNA Abs, NAbs and ADA isotyping for ONTs during development was also discussed. The consensus was that this is determined as part of risk assessment and driven by clinical signals. If there are no signals, testing is not needed. Technical difficulty to generate a positive control ADA and other challenges can be communicated to regulators.
Finally, ADA assessment strategy for oligonucleotides in case affinity-purified positive control (PC) antibody cannot be obtained was discussed. Affinity purification of antibodies for oligonucleotides pose unique challenges due to issues with immobilization of ONTs, and loss of binding affinity upon elution. There was agreement that the total IgG and serum from immunized animals can be used as positive control, as long as the relative concentration is determined. Sponsors are encouraged to communicate with regulators regarding technical difficulty to generate a purified PC, and other ADA assay related challenges [Citation38–40].
Lesson Learned on Cell & Gene Therapy Bioanalytical Strategy
Multiple CAR-T therapies have been approved in clinic. The clinical bioanalytical strategy has included the assessment of ligand binding based ADA assays to detect humoral immune response to the extra-cellular domain of CAR-T. There is also a risk to mount an innate and cellular immune response as the lentiviral and other agents used to engineer the CAR-T domains could leave residual contaminants. The ligand binding immunoassay methods for humoral immune response assessment require considerations related to assay reagents. For ADA assays, selection of soluble CAR protein fused to proper framework for right conformation, procurement for ADA positive controls is important for method development. For NAb assays, either the cell-based assay format or competitive ligand binding (CLB) assay format is acceptable. The CLB NAb assay method development would require critical reagents including soluble CAR protein, cancer target protein, and NAb positive controls. The bioanalytical strategy can be further refined by evaluating the impact of CAR ADAs on PK (cellular persistence vs. gene expression). Updated recommendations and lessons learned were discussed including areas of uncertainty and what data/assays are really needed.
To date, commercially approved CAR-T products have not provided cellular immune response data as part of the BLA package. However, the number of approved CAR-T programs are small with data that is still evolving. There is no suggestion or any clear correlation between clinical safety/efficacy to immune response to CAR. There are some potential scenarios where T cell response analysis could be needed: this may include to explain, i) any adverse event observed in the first few months when the humoral response has not kicked in, ii) explain impact on persistence in the absence of humoral, iii) evaluate any pre-existing cellular T-cell response memory for subjects where same or next generation CAR-Ts may be re-administered.
Some best practices would include performing a risk assessment based on the product design early in the development of molecule. By evaluating all identifiable risks and mitigating them proactively, a clinical immunogenicity strategy can be designed and shared with the regulators as part of INTERACT or a pre-IND submission.
Overall, the field of CAR-T needs more studies to assess correlation between anti-CAR humoral and cellular responses with safety and efficacy for both autologous and allogeneic cells. The assay designs may need more adaptations for complex engineered CAR-Ts with genome editing modifications as well as CAR-NK cells, iPSC derived, gamma/delta T cells. From the current available studies, there was an agreement that T cell responses may not be commonly measured for autologous CAR-T therapy. However, it is not clear if such cellular immune response measurements may be important for allogenic CAR-T therapy. The Class I & II dependent responses may matter in the context of allogenic CAR-T, but the data is insufficient to make any conclusions.
Similar discussions occurred on lessons learned for gene therapy where the first principle of AAV-based gene therapy is that every gene therapy is different - immunogenicity assessment strategy cannot be generalized across gene therapies. AAV mediated gene therapies represent a complex treatment modality with multiple components that may impact. Updated recommendations were provided based on current experience and case studies. Immunogenicity assessment strategy cannot be generalized across gene therapies. This is also true for AAV-based gene therapy as there are many different products. Several factors influence the immunogenicity of an AAV gene therapy: capsid serotype, promoter, CpG content and transgene. Early data production to justify immunogenicity strategy is also recommended. Early engagement with regulatory agencies is encouraged. However, producing early data is often challenging & in some circumstances, is only retrospectively informative [Citation30].
Vaccine Immunogenicity Strategies
Vaccine immunogenicity assays are often associated with critical endpoints in vaccine clinical trials and are part of the basis for vaccine licensure. They are also used to measure biomarkers (e.g., Susceptibility/risk biomarker (pre-vaccination serostatus), or pharmacodynamic/response biomarker (immune marker of protection). As such, their development, qualification and validation should follow the highest standards and be harmonized, as was highlighted during the drafting of the most recent White Papers in Bioanalysis [Citation27,Citation30,Citation46].
Paradoxically, despite their central role in the definition of optimal vaccine formulation, dose and scheduling, no specific guideline was released describing regulators' expectation with regards to vaccine immunogenicity assays validation. It is a common misconception that VIA development and validation should follow the guidelines edited for bioanalytical assays measuring drugs or biotherapeutics in human fluids [Citation47,Citation48] or for ADA assays. However, the bioanalysis of biomarkers and bioanalytical methods used for the assessment of vaccine immunogenicity assays are not within the scope of those guidelines due to the unique characteristics in their application (e.g. measuring activity of complex population of specific antibodies, produced by the human body as a subpopulation among many other specificities, covering a very broad range of response, often used over decades).
For these reasons, several vaccine manufacturers and CROs initiated a global effort to harmonize vaccine immunoassays development and validation through the drafting of a specific white paper aiming to clarify definitions, to make the link between assay performance characteristics and assay intended use, and to propose some ways of working throughout assay development, qualification and validation. Interestingly, the National Medical Product Administration from China, recently released an exposure draft for “Technical Guidelines for Studies on Antibody Analysis Methods in Vaccine Clinical Trials”. This is, to present knowledge, the first concrete initiative for vaccine immunogenicity assay specific guidelines.
Updated recommendations were provided while this dedicated harmonization WRIB White Paper is still in draft. In vaccine clinical trials, for vaccine immunogenicity assessment endpoints, measurement below the “cut-off” (LLOQ, technical limit) are often imputed to make use of all results yielding a positive response (above the Limit of Detection) but that is below the cut-off value to avoid truncating the data; one approach frequently used is to apply half the cut-off value. There was discussion, about imputation during assay validation. From experience, imputation artificially increases estimated variability of samples whose concentration/titer is close to cut off, potentially leading to overestimation of LLOQ. However, this apparent increase of assay variability may not represent a lack of performance of the assay but would need to be defined and empirically determined. Therefore, prior to validation, the recommendation, is to define the assay LLOQ by the lowest level with acceptable accuracy and precision. This is based on qualification studies. The LLOQ is then verified during validation with samples near and below this level to demonstrate when acceptable accuracy and precision are not achieved. Imputation of values for a validation study would not be appropriate.
Among the different sensitivity limits (LOB, LOD, LLOQ) that may be assessed during vaccine immunogenicity assay qualification and validation, the LOD was discussed. There was consensus that there is no value in determining the LOD/LOB when calculating Ab titers to determine vaccine efficacy.
Another discussion was on validation acceptance criteria considering both the intended use of the assay and the qualification data. Questions exist on how to weigh qualification data and intended use to define validation criteria (e.g., how to set validation criteria if the performance of the assay is better than what is needed to support the intended use). It was recommended to set the validation criteria based on the intended use. However, if there are significant differences, such as a shift in the precision determined during qualification vs. precision during validation, even if validation criteria are met, it is necessary to investigate why the assay is not performing as during qualification. This may be due to a number of factors, and importantly may signal a decrease in true assay performance that should not be ignored.
Vaccine Clinical Study Endpoints
Bioanalytical strategies for antibody assays used in vaccine regulatory filings is currently a very hot topic due to experience with COVID-19 vaccine trials. This included discussions of NAb assays, bioanalytical challenges and limitations of cell-based activity assays, and advantages of plate-based assays.
When the COVID-19 pandemic began, there were no binding or NAb detection assays available for use. These assays were developed in parallel to the clinical development program being initiated with assay development, qualification and validation occurring under rapid timelines. The majority of Regulatory Agencies relied on the functional Nab assays to quantitate the levels of functional Nab to the original SARS-CoV-2 strain. Many of these assays currently use pseudoviral constructs to enable testing in BSL2 facilities and have been compared to exploratory live viral assays in BSL3 laboratories. These pseudoviral NAb (PsvNeut) assays have continued to be used for the evaluation of vaccines against new SARS-CoV-2 variants as they have appeared and become prevalent in populations.
In parallel to these data sets, the industry has continued to generate binding antibody data using the LBA for all the emerging variants and has seen good correlation between those two assays. The PsvNeut assays are cell-based assays and are relatively low throughput and have the potential for longitudinal variability because of their reliance on transfected cells and other critical reagents such as the Pseudoviral particles. Ligand-binding assays (LBA), significantly reduce the possibility for longitudinal variability and have well significantly increased throughput compared to the PsvNeut assays. Regulatory Agencies have continued to request that development of the PsvNeut assays for new variants of concern use serum from confirmed variant infections, without prior infections and, in some cases, vaccination, a population that is increasingly becoming difficult to identify. These concerns limit the ability to rapidly deploy new assays in reaction to the appearance of variants of concern.
A case study was discussed on experience with PsvNeut assays and their correlation with LBA. Tens of thousands of samples have now been analyzed in multiple different PsvNeut Ab and LBA across different clinical programs for ancestral as well as variant strains. The two different assay formats remain highly comparable if not fully concordant across samples. Variability continues to be lowest for the LBA, compared to the cell based PsvNeut. It was discussed whether sponsors could use PseudoViral Neut assays for characterization of new variants of concern and confirm concordance with ligand binding assays using historical sample sets/controls. Ongoing clinical testing of those established variants could then use binding assays to measure Ab titers for clinical immune endpoints improving on variability and decreasing turnaround times.
LBA assays with a tighter profile than cell-based assays are being allowed by Regulatory Agencies but NAb assays still required for COVID-19. Regulatory Agencies continue to request Psv Neut assays for new variants. It may not be possible to confirm that a plate assay is sufficient for future variants, so agencies will take conservative approach. There was consensus that there is no objection to use total Ab if data shows good correlation and that it can predict for new variants.
Guidance for Fit-for-Purpose NGS Assay Selection & Validation
To develop gene and cell therapies such as investigational in vivo and ex vivo genome editing products as effective and safe therapeutic drugs, it is critical to have appropriate technologies to evaluate safety and efficacy [Citation49–51]. Next-generation sequencing (NGS) technologies provide methods to characterize the genomic changes induced by these therapies. Characterization of these products include identification and analysis of intended and potential unintended genomic changes that can occur due to cell engineering or genome editing. These methods range from amplicon sequencing to evaluate insertions and deletions (indels) at known genomic loci to discovery methods aimed at identifying potential unintended genomic changes. Unintended genomic changes can range from small indels to larger DNA structural variants (SVs) such as inversions, duplications and inter-chromosomal translocations.
Several novel case studies were reviewed demonstrating recent technological advancements with incorporation of prior experiences and challenges developing & implementing a comprehensive workflow for gene therapy. Several discovery methods have been developed to identify the potential for off-target editing (e.g., SITE-Seq, GUIDE-Seq, ONE-Seq, CIRCLE-Seq, RGEN-Seq) [Citation50–52]. Comprehensive off-target characterization consists of discovery and validation phases. The off-target discovery phase includes both in silico computational prediction and in vitro assays. Off-target editing discovery using a biochemical approach has demonstrated advantages to the widely used cell-based experimental technology [Citation53].
Multiplexed approaches can evaluate indels across many loci (e.g., rhAMPSeq and Hybrid Capture). Methods to assess chromosomal structural integrity (structural variants and large genomic changes) include short-read NGS technology characterization, long-read sequencing, and pinpoint DNA FISH direct visualization technology.
NGS is an important platform for detecting off-targeting effects. In vitro and in vivo assays may be used – but the full predictability is not known. For now, it was recommended to compare predictions to the highest dose in cells or, if possible, perform the analysis in vivo – this work is ongoing in industry. Overall, these applications of NGS need further discussion.
Important factors to consider in developing and validating NGS technologies in support of gene and cell therapy products were discussed. Key points to consider include understanding the value of proper fit-for-purpose analytical qualification/validation, employing a quality framework (e.g., reference samples, QC metrics, proficiency testing and reproducible data analysis) enhancing data quality and consistency (e.g., using automation to maintain chain of custody and decrease human error in pipetting) [Citation54–58]. A case study was shown with a process for implementing a clinical-grade RNASeq workflow for FFPE tissue testing and comparing RNASeq protocols across labs.
With the advent of various genomic assay platforms and increased accessibility of sequencing technologies to the drug discovery research community, it is important to understand that the use case for each application depends on the hypothesis and the intended usage. Whether the objective is to test a hypothesis or generate one, there are appropriate NGS based assays nowadays. Choosing the appropriate assay is important and may be dependent on the stage of clinical trial, intended analysis and sample type. For example, whole blood is the preferred sample type for germline profiling. Similarly, targeted PCR-based assays and targeted NGS panels are better suited for diagnostic and pathway-analysis assays. Historically, Taqman probe-based ddPCR or qPCR assays were preferred methods for variant calling (e.g., SNV / splice variant), allelic frequency determination and subject screening purposes. But whole exome sequencing (WES) and whole genome sequencing (WGS) might be more suitable depending on the gene-panel size and throughput requirement serving the hypothesis. Different matrix i.e. whole blood, FFPE blocks, and stool samples may need designated workflows to extract DNA or RNA samples prior to NGS analysis.
Assay validation is integral to the successful application of NGS analysis. Most vendor labs adhere to primary salient features such as reproducibility, robustness, sensitivity, and specificity. The validation plan also needs to emulate the real-world application – samples should be representative of the population heterogeneity so that it is captured within the dynamic range of the assay. Depending on the type of method, the analysis of method blanks/matrix blanks, standards, contrived samples, and spike-in materials should be used to calculate accuracy, bias, and precision. Not all methods will need the same set of determinants. Additional considerations like dedicated sample collection method, storage, and nucleic acid (DNA/RNA) sample preparation workflows will ensure better data quality. In vitro assays that use immortalized or primary cell-lines procured from vendors need to be checked periodically for genotypic drift, as mutations that are acquired during experimentation should not be perceived consequential towards the endpoint. A priori knowledge of population heterogeneity would ensure accurate separation of pathologically causal genotypes from healthy population. Additionally, sponsors should have access to the raw data generated at the vendor labs for independent analysis. Finally, choice of higher throughput assays like WGS and WES should be assessed depending on its impact on the hypothesis. Current cost of WGS might be higher than WES but genome-wide data can be significantly advantageous in the search for causal variants.
Recommendations on the validation guidelines for NGS assays that include software pipeline for genetic alteration calls were further discussed. These NGS assays are often home-grown with no control of versions over a trial, which can affect data. A fit-for-purpose validation package is recommended to understand the software capabilities. The extent of the software validation depends on the intended use of the assay. It was recommended to find and use bridging samples maintained over time if possible.
NanoString Technology in Gene Expression
Sponsors are increasingly using novel gene expression technologies for drug development beyond qPCR and ddPCR. NanoString Technology, a qRT-PCR alternative with automated tissue RNA extraction and gene expression analysis with low tissue input needs. A case study was discussed showing the use of NanoString technology for assessing disease and treatment related changes in clinical samples.
A gene expression-based biomarker strategy was developed to assess changes in gene expression in accessible tissues such as buccal cells and whole blood samples. Owing to the limited amount of RNA available in buccal cells, NanoString technology was used to assess disease or treatment–related changes of expression in a large set of genes using clinical samples. Considerations for method development and validation were presented. This case study demonstrated proper characterization of potential genes of interest and housekeeping genes should be performed in the desired tissue. A pilot experiment using naive matrix should be performed to define linearity range and level of expression in the tissue of interest. For each tissue, mRNA quantity should be chosen. Uniform timing of sample collection is an important consideration, due to circadian regulation of gene expression. LOQ based on %CV in dilutional linearity and precision and accuracy characterization can then be performed which in this case showed NanoString is an accurate method of quantifying gene expression level when compared to qRT-PCR. The results from clinical samples showed the technology allows for less intensive bioinformatic resource allocation and that differences in gene expression from multiple genes or interest can accurately inform on both disease and treatment effect.
Based on the case study, it was evaluated whether gene expression data and signatures of multiple genes, using novel technologies such as NanoString or traditional technologies can be used as a surrogate endpoint of efficacy in the context of a drug approval for gene and cell therapy and vaccines. There was agreement that this must be evaluated case by case regardless of technology. It was also agreed that there is risk if expression signature is not consistently seen.
Finally, the Regulators recommended again to reach out to their Agencies to discuss and to provide correlation of the data with clinical outcome.
Novel Platform for Infectivity Assays
Oncolytic viruses (OVs) comprise a diverse group of biologic agents that have been investigated as cancer immunotherapy platforms [Citation59]. Therapeutic efficacy of these replication competent OVs is generally established through specific mutations that genetically modify and engineer the virus to target and selectively kill tumor cells without causing harm to normal tissues [Citation60,Citation61]. The oncolytic efficacy of these OVs is typically enhanced through further genetic modifications to counter the immunosuppressive tumor microenvironment.
A case study was discussed evaluating one-such OV in clinical trials for the treatment of solid tumors following intravenous administration. Genetic manipulation of OVs classifies them as genetically modified organisms (GMOs) and the delivery of GMO's raises safety concerns related to the transmission of these viruses to untreated individuals as virus is shed through excreta, secreta, or through skin pustules, sores, or wounds [Citation62]. To understand and characterize these risks, shedding, and in the case of replication competent viruses, infectivity assays, must be conducted. Plaque assays are often used to characterize and quantify the amount of infectious virus in a sample; however, plaque assays are labor intensive, have a narrow quantitative range, and are highly variable with a relatively low throughput, all of which present challenges to validation and clinical testing implementation.
To reduce these risks, several novel infectivity platforms were investigated, and the xCelligence Real-time Quantitative Cell Analysis (RCTA) system from Agilent was chosen as the move-forward platform. The xCelligence RCTA system uses special 96-well plates coated with gold biosensors to monitor the impedance of electrical current. A reduction in impedance, measured in real-time, is indicative of cytopathic effect within the well and is quantified by the instrument using a cell index. Using a viral standard, a power fit model was used to correlate cell index to PFU/mL and a head-to-head comparison was performed between a standard plaque assay and the xCelligence RCTA system to demonstrate alignment between results. The strong correlation between these results supported further development and optimization of the xCelligence RCTA system for use in testing clinical trial samples.
The case study demonstrated the xCelligence RCTA system provides higher throughput, is less labor intensive, and has a broader quantitative range compared to standard plaque assays. These findings were used to support the fit-for-purpose validation of this assay and platform to support clinical trial testing for infectious virus in patient samples. Given the functionality of this platform, the xCelligence RCTA system has potential for broad virological applications.
This case study was the basis of discussion on the level of cross comparison required for confidence in novel assay platforms (e.g., a novel infectivity platform) and how to validate them. There was consensus that if a novel platform is used for a new trial, a fit-for-purpose validation of the assay on the novel platform is required before testing clinical samples. If available, the clinical samples from previous trials should be used as part of validation of this novel platform to establish confidence. A cross-validation of platforms is needed if data from different platforms is planned to be combined within a trial. Here, spiked-in samples or quality controls (high and low QC controls to cover the range) with pre-determined criteria may be used for cross comparison.
Bioanalytical PK Evaluation for siRNA using stem-loop RT-qPCR
While qPCR validation for regulated bioanalysis has been extensively discussed in previous White Papers in Bioanalysis [Citation24,Citation27,Citation30], the 2022 discussions were focused on the use of qPCR for PK, particularly for siRNA therapeutics.
Small interfering RNA (siRNA) therapies are ∼21 base pair double stranded RNAs that harness the endogenous RNA interference (RNAi) pathway to specifically target genes associated with disease. The approval of siRNA therapies ONPATTRO® (patisiran) and GIVLAARI® (givosiran) in 2018 and 2019 marked an important milestone in the 20-year effort to develop this new class of drug, and research and development of siRNA therapeutics continues to rapidly advance.
Multiple analytical tools are available to support the bioanalysis of siRNA therapeutics, including LCMS, hybridization ELISA (hELISA), and stem-loop RT-qPCR. LC-MS may be used for siRNA metabolite profiling and quantification of parent drug and metabolites, demonstrating suitable accuracy, precision, and specificity to support PK or toxicokinetic evaluations. hELISA and RT-qPCR are alternative methods that may be utilized in cases in which a higher degree of sensitivity is required. hELISA is a ligand binding-based method that is often designed using capture and detection probes that specifically anneal to the target siRNA through Watson-Crick base pairing. RT-qPCR methods utilize similar target-specific oligonucleotide primers and probes but include reverse transcriptase and DNA polymerase enzymes to first convert the RNA to cDNA, followed by exponential amplification of the cDNA product in a RT-PCR reaction.
While the RT-qPCR method demonstrates high sensitivity, this method shows limitations in distinguishing between parent drug and metabolites. A case study was discussed in which a high-throughput and sensitive RT-qPCR method was developed to measure siRNA concentrations in biofluids, with the reported concentrations representing the sum of the parent plus metabolites of the antisense strand of the siRNA. This method utilized primers that were complementary to the 5’ and 3′ end of the siRNA antisense strand. While the method demonstrated accuracy for detecting both parent and 3′ metabolites, the method was unable to accurately quantify 5′ deaminated metabolites due to the mismatch between the primer and target that resulted from the deamination. While the stem-loop RT-qPCR method may have useful applications in quantifying low drug levels, this novel and important information on the limitation of this method for quantifying 5′ metabolites should be considered when selecting the appropriate tool for siRNA bioanalysis for each application.
This case study led to a discussion on guidelines for evaluating the performance of PCR methods designed to evaluate copy number in support of gene therapies and the advantages and disadvantages of available technologies for PK analysis of nucleic acid therapies. The 2020 and 2021 White Paper in Bioanalysis qPCR recommendations [Citation30] applied for assay method development and FFP Validation criteria are still applicable in other uses of qPCR such as PK analysis. Applicability of stem loop qPCR can depend on the size of the modifications and if it fits the purpose of the results and meeting existing recommendations. Due to limitations, it was suggested that alternative methods to qPCR such as mass spectrometry assays with lower CVs might be used.
qPCR & ddPCR Method Development & Validation
As mentioned in the previous paragraph, the 2020 and 2021 White Papers in Bioanalysis featured extensive discussion and recommendations for qPCR and ddPCR validation for gene and cell therapy and vaccines [Citation27,Citation30]. New case studies were discussed in 2022, to further support these internationally recognized recommendations. Moreover, the recently published White Paper from the Global CRO Council in Bioanalysis (GCC) [Citation63] was reviewed and discussed.
Several discussions pertained to reporting guidelines for sensitivity during development, validation, and sample analysis for gene and cell therapy and whether there are unique validation parameters/ approaches and data reporting unites for gene therapy. Specially, it was discussed whether conversion factors or copy/µg nucleic acid can be used in place of volume-based units to assess biodistribution. It was recommended that general existing qPCR and ddPCR validation guidelines should be used for biodistribution. For data reporting, requirements do not vary by method used and a justification for choice of units should be provided. Specifically, copy/µg nucleic acid is appropriate for biodistribution studies in tissue if tissue type is specified. The use of other units or method (i.e., copy/mg tissue, use of conversion equations) needs to be further discussed for the fit-for-purpose validation in in vivo and ex vivo gene therapy [Citation64,Citation65].
Cut-offs for positivity for pathogens in vaccine trials to support efficacy endpoints was further discussed. Since qPCR is very sensitive and a clinically relevant cutoff may not be available, it was discussed whether LOD is a better cutoff than zero. There are several technical considerations to this topic. It is important to understand parameters that affect assay performance (i.e., background/ noise in the absence of matrix, CT, sample type, collection method) and use of negative and positive controls if available. It is also possible to correlate titer to amount for infection/ clinical significance - but usually this is difficult to establish. Controls and diluted commercial samples (if available) can also be characterized in an orthogonal functional assay. It was agreed that LOD is reasonable if an orthogonal method or clinically relevant cut-off is not available. It is important to include appropriate controls to show that LOD is robust. It was agreed that clinical/ treatment implications of this approach should be further discussed at next WRIB.
Another discussion, on LOD was whether the standard 50 cp/μg of DNA is the optimal measure of sensitivity and if so, what is the rationale. The LOD determination in PCR assay is dependent on many factors i.e., the primer/probe performance (i.e., the least copies that can be reliably distinguished from background) and the amount of input DNA used in the reaction. As discussed above, there were cases when LOD of 50 cp/μg of DNA is difficult to achieve. As an alternative, the expert panel shared experience that they have demonstrated the LOD determination in water, as opposed to blank matrix, to show assay sensitivity and specificity. Ultimately, the determination of LOD during validation should be justifiable.
Post-treatment samples may contain detectable contamination/ background i.e., the DNA template as well as in vivo expressed gRNA when analyzed by bDNA or RT-PCR. It was discussed the best practices when presenting/reporting data in the presence of background or contaminant. It is essential to reduce contamination by using DNAse and RNAse to show specificity of assay. Characterization of the background/ contaminant may be performed i.e., run samples on gel to understand the source, consistency. Correlation of the background level to the clinical outcome or infectivity is informative. Normalization to a reference housekeeping gene at DNA level may be used in the presence of contaminant. Overall, the presence of background/contaminant may be acceptable in a validated assay with justification and risk assessment.
Approaches for matrix selection and instrument reproducibility were also discussed. For example, it was discussed about the experience with matrix selection for qPCR methods and the pros and cons of using plasma vs. PAXgene tube. It was agreed that plasma may give low RNA yield but in certain cases PAXgene tubes and associated extraction methods (e.g., RNA encapsulated in nanoparticle) may cause difficulties and low yields. The recommendation was that the choice of matrix should be made on a case-by-case basis. Carefully consider selection of cell free vs. cell containing matrices. Fixative must be used when assessing RNA expression. Importantly, switching platform (or matrix) will require re-validation, and controls should represent/mimic samples.
The final discussion was the assessment of instrument validation and the impact to the results [Citation63]. Compared to qPCR, ddPCR instrumentation includes additional complexity in sample partitioning and homogeneity of the droplet size. It was agreed that IQ/OQ of instruments by vendor alone may be insufficient. Additional analysis i.e., statistics of partition, may be needed to monitor instrument performance and robustness. The overall recommendation was to use assay quality controls (high and low copies) to monitor run acceptance and consult with vendor for their expertise.
bDNA for CRISPR-Cas9 Analysis of sgRNA
Previous White Paper in Bioanalysis recommendations focused on bioanalytical strategies and risk assessment of off-target toxicity between ex vivo and in vivo administered CRISPR/Cas9 therapies [Citation27,Citation30,Citation46]. Updated guidance was provided on exclusion criteria based on pre-existing antibodies against Cas9 protein when selecting patients for in vivo use of CRISPR Cas9. Previous White Papers explained pre-screening for Cas9 protein antibodies may not be feasible for orphan or rare disease populations.
For CRISPR-Cas9 therapies, the impact of pre-existing immunity against Cas9 protein might be very important depending on the route of administration and target site. The sponsor may need to establish exclusion criteria in early phase study to ensure patient safety or to justify the use of CRISPR-Cas9 therapies in the presence of pre-existing immunity.
Data may also be collected to correlate the level of pre-existing antibodies and/or T cell responses with the efficacy of drug product. It was recommended to use pre-clinical data to relate titers to dose/ efficacy and screen a subset of clinical samples pre-dose. This evaluation is case by case depending on efficacy, dosing, distribution in body/ delivery method. Ultimately, exclusion criteria are a clinical decision (not bioanalytical). The priority of Phase 1 is to determine patient safety and sponsors should be prepared to address immunogenicity.
RECOMMENDATIONS
Below is a summary of the recommendations made during the 16th WRIB.
LNP Immunogenicity
Anti-PEG Ab have been detected in patients who received PEGylated biologics. Low MW PEG used in LNP can reduce immunogenicity risk.
Perform risk assessment and then monitor in a relevant preclinical model to see if an assessment is needed in clinic.
Samples can be banked in early-stage clinical studies, and may be analysed if there is a clinical safety signal that may be suggestive of immunogenicity
Cell Therapy Immunogenicity Risk Assessment
Perform risk assessment and design assays to assess cut-off values for residuals based on the specific cell product. Ex vivo cell therapy and in vivo gene therapy are expected to have different risks.
Risk assessment needs to be tailored for the specific cell product and disease indication and cannot be applied to all cell therapies as an overall strategy.
BLA guidance is still relevant for screening, confirmatory, and titer tier-based strategy for monitoring the humoral antibody response. If seroconversion rate is very low or very high, scientific rationale is needed to set up a more stringent screen cut point, skip the confirmation, and perform titer determination.
Viral Vectors Immunogenicity
Have a reliable strategy to identify negative samples; if not possible, use specific depleted sera (capsid depletion preferred).
TAb assay is suggested due to many advantages over cell based NAb assay, particularly relevant to sensitivity, specificity and reproducibility.
Use TAb (or NAb) for patients' clinical trial (pivotal study) eligibility. CDx and PMA (pre-market approval by FDA CDRH) are required by Regulatory Agencies.
CDx is for enrollment/diagnosis and monitor response to treatment for adjusting future treatments, generally not used for PK studies.
Bridging LBA to assess ADA response to CAR-T
Both bridging LBA and Flow Cytometry assays have been used. It is the sponsor's responsibility to select the suitable assay based on feasibility and intended use.
Regulatory Agencies are flexible and glad to accept bridging LBA or Flow Cytometry assays if justified based on quality data.
Immunogenicity Assessment for Oligonucleotide-based Therapeutics
Platform based approach and literature can be used to inform immunogenicity risk assessment.
As determined by the risk assessment, it may be adequate to bank samples from nonclinical and Phase 1/ First-in-human [FIH] studies for later testing if needed. Risks are product and disease specific. If there are no clinical signals, testing for dsDNA Abs, NAbs (neutralizing antibodies) and ADA isotyping is not needed.
In case affinity-purified positive control (PC) antibody cannot be obtained, total IgG and also serum from immunized animals can be used as PC as long as relative concentration is determined.
Lesson Learned on Cell & Gene Therapy Bioanalytical Strategy
Perform a risk assessment strategy based on the product design.
Proactive data generation for preclinical risk assessment to develop an immunogenicity strategy.
Discuss with agency on a case-by-case basis to get agreement.
T cell responses are not commonly measured for autologous CAR T therapy.
Immunogenicity assessment strategy cannot be generalized across gene therapies. This is also true to AAV-based gene therapy with next generation of gene therapies
Vaccine Immunogenicity Strategies
There is limited value to LOD when calculating titers.
Favor intended use over qualification data when setting the validation criteria.
Vaccine Clinical Study Endpoints
Regulatory Agencies require Psv Neut assays for new Covid-19 variants.
It is possible to use total Ab and plate-based LBA as long as data shows good correlation and that it can predict for new variants
Guidance for Fit-for-Purpose NGS Assay Selection & Validation
NGS is an important platform for detecting off targeting effects. In vitro and in vivo assays may be used – but they are not fully predictive.
Fit-for-purpose validation package is recommended to understand the software capabilities (e.g., analysis pipeline). The extent of the software validation depends on the intended use of the assay.
NanoString Technology in Gene Expression
The possibility to use gene expression data as a surrogate endpoint needs to be evaluated case by case with discussion with Regulatory Agencies and data showing correlation with clinical outcomes.
Novel Platform for Infectivity Assays
If only using the new platform, a fit-for-purpose validation of the assay on the novel platform is required before testing samples from a new trial.
If data from both a traditional and new platform are combined, it is necessary to perform a cross validation.
Bioanalytical PK Evaluation for siRNA using stem-loop RT-qPCR
qPCR is applicable to regulated BA for PK evaluation and recommendations from 2020 and 2021 White Papers in Bioanalysis are applicable.
qPCR & ddPCR Method Development & Validation
For in vivo gene therapy biodistribution studies in tissue, copy/μg nucleic acid is appropriate.
For endpoint in vaccine trials (not IVD) with qPCR assay, LOD is reasonable if orthogonal method or clinically relevant cut-off is not available. Include appropriate controls to show LOD is robust.
When reporting background in qPCR (e.g., AAV DNA template and gRNA) characterize background signal. Background may beacceptable if background is explained.
Orthogonal assessment of potential contamination
Determine if signal and background is robust.
Correlate with clinical outcome.
Choice of matrix (e.g., plasma vs. PAXgene) should be made on a case-by-case basis. Carefully consider selection of cell free vs. cell containing matrices. Fixative must be used when assessing RNA expression.
To monitor instrument reproducibility (e.g., ddPCR) use and monitor run acceptance by controls with known high and low copies. Consult with vendor.
bDNA for CRISPR-Cas9 Analysis of sgRNA
Exclusion criteria may need to be established in early phase study to ensure patient safety.
Ultimately, exclusion criteria are a clinical decision (not bioanalytical). The priority of Phase 1 is to determine safety and sponsors should be prepared to address immunogenicity.
SECTION 2 – Immunogenicity of Biotherapeutics
Albert Torri24, Susan Kirshner19, Kimberly Maxfield19, Joao Pedras-Vasconcelos19, Mohsen Rajabi Abhari19, Daniela Verthelyi19, Eric Brodsky19,‡, Montserrat Carrasco-Triguero25, John Kamerud17, Matthew Andisik24, Daniel Baltrukonis17, Nicoletta Bivi26, Isabelle Cludts9, Kelly Coble27, Boris Gorovits28, George R Gunn29, Swati Gupta30, Anders Holm Millner31, Akiko Ishii-Watabe11, Alison Joyce17, Sumit Kar12, Robert J Kubiak21, Seema Kumar32, Karen Liao33, Mohanraj Manangeeswaran19, Michael Partridge24, Samuel Pine34, Johann Poetzl35, Manoj Rajadhyaksha36, Michele Rasamoelisolo37, Susan Richards38, Yuan Song39, Steven Swanson39, Seth Thacker19, Meenu Wadhwa9, Andreas Wolf40, Lucia Zhang10 & Lin Zhou19
Authors are presented in alphabetical order of their last name, with the exception of the first 9 authors who were session chairs, working dinner facilitators or major contributors.
The affiliations can be found at the beginning of the article.
‡Co-author for the New FDA Draft Guidance on Immunogenicity Information in the U.S. Prescribing Information section
HOT TOPICS & CONSOLIDATED QUESTIONS COLLECTED FROM THE GLOBAL BIOANALYTICAL COMMUNITY
The topics detailed below were considered as the most relevant “hot topics” based on feedback collected from the 15th WRIB attendees. They were reviewed and consolidated by globally recognized opinion leaders before being submitted for discussion during the 16th WRIB. The background on each issue, discussions, consensus and conclusions are in the next section and a summary of the key recommendations is provided in the final section of this manuscript.
New FDA Draft Guidance on Immunogenicity Information in the U.S. Prescribing Information
What is the impact of this guidance on the industry? What is the industry reaction to this guidance?
Immunogenicity & Bioanalysis for Drugs that have a Prolongation Effect in vivo
For prolongation effect and half-life of multi-domain drugs, what are the current expectations for evaluating ADA to specific domains in a drug? Is pos/neg for domain reactivity sufficient? Is reporting total ADA incidence in the labeling sufficient? NAb for multi-domain drugs: Is evaluation of neutralization potential related to MOA of the intact drug sufficient and reported as pos/neg? If different domains should be considered, then which ones and why?
Anti-PEG antibodies: What is the current thinking for evaluation of antibodies to PEG? Can anti-PEG antibodies be better evaluated and should specifically be reported in labeling? Challenges in anti-PEG antibody detection/assessment: Should there be better differentiation of anti-IgG vs IgM anti-PEG antibodies. This could contribute to safety, hypersensitivity reactions due to different ability to bind complement.
“Inconclusive” label for samples (ADA negative but drug levels still high): How often is this used? Should we better align ADA and drug level data that is shared to provide a final interpretation for the sample? E.g., in the same listing when doing ADA/PK evaluation?
Affinity of ADA in Clinical Samples
What is viewed as a potential value of evaluating ADA affinity? What type of studies may benefit from developing methodologies designed to determine ADA affinity? What potential technologies may be advanced to develop for ADA affinity? When is it necessary to carry out further characterization of the ADA response beyond ADA detection and NAb assessment? E.g., isotyping, relative affinity, concentration?
What are the barriers/challenges to deep characterization of the ADA response beyond ADA detection and NAb assessment?
Risk-based Approaches, Prediction & Mitigation
In vitro assays as predictive tools are used for CMC and other applications – can these tools tell us about post translational modifications, reactivity, and immunogenicity risk? What is the acceptability of using predictive tools for quality attributes? What are the considerations of utilizing complex in-vitro organoid models to assess risks as a substitute for conducting nonclinical studies? What are guidelines and value for the use of in-vitro tools in biosimilar development? Would FDA consider in vitro / in silico immunogenicity prediction data being supportive and of value to allow a targeted biosimilar development and corresponding clinical study data? Further, could in vitro / in silico immunogenicity prediction data be supportive to open a discussion on “interchangeability” topic, i.e., if an additional clinical study as currently recommended by FDA would be required or could be waived to get interchangeability designation for biosimilars based on comprehensive immunogenicity prediction data? Flow cytometry is a common assay used in in vitro prediction of immunogenicity risk assessment (e.g., T cell proliferation assays). What level of validation is appropriate for these assays? There is lack of robustness/repeatability for many in vitro risk mitigation methods. What is the practical predictive value for state-of-the-art methods? Is it better for some modalities than others?
Characterization of “high” Incidence Clinical ADA beyond ADA & NAb Assay Testing
What clinical ADA/safety/efficacy/exposure results would trigger additional ADA characterization assessments to be conducted? Should these additional characterization assessments be included in the BLA submission package or held internally and used when needed to answer regulatory questions?
T-cell Engager (BiTE) Immunogenicity & Associated Cytokine Release
What have we learned about immunogenicity and route of administrations having an impact for T-cell Engagers? What have we learned about cytokine measurement for BiTE? What is the translatability of NHP cytokine release? How many cytokines should be measured and how sensitive?
Target Interference on Screening Assays Cut Point & Importance of Risk Assessment for pH Sensitive Multi-domain Biotherapeutic (MDB)
How do you address assay interference in immunogenicity assays, particularly in the context of MDBs that target multimeric/multivalent membrane-bound proteins/receptors? What are challenges and mitigation strategies for identifying appropriate target protein (endogenous vs recombinant vs synthetic peptide) to test target interference in the assay? How do you characterize assay performance? What is the extent of reagent characterization needed, particularly for hard to express membrane bound target proteins/receptors?
Preclinical & Clinical Harmonization & Enhanced Tiered & Cut Point Approaches
When is ADA testing needed for biopharmaceutical GLP studies? ADA results are used to inform regarding potential cause for observations including altered drug exposure and some safety findings. Since these findings are informative and not usually definitive, what level of compliance is required? Can immunogenicity endpoints be performed as exploratory and/or non-GLP study contributions for GLP studies? What are the recommendations on what is needed for ADA method cross-validation and acceptance criteria? If ADA screen and confirmatory data (validation and/or sample analyses) tells us the ADA assay is specific, what is industry and health authorities' perspective on not performing the confirmatory tier for clinical assays? If a confirmatory tier is no longer needed and only screening is performed, what would be an appropriate and accepted false positive rate for the screen cut point?
NAb Assays Integrated Approach
In which situations would using PK and PD data to determine neutralization, without an actual NAb assay, be acceptable? Is domain mapping needed when a sensitive and drug tolerant NAb is available?
ADA Assay Comparison & Monitoring
How can data from the same assay used in different labs internationally be compared? Is there a strategy to pool the data? Should ADA assay monitoring criteria be built into plate acceptance? How can limits be set for new assays vs. legacy assays? Where do the regulatory agencies stand on this topic?
DISCUSSIONS, CONSENSUS & CONCLUSIONS
New FDA Draft Guidance on Immunogenicity Information in the U.S. Prescribing Information
The FDA issued a new draft guidance: Immunogenicity Information in Human Prescription Therapeutic Protein and Select Drug Product Labeling — Content and Format in February 2022 that provides recommendations to sponsors on how to incorporate immunogenicity information into the labeling of therapeutic protein products and select drug products that have immunogenicity assessments [Citation39]. This draft guidance recommends including a new dedicated subsection, 12.6 Immunogenicity in the Clinical Pharmacology section, to improve the ability of healthcare practitioners to access immunogenicity information in labeling. Historically, most immunogenicity information has been described in the Adverse Reactions section, not the Clinical Pharmacology section.
The following examples were mentioned by the industry where historically the content, format, and organization of immunogenicity information in product labeling has been reported differently for therapeutic protein products within the same class and products with the same target:
The adalimumab labeling describes a 5% ADA incidence in studies of adalimumab-treated patients with rheumatoid arthritis (RA). By comparison, ADA incidence in the etanercept labeling in etanercept-treated patients with RA (3.6% to 8.7%) and in the golimumab labeling in golimumab-treated patients with RA (4% or 16% depending on the assay), is comparable or higher than the ADA incidence reported in adalimumab-treated patients with RA in the adalimumab labeling. However, higher ADA incidences may or may not be associated with clinically significant effects. Furthermore, the golimumab labeling reports ADA using both standard and drug tolerant assays, but the adalimumab and infliximab labeling do not.
The pembrolizumab ADA assay has poor drug tolerance and a substantial portion of pembrolizumab-treated patients were unevaluable for immunogenicity which appears to be a different standard compared to how immunogenicity information was included in the golimumab labeling. Additionally, the pembrolizumab labeling includes immunogenicity data from patients who received weight-based dosing, patients who received 200 mg every 3 weeks, but no data from patients who received every 6-week dosing (which is approved in adults). Furthermore, immunogenicity data in the pembrolizumab labeling is not reported by indication whereas the labeling for atezolizumab and nivolumab have ADA incidence listed by indication. Finally, the Immunogenicity subsection of the atezolizumab labeling is more than one page. However, publications indicate atezolizumab ADA is not clinically meaningful and should not impact treatment decisions [Citation66]
Additionally, according to a recent FDA analysis, over 30% of labeling with immunogenicity assessments did not include any statements regarding the immunogenicity impact on safety or effectiveness (30% of these labeling did not state whether there was an observed or potential clinical impact, there was no observed clinical impact, or the clinical impact was unknown) [Citation55]. This draft guidance recommends that the labeling for therapeutic proteins and small molecules with immunogenicity assessments clarify whether ADA is clinically significant (e.g., ADA has a clinically significant effect on safety or effectiveness) or ADA is not clinically significant.
During the discussion, the FDA provided background on the writing of the guidance as a technical guidance. The FDA reviewed all post-marketing requirements (PMRs) and post-marketing commitments (PMCs) from the last several years. More than half had PMR or PMC related to improving immunogenicity assays. Results indicated that many historical submissions had suboptimal assay development [Citation67].
If the methodology for the immunogenicity evaluation was adequate, the draft guidance recommends including:
The following statement in the Immunogenicity subsection because cross study comparisons regarding ADA results can be problematic:
“The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons. of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of [proper name] or of other [core name] products.”
A statement in the Immunogenicity subsection on whether the presence of ADA was associated with a change in PK, PD, effectiveness, and/or safety. If applicable, adverse reactions that are associated with ADA will still be included in the Adverse Reactions section of the labeling; however, if ADA were not associated with adverse reactions, ADA information generally should not appear in the Adverse Reactions section. If data are sufficient to support a determination that observed ADA are not clinically significant, the Immunogenicity subsection should include a statement about the lack of clinically significant effect of ADA.
When the methodology for the immunogenicity evaluation is not adequate, the following or similar language should be included in the Immunogenicity subsection: “There is insufficient information to characterize the ADA response to [proper name] and the effects of ADA on PK, PD, safety, or effectiveness of [core name] products.”
Case studies of clinically significant ADA were discussed including:
Pure red cell aplasia due to ADA against Epoetin alfa that cross-reacted and neutralized endogenous erythropoietin [Citation68].
Reduced adalimumab concentrations and loss of efficacy due to immunogenicity [Citation69]
In a clinical trial, treatment with a megakaryocyte growth and development factor induced NAbthat cross-reacted with endogenous thrombopoietin causing dramatic reduction in platelets [Citation70]
The introduction of the draft guidance will potentially lead to more uniformity with including immunogenicity information in labeling across product classes and therapeutic areas, as appropriate. However, it is unknown how some items will be handled. For example, the draft guidance does not address how to incorporate immunogenicity data from approved products with multiple indications and complicated studies.
Cases were discussed that highlighted that immunogenicity information in labeling should provide information that is useful to physicians in making treatment decisions. For example, the adalimumab ADA incidence and clinical impact [Citation69] is being used to devise treatment algorithms in clinical practice [Citation71]. There is strong need for consistent presentation of data, as appropriate, across modalities (e.g., mAbs), therapeutic areas, and products with the same target (e.g., TNFi, PD-1/PD-L1), and reporting multiple ADA rates – regular and/or drug tolerant.
The industry perspective on the draft guidance was thoroughly discussed. As discussed above, the draft guidance provides suggestions on how to include and when to update the immunogenicity information in the labeling. The clarity and consistency this guidance will bring forth is welcomed and encouraged by industry. The draft guidance will provide clinicians with more information to be able to interpret the immunogenicity data. More information is always better for providers to manage and discuss risks-benefits of drugs and monitoring needs for their patients. It is envisioned that with this information, providers may be better able to monitor for possible immunogenicity-related adverse effects and choose the product with the best benefit-risk profile for their patients. However, the context in which all this information will be assessed and interpreted by healthcare providers should be considered. Do the providers who prescribe the drugs with immunogenicity information in labeling have the knowledge and understanding of the nuances of how ADAs and NAbs are detected to interpret immunogenicity information from different assays? What are the critical differences between the ADA/NAb assays of two products in the same class or with the same target? How important is it to have this understanding to adequately discuss risks and benefits of the drug based on the immunogenicity information provided in the labeling? The draft guidance is a step in the right direction. However, additional practical considerations may be needed for providers, to derive the full benefits of the changes suggested in the draft guidance.
The guidance also makes a distinction between reporting immunogenicity using “inadequate” and “adequate” methodology. However, it does not specify what makes a methodology adequate. Observing high ADA incidence combined with pronounced impact of ADA on PK/PD and safety support the adequacy of immunogenicity assessments. On the other hand, detection of ADA in a small fraction of study subjects with no identified clinical impact may prompt concerns about suitability of bioanalytical methods since absence of evidence for ADA presence is not evidence of ADA absence. Methodology for immunogenicity evaluation should be capable to obtain sufficient information to characterize ADA response and its effects on PK, PD, safety, and/or effectiveness. Commonly, the adequacy of immunogenicity assays is judged based on their ability to detect clinically impactful ADA concentrations, if present, at drug levels expected to be present at relevant sample collection time points. While the impactful ADA concentration is typically set at 100 ng/mL this may not necessarily apply to all drug development programs. The ability of ADA to affect PK and PD is likely dependent on the drug concentration and the stoichiometry of binding between ADA and the drug. Failure to meet this expectation may be incorrectly interpreted as a flaw in the bioanalytical methodology; however, it may be a result of ADA incidence comparable to or lower than the false positive rates used for cut-off point determination.
A broad outline of criteria was discussed that could be used to assess the adequacy of immunogenicity methodology as related to immunogenicity information in the labeling. There is no universal definition of adequate methodology for immunogenicity assessments. The same ADA concentration (e.g., 100 ng/mL) may have very different impact on PK/PD/efficacy/safety depending on the drug program. Sensitivity and drug tolerance are mostly characteristics of the positive control antibodies and less of the analytical method. Adequacy of methodology for immunogenicity assessment should be evaluated using totality of data and immunogenicity information in the labeling should evolve as new data become available [Citation61–63,Citation65].
Industry also stated that that draft guidance does not clarify if detailed information about the assay type should be included in the Immunogenicity subsection and recommended that the proprietary name (instead of the proper name) be used in the immunogenicity clarification statement. Additionally, the panel though it would be helpful if the final guidance discusses when baseline ADA information should be included in the Immunogenicity subsection or should the focus in this subsection be on treatment-emergent ADA data (ADA positivity after drug initiation).
The draft guidance makes a distinction between situations when the assay methodology is inadequate versus situations when assay methodology is adequate but there is insufficient data (e.g., low ADA incidence) to determine whether ADA affected the safety or effectiveness of the product. The panel noted that the examples in the draft guidance do not include titer information, and the FDA stated that they will consider including information about titer in the final guidance. The titer levels that are clinically impactful are determined based on the integrated analyses of immunogenicity, PK, and clinical data. Another key component is to classify patients' immune responses as clinically significant or not clinically significant. This will involve evaluating effects of ADA on PK, PD, efficacy, and safety. This depends on if drug has a narrow therapeutic index. The treatment period for immunogenicity assessment may be difficult to determine in some programs (e.g., oncology studies have different treatment length depending on disease progression). Therefore, there may be a need to report immunogenicity results from each study if there are different durations of drug exposure, immunogenicity methods used, or dosage regimens rather than pooling immunogenicity results from different studies.
Finally, when this guidance is finalized, all labeling for therapeutic protein products and drug products that have immunogenicity assessments should be updated with these recommendations, which will have a significant impact on submission work. Updating labeling with immunogenicity information will take a long time with significant efforts and challenges. Industry is encouraged to start revising the labeling proactively.
Immunogenicity & Bioanalysis for Drugs that have a Prolongation Effect in vivo
Currently, factors that influence developing ADA are better understood, how to measure ADA is increasingly harmonized and a risk-based approach used to develop a clinical immunogenicity assessment program. However, more sophisticated drug development requires a broader view encompassing protein characterization as well as clinical pharmacology in the overall immunogenicity strategy. This is most evident in the bioengineering approaches that are increasingly utilized to develop modified biotherapeutics having improved properties that would benefit patients. Key areas of interest include designing into drugs properties that would extend the drug half-life, enhance bioavailability, and facilitate multi-target binding. There are several strategies that can be used to extend half-life for proteins and peptides which generally involve FcRn recycling or by acting as a bulking agent. Such approaches include PEGylation, glycoengineering, Fc-fusion proteins (e.g., etanercept, efmoroctocog alfa), albumin fusions (e.g., albiglutide), unstructured protein polymers and drugs that are single-chain chemically linked and use albumin as with nanobodies.
However, such modifications may also result in the development of new conformational areas thereby creating neoepitopes or disrupt existing ones, in both cases influencing antibody formation and detection. In addition, the use of glycoengineering, an approach that is used to enhance bioavailability, may contribute to an antigenic masking effect, depending on epitope location. Overall, the prolongation effect generally results in increased exposure to the drug by the immune system. This may further facilitate soluble immune complex formation resulting in a greater need for sensitive, drug tolerant assays and an extended duration sampling strategy. An understanding of the structural changes related to half-life extension, improved bioavailability and the biologic properties associated with its components are important aspects to consider during immunogenicity assay development as well as relevant for data reporting and interpretation.
The case studies discussed demonstrated recent bioengineering approaches and relevant aspects related to immunogenicity bioanalysis and interpretation. Anti-PEG antibodies are an important consideration for PEGylated molecules. Development of antibodies to PEG appears to depend on linked drug, PEG size and chemical composition and prevalence of pre-existing anti-PEG antibodies in the general population is increasing (up to 25%) [Citation72,Citation73]. Prevalence of reported antibodies to PEG highly depends on subjects and assays used. Therefore, bioanalysis challenges and clinical consequences should be considered. For example, ELISA approaches have limitations that may result in inadequate immune complex dissociation [Citation74]. Anti-PEG antibodies can lead to accelerated blood clearance phenomenon, anaphylactic reaction on first dose and complement activation [Citation75].
Current approaches for evaluation of antibodies to PEG were discussed and if anti-PEG antibodies could be better evaluated and should specifically be reported in labeling. The consensus opinion was that antibodies to PEG should be evaluated but not necessarily reported in labeling. Current practices for evaluating antibodies to PEG appear to be sufficient. Differentiation of IgG and IgM is possible by ELISAs, but typically not done and not required. Timing of response can be used to differentiate between IgG and IgM. More data around analysis methods and impact needs to be shared and published.
Another discussion topic for multi-domain drugs was about the current expectations for evaluating ADA to specific domains in a drug and whether pos/neg for domain reactivity is sufficient. It is usually best to characterize at least some of the domains with a tiered approach, but this characterization does not need titer assessment (i.e., can be yes/no). Labelling is a separate consideration. It is informative to have multi-domain characterization for regulator review and for understanding the product but it is not part of the labeling. PEG is an exception where it is definitely needed. The recommendation was that the assessment is based on risk: if there is no clinical signal, it may not be necessary to define domain specificity, and reporting total ADA is sufficient. Discussion with regulators before application is encouraged.
NAb for multi-domain drugs is another consideration to evaluate. The reactivity depends on intact molecule. Therefore, mechanism of action NAb assay is typically sufficient. Separate assays for each domain are not usually necessary and should depend on risk.
The final discussion was about the use of “Inconclusive” label for ADA negative samples with drug levels that exceeded the drug tolerance of the ADA assay and the value of aligning ADA and drug level data that is shared to provide a final interpretation for the sample. Many/most companies do not use the term “inconclusive” to categorize ADA. It is challenging because groups in some companies do not see ADA and PK data together. Overall, it is important to report the drug tolerance of the ADA assay determined during validation. Seeing the PK and ADA data together is important for interpretation of a negative result, and pharmaceutical companies should provide both PK and ADA data when submitting data listings. Regulators currently conduct more extensive review of the data.
Affinity of ADA in Clinical Samples
Characterization of clinical ADA responses to biotherapeutics can be important for understanding the consequences of immunogenicity. ADA are expected to be polyclonal, with composition and affinities that evolve over time. Measuring ADA binding affinity can be complicated by the polyclonal nature of response, residual drug in sample and low ADA levels.
A case study was discussed of a novel workflow to determine the apparent ADA affinity (KD) against a monoclonal antibody biotherapeutic [Citation76]. An affinity capture elution pre-treatment step was used to isolate ADA and remove residual drug interference from samples. Solution-phase equilibrium incubation was performed using drug and sample ADA as fixed and variable binding interactants, respectively. Unbound ADA concentration was measured using a Singulex Erenna ligand-binding assay (LBA) method and apparent ADA KD values were calculated using a custom algorithm [Citation77]. KD values determined for ADA positive samples showed good correlation with other immunogenicity parameters, including titers and NAb activity with a general increase in affinity over time, indicative of a maturing immune response. Presence of high affinity ADA was observed in later timepoint samples that also scored high in the routine ADA assay. The caveats of this approach were that a custom algorithm was used and that KD of a polyclonal is an overall average.
High affinity of ADA immunoglobulins for the high titer samples were observed in the reported study. The expert panel discussed these findings and the potential value of evaluating ADA affinity. Overall, the consensus view was that there is limited added value in trying to determine affinity though it may inform models internally. The panel did not have examples where this was necessary. In addition, it is not necessary to carry out further characterization of the ADA response beyond ADA detection and NAb assessment unless there is a risk-based need. ADA affinity can be very difficult to perform and provide limited value.
Risk-based Approaches, Prediction & Mitigation
The development of unwanted immune response to protein therapeutics, gene therapies and cellular therapies might pose a risk. A weight of evidence approach is taken for immunogenicity risk assessment to evaluate risk by using different in vitro tools from the toolbox at the early stages of biologics development. Based on this, appropriate mitigation and de-risking strategies are implemented to have successful clinical development. Per the EU EMA, non-clinical in vitro or in vivo immunogenicity studies are not required but consideration should be given to the use of emerging technologies (novel in silico, in vitro and in vivo models). Methods include in silico screening (MHCII epitopes, humanness, developability), in vitro / ex vivo screening (MAPPs, cytokine secretion/ release, PBMC proliferation and binding, platelet aggregation), non-clinical ADA assays, and patient HLA typing.
Case studies were discussed that explore the utilization of in vitro tools for assessing immunogenicity risk for ocular therapeutics and aesthetic devices. Although the eye is regarded as an immune privileged site, this does not mean that an immune response cannot occur in the eye. Introduction of biologics in the ocular compartment can break the tolerance and trigger humoral responses under pro-inflammatory conditions [Citation78,Citation79]. In addition to the basic principles of immunology that govern T and B cell responses to foreign and self-proteins, the implications of extrinsic factors (product or host cell related impurities) that are particular to the therapeutic protein or its administration can act as adjuvant to influence immunogenicity or intra ocular inflammation for the intravitreal drugs. The ocular immunity is a spatial phenomenon and is dependent upon co-localization of immune cells with antigen presentation and co-stimulation forming the link between innate and adaptive immunity. Thus, apart from sequence specific liabilities, risk assessment of intravitreal drugs requires us to look for innate response (due to HCP, endotoxin, aggregates, excipients), impacts due to limited volume with high concentration, precipitation of biologics, toxicity, pH, presence of sub visible particles, silicone oil from syringes/needle etc. [Citation80–82].
Implanted and injected fillers can trigger a wide variety of adverse reactions and require a risk assessment just like any other therapeutics based on their origin and average persistence in the tissue. Fillers may also elicit an APC-mediated adaptive immune response which can be assessed using in vitro tools to characterize the risk.
It was agreed these case studies demonstrate that data from multiple orthogonal methods should be integrated for the immunogenicity risk assessment. The in vitro immunogenicity risk assessment tools that assess innate immune activation, MHC presentation, and antigen-specific T cell or non-T cell responses can be used to inform an immunogenicity risk assessment.
Another topic was the use of complex in-vitro organoid models to assess risks as a substitute for conducting nonclinical studies. Sponsors are exploring these tools and applications for assessing the immunotoxicity risk. Given that the predictive power of these models is uncertain, it is premature to use them for replacing pre-clinical data, however the FDA is receptive to receiving and considering data using these models.
It was discussed if in vitro assays as predictive tools can inform about post translational modifications, reactivity, and immunogenicity risk. There was agreement that these assays are expected to have a large impact to characterize the amount and types of impurities and post-translational modifications. However, qualification of these assays is important to build out with developed standards. There is a large variety of assays used and many vendors and internally produced standards are being used. These methods are not required and not common practice yet, but use of these tools is being considered by industry and regulators [Citation39].
For biosimilar development, the immunogenicity of the molecule per se is already known from the reference product and can be considered for an immunogenicity risk assessment of biosimilars. Minor modifications (e.g., in glycosylation profile, formulation used, or level of impurities) for the biosimilar compared to reference product are allowed, but their impact on immunogenicity needs to be assessed to demonstrate that observed changes in the biosimilar molecule will not induce unwanted immune responses. Beside extensive analytical characterization tools, methods used for the prediction of immunogenicity become of more and more interest in biosimilar development. Of note, the sensitivity of in vitro and in silico predictive methodologies needs to be factored in when evaluating results derived from such kind of tools. Even though in vitro and in silico immunogenicity assessment cannot definitively predict ADA generation, they could inform and assessment of immunogenicity risk relative to a product that has undergone extensive clinical assessment. Accordingly, information generated from such assays can help to support the biosimilar immunogenicity risk assessment.
Case studies using in vitro / ex vivo and in silico immunogenicity predictive methods in biosimilar development were discussed, including the use of NetMHCIIpan-2.0 analysis to predict HLA binding to drug product variants and the impact of formulations on immunogenic potential to be tested in MAPPS and DC assay. Data derived from those methods are used for the evaluation of differences in the biosimilar product compared to its reference product but may not be sensitive enough to resolve small difference that may still be biologically relevant. Immunogenicity potential assessment can help to de-risk biosimilar development programs and enable a more targeted biosimilar development based on a thorough understanding of the overall immunogenicity potential of the drug product.
The use of in vitro / in silico immunogenicity prediction data as being supportive and of value to “Interchangeability” was discussed versus performing an additional clinical study as currently recommended by US FDA to get interchangeability designation for biosimilars. In vitro and in silico studies for interchangeability could provide value since these studies in clinic can take years. Regulatory Agencies are very supportive of this work and would be open to consider interchangeability using in vitro or in silico prediction, but there are very few examples so far and more data is needed.
While the in vitro methods must be reliable in order to establish scientifically sound assessments, such methods do not require validation. Ultimately, clinical immunogenicity data generated during the clinical phases of the development are mandatory to evaluate the clinical relevance of immune responses. On the other hand, if these data were used to support a submission such as an ANDA, a validation of the in vitro methods would be required. To date, no regulatory recommendations are available that described the method development and validation rigor required for such in vitro methods. A case study was discussed of a dendritic cell: CD4+ T cell co-culture proliferation flow cytometry-based assay intended to predict the immunogenicity risk of peptide-related impurities that may be present in the peptide therapeutic proposed drug products. It was validated for its context of use and implemented in the laboratory based on the Clinical and Laboratory Standard Institutes guideline H62 – Validation of Assays performed by Flow Cytometry [Citation83].
It was discussed the use of flow cytometry for in vitro prediction of immunogenicity risk assessment (e.g., T cell proliferation assays) and the level of validation appropriate for these assays. Consensus was reached that validation is required to ensure the method fits for the purpose. There are many manuscripts for flow cytometry for other applications and suggestions for assay validation similar to those used in the presented case study above. Similar to clinical immunogenicity methods, the absence of assay harmonization and standardization is a challenge for in vitro flow cytometry assays used to predict immunogenicity [Citation84]. Therefore, the recommendation was to build more experience, publications, working group and case studies are necessary to expand knowledge/ data on this application and Regulatory Agencies would be willing to discuss this together.
It was agreed that overall, there is lack of robustness/repeatability for many in vitro risk mitigation methods. There is no standardization yet, and so far, no strong predictive value. These methods are mostly used for early selection vs. prediction or clinical follow up. There are limitations of assessing robustness (appropriate control/ reference material, master pools of donor cells, how many donors, practicality, amount of resources and overall value). Differences in modalities have not been defined. Therefore, the recommendation was to continue to develop methodologies and standardization protocols to show their value of them. As mentioned previously, there is no requirement by regulators for these when used as a research tool.
Characterization of “high” Incidence Clinical ADA beyond ADA & NAb Assay Testing
US FDA regulatory guidances regarding additional characterization of ADA beyond testing for ADA and NAb suggest that it may be useful to perform additional evaluation of potential impacts of ADA on PK, PD, safety, and efficacy [Citation39]. Depending on the size of the clinical trials and observed clinical ADA data, additional characterization may assist in the interpretation of impact of ADA.
This guidance was used as a starting point for a case study demonstrating the potential impact of the clinical ADA and the determination of the threshold of impactful clinical ADA titer observed in two pivotal clinical trials. A series of orthogonal methods was developed to further characterize ADA observed in two pivotal clinical trials supporting a mAb therapeutic with Orphan Drug Designation - available immunogenicity data were slim due to small size of the two trials. The case studies highlighted the methodologies developed to further characterize and assess impacts of clinical ADA and showed how the data was used during receipt of regulatory questions and helped the sponsor to provide answers following submission.
Subsequent characterization of the observed ADA/NAb from pivotal clinical trials in preparation for submission included assessment of ADA/NAb impact on PK (e.g., drug interference testing and clearance determined with PK assay measuring free drug concentration in clinical samples), orthogonal titer assessment to confirm magnitude of titer observed in bridging assay (i.e., ensure no artifact was present in the bridging electrochemiluminescent (ECL) ADA assay) and isotyping of ADA samples using LCMS-based methods to better understand the maturation of the immune response in Generalized Pustular Psoriasis (GPP) patients. Given the observed clinical ADA and the small GPP patient population size, additional characterization of the ADA response beyond the ADA and NAb assays assisted in the interpretation of ADA impact. The ability to understand the difference between ADA interference in the PK assay and clearance from the systemic circulation and providing orthogonal assessments to support the observed titer in the bridging assay were very useful in internal discussions. The isotyping methods provided supporting data to help understand the clinical ADA.
The discussions evaluated what clinical ADA/safety/efficacy/exposure results would trigger additional ADA characterization assessments to be conducted based on these case studies and whether these additional characterization assessments be included in the BLA submission package or held internally and used when needed to answer regulatory questions. Additional immunogenicity characterization (beyond NAb, and titer) is rarely performed by some sponsors unless there are clinical concerns. These concerns may include significant unexplained increase or decrease in exposure, loss of efficacy not linked to PK, and unexplained safety issues. These additional analyses should be discussed with the agency if they prove informative and are evaluated on a case-by-case basis.
T-cell Engager (BiTE) Immunogenicity & Associated Cytokine Release
A case study with Pasotuxizumab was discussed which is a T-cell engager (BiTE) molecule with specificities for PSMA and CD3 and was investigated in a clinical phase I study in patients with castration-resistant prostate cancer. To monitor potential immunogenicity in the clinical study an ADA assay and a NAb assay were developed and validated. The NAb assay is cell based and uses two cell lines to reflect the mode of action of the drug. Features of the immunogenicity assays and the corresponding data were discussed.
Pasotuxizumab is a BiTE molecule of the first generation with a molecular weight below the renal filtration cutoff and a serum half-life of a few hours. To achieve appropriate exposure in patients, the administration via subcutaneous injection or via continuous intravenous infusion were tested. There was a pronounced difference in clinical immunogenicity against Pasotuxizumab based on the route of administration within the clinical trial. The mode of action of T-cell engagers is based on the dual binding of an effector cell (T-cell) and a target cell (tumor cell) to the therapeutic molecule, leading to T-cell activation and subsequent lysis of the tumor cell.
Cytokine release is part of the mode of action of t-cell engagers. Dual binding of a tumor cell and a T-cell leads to formation of a cytolytic synapse and to cytokine release. The clinical cytokine release syndrome (CRS) is classified according to the “Lee criteria” and is a potential dose limiting toxicity [Citation85]. Cytokine release syndrome is a known side effect for T-cell engager treatments in the clinic and monitoring of cytokine concentrations in patient serum is routinely performed.
The analytical assay to monitor cytokine levels in patients treated with Pasotuxizumab and the corresponding validation data as well as the clinical data was discussed. Various multiplex assay systems are available. Serum concentrations of cytokines in response to BiTE treatment are dependent on tumor target binding and follow a characteristic pattern. The strategy used for the clinical characterization of Pasotuxizumab immunogenicity and cytokine release was discussed in the context of the corresponding non-clinical data obtained from toxicity studies in cynomolgus monkeys and lessons learned for further development of T-cell engaging drugs.
Based on the case study and other examples, discussions were held on the learnings about immunogenicity and route of administrations having an impact for T-cell engagers. There was consensus that the route of administration may be important. There are case studies where route of administration had a large impact on cytokine release. Cytokine release is a central safety consideration for BiTEs. Good correlation has not been seen between preclinical cytokines and clinical CRS/immunogenicity. Clinical criteria for CRS (and the specific cytokines) are different from preclinical cytokine data. Sponsors may need to develop custom assays for specific species. Looking at a limited number of cytokines may be informative but no consensus on how many to do or how informative it might be. Overall recommendations were to measure some cytokines, but there are not specific recommendations yet on number needed or sensitivity required. More data is needed.
Target Interference on Screening Assays Cut Point & Importance of Risk Assessment for pH Sensitive Multi-domain Biotherapeutic (MDB)
Multi-domain biotherapeutics (MDBs) often contain complex structures, multiple functional domains, and multi-step pharmacological mechanism of action (MoA). Due to their structural and functional complexity and the risk-based immunogenicity strategy, the sheer number of immunogenicity assays required to support clinical development of MDBs can be higher than single-domain biotherapeutics. The increased number of assays also increases the likelihood of encountering immunogenicity assay related challenges such as, soluble target interference, drug tolerance, pre-existing reactivity etc. Additionally, these assays require specialized critical reagents (e.g., domain-specific positive control, domain-specific competitor molecules or domain-specific labeled MDB component reagents).
Previous White Papers in Bioanalysis [Citation7,Citation10] discussed target interference in immunogenicity assays and potential to produce false-positive results due to the bridging of dimeric or multimeric targets. Previous recommendations were to examine and mitigate interferences mediated by circulating target which may require greater characterization beyond the typical two-tier analysis. Evaluating circulating target interference with recombinant target may not accurately reflect the effects of endogenous target. The 2015 White Paper in Bioanalysis discussed approaches to mitigate interferences - acidification, blocking/binding agents and sample pre-treatment to remove drug or target [Citation10]. Drug/target may interfere in the ADA assay but at the same time, ADA/target interference may impact the PK assay thus making data interpretation difficult. Another previous recommendation was to use integrated data interpretation from other available data (PK, target measurement assay etc.).
Updated case studies highlighting some of these unique bioanalytical challenges for MDB immunogenicity assays were discussed. It was agreed that MDBs pose unique and/or additional immunogenicity assay challenges that require additional scientific considerations. Target biology/ structure and MoA of MDB should be taken into consideration for designing appropriate bioanalytical assay strategy. Recombinant target may not (or fully) replicate endogenous target binding in the assay conditions. The importance of knowing your assay was agreed.
Updated recommendations were provided for addressing assay interference in immunogenicity assays, particularly in the context of MDBs that target multimeric/multivalent membrane-bound proteins/receptors. There should be no target inference if the target is not present in soluble form. However, there are cases where shedding can cause target interference and signal from target should be blocked. The biggest challenge is sourcing the necessary reagents (a protein that binds to the target or a specific anti-target antibody to block the target or alternatively, assay conditions that reduce target binding).
Another discussion point was the challenges and mitigation strategies for identifying appropriate target protein (endogenous vs recombinant vs synthetic peptide) to test target interference in the assay. The biggest challenge is getting/generating the reagents that mimic the endogenous protein.
Endogenous proteins are typically very difficult to purify at amounts needed. Recombinant proteins and synthetic peptides do not always behave like the endogenous protein. If available, compare results from patients with and without target expression to test interference. Characterization is important to show that reagent generates relevant activity.
The recommendations on chasing sensitivity, drug tolerance, target interference, and cut point against individual functional domains in MDB immunogenicity assays were also revisited. It was noted that the assays are not always developed for each functional domain. When they are, they are typically used for characterization, and as such sensitivity, drug tolerance, and interference are not usually determined. Trying to determine these parameters would be very challenging and very resource intensive, and controls against different domains would be needed. The recommendation was to develop fit for purpose characterization assays as needed.
Finally, recommendations were provided on the impact of ADA/target on PK assay, and impact of drug/ADA on biomarker (target) assay that makes integrated data interpretation difficult. This is a problem in complex biological systems and difficult to chase. The current practice of most is to restrict testing to drug interference in ADA and target assays, and target interference in PK assays unless there is a compelling need for additional characterization usually based on a specific risk concern. The recommendation is to take a risk-based approach and follow current practice unless there is specific concern.
Preclinical & Clinical Harmonization & Enhanced Tiered & Cut Point Approaches
GLP toxicology studies are a key component in progressing biotherapeutics from discovery to clinical trials. The purpose of these studies is to explore the relationship between therapeutic dose and adverse effects. Administration of human or humanized biologic therapeutics to animals in a GLP study may induce immunity in a subset of treated animals. This immunity is likely to manifest as ADA, the impact of which may be minimal, modify drug clearance (accelerate or sustain) or may contribute to safety findings, including immune complex disease. Preclinical immunogenicity was discussed in the 2021 White Paper in Bioanalysis in the context of the China NMPA Immunogenicity Guidance. The discussion covered regulatory expectations and common practice across industry. In general, regulatory guidance documents refer to the limited relevance of immunogenicity in the preclinical setting, i.e., to contribute to TK/PD and safety finding discussions without direct translation to human immunogenicity. Apart from an expectation of method validation, the guidance around compliance is generally limited [Citation38,Citation40,Citation86]. Across industry there are a variety of approaches used for preclinical immunogenicity testing with regards to the level of compliance, analytical strategy, assay format and testing scheme.
The discussion was continued of when ADA testing is needed for biopharmaceutical GLP studies. ADA results are used to inform regarding potential cause for observations including altered drug exposure and some safety findings. Since these findings are informative and not usually definitive, experts discussed what level of compliance is required and if immunogenicity endpoints can be performed as exploratory and/or non-GLP study contributions for GLP studies. Based on the experience of the expert panel, especially for lower risk molecules, preclinical ADA is typically not done if not necessary. Often sponsors have non-GLP assays ready. A confirmatory step is no longer performed and not required. Most sponsors are using S:N instead of titer. The updated recommendation was that ADA testing necessity is risk based. Sponsors should look at the PK data for exposure, and safety. If ADA characterization is necessary based on exposure and safety data, it can be done using non-GLP assays and samples should be collected as recommended by ICH S6 (R1) [Citation86].
Acceptance criteria for ADA method cross-validation was evaluated. Most variability is in negative controls since signal for PCs are much higher. The cut point will define key assay characteristics and study specific cut points can help to address these concerns. Different CRO labs interpret and perform cross validation differently. This is most challenging for assays and samples analyzed outside the US, particularly China. One suggestion from the expert panel was to utilize CLSI guidelines for cross-validation – but applying diagnostics standards is difficult for ADA because of lack of reference standards and quasi-quantitative assay nature. It was also brought up to use ISR based approaches for reproducibility, but expert panel did not think this is feasible for immunogenicity. Many sponsors are trying to develop better methods for cross-validation. The recommendation was that if results differ between labs (i.e. due to different populations for cut-point), the data need to be explained instead of performing a revalidation. Use cross-validation as a characterization tool, not a pass-fail tool.
Perspectives were provided on not performing the confirmatory tier for clinical assays if ADA screen and confirmatory data (validation and/or sample analyses) show the ADA assay is specific. Discussion with Regulatory Agencies is encouraged should this alternative approach be followed. Currently, Regulatory Agencies recommend a confirmatory tier in the tiered approach of assessing immunogenicity risk.
It has been discussed that even if S/N has gained momentum preclinically it has not in the clinical environment. Regulatory Agencies consider the use of S/N case by case if limitations of replacing titer with S/N can be reasonably addressed. Until the best practice is established, the use of titer data to assess clinical risk of immunogenicity is preferred.
NAb Assays Integrated Approach
The use of an integrated data approach versus in vitro NAb to determine the presence and impact of NAbs was previously discussed in depth and recommendations have stated that translation of in vitro NAb data to in vivo neutralizing effect can be complex [Citation17,Citation19,Citation27,Citation30,Citation46].
Dichotomic classification of samples as being POS/NEG for NAb is not always conclusive and knowledge is lacking about the clinically relevant levels of NAbs. Integrated approaches evaluate impact on exposure, efficacy and safety by correlating levels and persistence of binding ADA to PK, PD, biomarkers and safety events, and can also include relevant in vitro NAb assay. The debate is ongoing on the utility of data from in vitro NAb compared to the PK/PD/ADA integrated approach. The integrated approach should be part of the investigation of the consequence of ADA regardless of the presence of an in vitro NAb assay.
It was concluded that NAb assays are still to be routinely performed if possible. For high-risk drugs, it should be done early during drug development. For low-risk drugs, it may only be needed for pivotal trials. If there is a potential safety concern, then a NAb assay is required. US FDA will always defer to the most conservative position, especially for new molecular entities. Health Canada recommends that doing the NAb assay is the default position, but if there is a case for using PK/PD/ADA data, the Agency should be contacted to discuss. However, it was argued again that more often than not, NAb assays do not provide enough additional information to justify the labor required to develop and validate the assay and test clinical samples.
ADA Assay Comparison & Monitoring
As stated in the 2021 White Paper in Bioanalysis [Citation30], ADA assays play a key role on the road from drug development to drug approval, providing valuable insight into immune responses potentially developed against the administered drug and helping to evaluate any clinical relevancy or implications with respect to the safety. In the large molecule world, ADA assays have continued to evolve as industry has introduced more complex drug candidates while pursuing increasingly challenging targets. Developing and validating an ADA assay that is robust enough to withstand the test of time during clinical drug development, where even a single study can span multiple years is challenging.
Once a robust assay is available and is being used over a long period of time, the focus shifts on how to best monitor its performance. To do that, fluctuations in assay signal or signal to noise ratio (S/N) are followed over time as a measure of assay performance. The a priori establishment of acceptance criteria on the performance of a specific set of samples or QCs (usually a negative control, NC, and two or more concentration levels of a positive control, PCs) to be incorporated in every plate has been proposed as a way to statistically assess the behavior of the assay over time. Establishing a priori criteria can help remove the ambiguity that exists when anomalous signal is observed, thus reducing the number of judgements calls the data reviewer is required to make.
Ideally, these monitoring limits should be established in a way that accurately reflects the expected performance of the assay, including accounting for the inherit variability that exists. The goal is not to use criteria that are too conservative, but rather to provide guardrails that ensure that the assay does not veer too far from expected historical data.
Whether or not ADA assay monitoring criteria should be built into plate acceptance was discussed based on a case where EMA asked that monitoring criteria be implemented. It was agreed that no standard approach currently exists to determine criteria, but useful guideposts can be provided by a statistical evaluation of the QCs. It was recommended that if it was determined that acceptance criteria were required, it was important to set limits using both validation and production runs so that QC ranges reflect assay performance over a sufficiently long period of time. However, it was also discussed that applying a priori criteria that are inappropriately set can result in plate failures due to small changes in QC signal, leading to rejection of otherwise valid data.
As more and more clinical trials are conducted in different countries and companies are facing increasing difficulty in shipping study samples out of certain countries, many companies have been conducting ADA sample testing at local labs where the samples are being collected. FDA issued guidance in 2019 requesting sponsors to demonstrate reproducibility of an ADA assay run by two or more independent laboratories during a study. ADA assay comparability strategy should be based on an integrated approach driven by science, immunogenicity risk assessment including clinical impact. It aims at characterizing/evaluating performance of one assay at two labs or two assays at the same lab.
ADA assay comparability strategy should include both technical and clinical assessment. Sensitivity, drug tolerance and precision need to be evaluated in order to establish comparable assay performance (technical assessment) as shown in . Two One-Sided T-Tests (TOST) is proposed to be used as a statistical tool and Maximum Allowable Difference (MAD) as acceptance criteria for evaluating technical aspect of ADA assay comparability. This approach has been used by commercial groups when transferring quality control (QC) assays between contract manufacturing sites. If the absolute difference in population means and the confidence intervals are less than the MAD, two groups are considered comparable. Clinical comparability assessment should be categorized into basic, in-depth or alternative assessment, depending on molecule's immunogenicity risk/impact and sample/matrix availability. Clinical comparability should be performed per method and not per clinical study (). Confusion matrix and Cohen's Kappa coefficient are recommended to be used for the assessment. Basic clinical assessment using spiked QC samples applies to all cases and should be part of ADA assay cross-validation between two labs or two methods. In-depth clinical assessment only applies to molecules with medium and high immunogenicity risk and should be conducted only one time within a study. In-depth clinical assessment is recommended to be conducted at both sample level (characterize the assay) and patient level (determine assay comparability). Alternative clinical assessment is driven by availability of the incurred study samples. Several options, such as waiting for future availability of the incurred samples, combining spiked QCs and incurred samples, or spiking QC samples in a surrogate matrix or disease state, etc., can be explored (). Number of samples and acceptance criteria for ADA assay comparability assessment are still evolving as more data from is being gathered.
RECOMMENDATIONS
Below is a summary of the recommendations made during the 16th WRIB:
New FDA Draft Guidance on Immunogenicity Information in the U.S. Prescribing Information
This new draft guidance will provide clinicians with more information to be able to interpret immunogenicity data.
Draft guidance is a great first step and provides help, but there are still lingering questions about how to include immunogenicity information in labeling (e.g., reporting immunogenicity by pooling study data or by patient population, whether detailed information about the assay type should be included, when it would be appropriate to report baseline immunogenicity prior to drug initiation).
When this guidance is final, labeling for therapeutic protein products and drug products with immunogenicity assessments should follow the recommendations in the guidance, which will have a significant impact on submissions.
Updating labeling will take a long time with significant efforts and challenges.
Industry is encouraged to start revising the labeling proactively.
Immunogenicity & Bioanalysis for Drugs that have a Prolongation Effect in vivo
Reporting total ADA incidence in the label for multi-domain therapeutics is based on risk: If there is no clinical signal, reporting total ADA is enough. Have discussion with regulators before application.
Mechanism of action NAb assay is typically sufficient. Separate assays for each domain are not usually necessary. May depend on risk.
Analyze for anti-PEG antibodies but it does not need to be isotype specific nor necessarily part of the label
There is no requirement to characterize responses as inconclusive rather than negative. Provide both the PK and ADA in the listing.
Affinity of ADA in Clinical Samples
There is limited added value in trying to determine affinity though it may inform models internally. No examples available in the industry where this was necessary.
It is not necessary to carry out further characterization of the ADA response beyond ADA detection and NAb assesment unless there is a risk-based need. ADA affinity can be very difficult to perform and provide limited value.
Risk-based Approaches, Prediction & Mitigation
In vitro assays as predictive methods are not required and not common practice for follow on recombinant peptides yet, but use of these tools is being considered by industry and regulators.
In vitro methods such as organoid models need more experience and data comparison to pre-clinical data. Regulatory Agencies are interested in seeing more data
Regulatory Agencies would be also receptive to data derived by in vitro tools for targeted biosimilar development (including for interchangeability designation), but there is no expectation to provide such data in general. More experience and data is needed for providing guidelines
Characterization of “high” Incidence Clinical ADA beyond ADA & NAb Assay Testing
Additional immunogenicity characterization (beyond ADA, NAb, and titer) normally are not performed unless there are clinical concerns. These concerns may include:
Significant unexplained increase or decrease in exposure
Loss of efficacy not linked to PK
Unexplained safety issues
This additional analysis should be discussed with the Regulatory Agencies if they prove informative. Evaluate need on a case-by-case basis and discuss relevant results with Agencies
T-cell Engager (BiTE) Immunogenicity & Associated Cytokine Release
Route of administration may be important.
May need to develop custom assays for specific species for cytokine release. Looking at a limited number of cytokines may be informative.
It is important to measure cytokines, but there is not specific guidance yet on number needed or sensitivity required.
Target Interference on Screening Assays Cut Point & Importance of Risk Assessment for pH Sensitive Multi-domain Biotherapeutic (MDB)
Regarding assay interference of MDBs that target multimeric/multivalent membrane-bound proteins/receptors, no target inference is expected if there is no soluble form. However, there are cases where shedding can cause target interference and signal from target should be blocked.
Obtaining reagents is challenging for identifying appropriate target protein (endogenous vs recombinant vs synthetic peptide) to test target interference. Results from patients without target expression should be compared and characterize reagent as thoroughly as possible
Preclinical & Clinical Harmonization & Enhanced Tiered & Cut Point Approaches
Sponsors should look at the PK data for exposure, and safety. If ADA characterization is necessary based on exposure and safety data, it can be done non-GLP. Samples should be collected as recommended by ICH S6 (R1).
If assays perform differently between labs (i.e., due to different populations for cut-point), the data should be explained. Cross-validation should be used as a characterization tool, not a pass-fail tool.
NAb Assays Integrated Approach
NAb assays should be developed if possible.
Regulatory Agencies always defer to most conservative position, especially for new molecular entity. NAb assays are used to see if the possibility of a rare adverse event or loss in efficacy is likely.
Health Canada recommends that doing the NAb assay is the default, but if the sponsors have a case for using PK/PD/ADA data, they should contact the Agencies in advance to discuss.
Domain mapping may be performed for ADA assays, but not typically done for NAb assays.
ADA Assay Comparison & Monitoring
It is part of the assay life cycle management if an assay has been modified or evolved overtime. Thus, it may not be necessary to compare subsequent iterations of the same assay; it may be sufficient to use data collected using the latest iteration of the assay, which is presumably also the most optimized.
Assay monitoring should be performed for all ADA assays. There are no standard approaches. However, the use of acceptance criteria based on the performance of controls to be incorporated in every plate is advised.
Validation and production runs should be used when setting criteria to monitor ADA assay so that criteria may be more reliable over the long term.
Acknowledgements
US FDA, Europe EMA, UK MHRA, Norway NoMA, Brazil ANVISA, Health Canada, Japan MHLW and WHO for supporting this workshop
All Session Chairs & Working Dinner Facilitators for chairing the workshop and the White Paper discussions: Dr. Chris Beaver (Syneos), Dr. Arindam Dasgupta (US FDA), Dr. Fabio Garofolo (BRI Frontage), Ms. Dina Goykhman (Merck), Dr. James Huleatt (Sanofi), Dr. Akiko Ishii-Watabe (Japan MHLW / ICH M10 EWG), Mr. Gregor Jordan (Roche), Dr. John Kamerud (Pfizer), Dr. Steve Keller (AbbVie), Dr. Lina Loo (Vertex), Mr. Fred McCush (Pfizer), Mr. Luis Mendez (Merck), Ms. Dulcyane Neiva Mendes Fernandes (Brazil ANVISA / ICH M10 EWG), Dr. Luying Pan (Takeda), Mr. Noah Post (Ionis), Dr. Mohsen Rajabi Abhari (US FDA), Dr. Yoshiro Saito (Japan MHLW / ICH M10 EWG), Dr. Daniel Spellman (Merck), Dr. Giane Sumner (Regeneron), Dr. Matthew Szapacs (Abbvie), Dr. Albert Torri (Regeneron), Dr. Montserrat Carrasco-Triguero (Sangamo), Dr. Elizabeth Verburg (Lilly), Dr. LaKenya Williams (BMS), Dr. Karl Walravens (GSK), Dr. Yongjun Xue (BMS)
All the workshop attendees and members of the Global Bioanalytical Community who have sent comments and suggestions to the workshop to complete this White Paper
Future Science Group as a trusted partner
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Savoie N , BoothBP, BradleyTet al. The 2nd Calibration and Validation Group Workshop on Recent Issues in Good Laboratory Practice Bioanalysis. Bioanalysis. (2009).
- Savoie N , GarofoloF, Van AmsterdamPet al. 2009 White Paper on Recent Issues in Regulated Bioanalysis from the 3rd Calibration and Validation Group Workshop. Bioanalysis.2(1), (2010).
- Savoie N , GarofoloF, Van AmsterdamPet al. 2010 White Paper on Recent Issues in Regulated Bioanalysis & Global Harmonization of Bioanalytical Guidance. Bioanalysis.2(12), (2010).
- Garofolo F , RocciML, DumontIet al. 2011 White Paper on Recent Issues in Bioanalysis and Regulatory Findings from Audits and Inspections. Bioanalysis. (2011).
- Desilva B , GarofoloF, RocciMet al. 2012 White Paper on Recent Issues in Bioanalysis and Alignment of Multiple Guidelines. Bioanalysis.4(18), (2012).
- Stevenson L , GarofoloF, DesilvaBet al. 2013 White Paper on Recent Issues in Bioanalysis: “Hybrid” – The Best of LBA and LCMS. Bioanalysis.5(23), (2013).
- Stevenson L , AmaravadiL, MylerHet al. 2014 White Paper on Recent Issues in Bioanalysis: A Full Immersion in Bioanalysis (Part 3 – LBA and Immunogenicity). Bioanalysis.6(24), (2014).
- Fluhler E , HayesR, GarofoloFet al. 2014 White Paper on Recent Issues in Bioanalysis: A Full Immersion in Bioanalysis (Part 1 – Small Molecules by LCMS). Bioanalysis. (2014).
- Dufield D , NeubertH, GarofoloFet al. 2014 White Paper on Recent Issues in Bioanalysis: A Full Immersion in Bioanalysis (Part 2 – Hybrid LBA/LCMS, ELN & Regulatory Agencies' Input). Bioanalysis. (2014).
- Amaravadi L , SongA, MylerHet al. 2015 White Paper on Recent Issues in Bioanalysis: Focus on New Technologies and Biomarkers (Part 3-LBA, Biomarkers and Immunogenicity). Bioanalysis. (2015).
- Welink J , FluhlerE, HughesNet al. 2015 White Paper on Recent Issues in Bioanalysis: Focus on New Technologies and Biomarkers (Part 1 – Small Molecules by LCMS). Bioanalysis.7(22), (2015).
- Ackermann B , NeubertH, HughesNet al. 2015 White Paper on Recent Issues in Bioanalysis: Focus on New Technologies and Biomarkers (Part 2 – Hybrid LBA/LCMS and Input from Regulatory Agencies). Bioanalysis.7(23), (2015).
- Richards S , AmaravadiL, PillutlaRet al. 2016 White Paper on Recent Issues in Bioanalysis: Focus on Biomarker Assay Validation (BAV): (Part 3 – LBA, Biomarkers and Immunogenicity). Bioanalysis.8(23), (2016).
- Song A , LeeA, GarofoloFet al. 2016 White Paper on Recent Issues in Bioanalysis: Focus on Biomarker Assay Validation (BAV): (Part 2 – Hybrid LBA/LCMS and Input from Regulatory Agencies). Bioanalysis.8(23), (2016).
- Yang E , WelinkJ, CapeSet al. 2016 White Paper on Recent Issues in Bioanalysis: Focus on Biomarker Assay Validation (BAV) (Part 1 – Small Molecules, Peptides and Small Molecule Biomarkers by LCMS). Bioanalysis.8(22), (2016).
- Welink J , YangE, HughesNet al. 2017 White Paper on Recent Issues in Bioanalysis: Aren't BMV Guidance/Guidelines “Scientific”? (Part 1 – LCMS: Small Molecules, Peptides and Small Molecule Biomarkers). Bioanalysis.9(22), (2017).
- Gupta S , RichardsS, AmaravadiLet al. 2017 White Paper on Recent Issues in Bioanalysis: A Global Perspective on Immunogenicity Guidelines & Biomarker Assay Performance (Part 3 – LBA: Immunogenicity, Biomarkers and PK Assays). Bioanalysis. (2017).
- Neubert H , SongA, LeeAet al. 2017 White paper on Recent Issues in Bioanalysis: Rise of Hybrid LBA/LCMS Immunogenicity Assays (part 2: Hybrid LBA/LCMS Biotherapeutics, Biomarkers & Immunogenicity Assays and Regulatory Agencies' Inputs). Bioanalysis.9(23), (2017).
- Stevenson L , RichardsS, PillutlaRet al. 2018 White Paper on Recent Issues in Bioanalysis: Focus on Flow Cytometry, Gene Therapy, Cut Points and Key Clarifications on BAV (Part 3 – LBA/Cell-Based Assays: Immunogenicity, Biomarkers and PK Assays). Bioanalysis.10(24), 1973–2001 (2018).
- Welink J , XuY, YangEet al. 2018 White Paper on Recent Issues in Bioanalysis: “A Global Bioanalytical Community Perspective on Last Decade of Incurred Samples Reanalysis (ISR)” (Part 1 – Small Molecule Regulated Bioanalysis, Small Molecule Biomarkers, Peptides & Oligonucleotide Bioanalysis). Bioanalysis.10(22), (2018).
- Neubert H , OlahT, LeeAet al. 2018 White Paper on Recent Issues in Bioanalysis: Focus on Immunogenicity Assays by Hybrid LBA/LCMS and Regulatory Feedback Part 2 - PK, PD & ADA Assays by Hybrid LBA/LCMS & Regulatory Agencies' Inputs on Bioanalysis, Biomarkers and Immunogenicity. Bioanalysis. (2018).
- Garofolo W , SavoieN. The Decennial Index of the White Papers in Bioanalysis: ‘A Decade of Recommendations (2007–2016).’Bioanalysis. [ Internet]. 9(21), 1681–1704 (2017). Available from: 10.4155/bio-2017-4979.
- Booth B , StevensonL, PillutlaRet al. 2019 White Paper on Recent Issues in Bioanalysis: FDA BMV Guidance, ICH M10 BMV Guideline and Regulatory Inputs (Part 2-Recommendations on 2018 FDA BMV Guidance, 2019 ICH M10 BMV Draft Guideline and Regulatory Agencies' Input on Bioanalysis, Biomarkers and Immunogenicity). Bioanalysis. (2019).
- Piccoli S , MehtaD, VitalitiAet al. 2019 White Paper on Recent Issues in Bioanalysis: FDA Immunogenicity Guidance, Gene Therapy, Critical Reagents, Biomarkers and Flow Cytometry Validation (Part 3-Recommendations on 2019 FDA Immunogenicity Guidance, Gene Therapy Bioanalytical Challenges, Strategies for Critical Reagent Management, Biomarker Assay Validation, Flow Cytometry Validation & CLSI H62). Bioanalysis.Future Medicine Ltd., 2207–2244 (2019).
- Fandozzi C , EvansC, WilsonAet al. 2019 White Paper on Recent Issues in Bioanalysis: Chromatographic Assays (part 1 – Innovation in Small Molecules and Oligonucleotides & Mass Spectrometric Method Development Strategies for Large Molecule Bioanalysis). Bioanalysis. (2019).
- Neubert H , AlleySC, LeeAet al. 2020 White Paper on Recent Issues in Bioanalysis: BMV of Hybrid Assays, Acoustic MS, HRMS, Data Integrity, Endogenous Compounds, Microsampling and Microbiome (Part 1 – Recommendations on Industry/Regulators Consensus on BMV of Biotherapeutics by LCMS, Advanced Application in Hybrid Assays, Regulatory Challenges in Mass Spec, Innovation in Small Molecules, Peptides and Oligos). Bioanalysis.13(4), (2021).
- Corsaro B , YangT, MurphyRet al. 2020 White Paper on Recent Issues in Bioanalysis: Vaccine Assay Validation, qPCR Assay Validation, QC for CAR-T Flow Cytometry, NAb Assay Harmonization and ELISpot Validation (Part 3 – Recommendations on Immunogenicity Assay Strategies, NAb Assays, Biosimilars and FDA/EMA Immunogenicity Guidance/Guideline, Gene & Cell Therapy and Vaccine Assays). Bioanalysis.13(6), 415–463 (2021).
- Spitz S , ZhangY, FischerSet al. 2020 White Paper on Recent Issues in Bioanalysis: BAV Guidance, CLSI H62, Biotherapeutics Stability, Parallelism Testing, CyTOF and Regulatory Feedback (Part 2A – Recommendations on Biotherapeutics Stability, PK LBA Regulated Bioanalysis, Biomarkers Assays, Cytometry Validation & Innovation Part 2B - Regulatory Agencies' Inputs on Bioanalysis, Biomarkers, Immunogenicity, Gene & Cell Therapy and Vaccine). Bioanalysis.13(5), (2021).
- Hersey S , KellerS, MathewsJet al. 2021 White Paper on Recent Issues in Bioanalysis: ISR for Biomarkers, Liquid Biopsies, Spectral Cytometry, Inhalation/Oral & Multispecific Biotherapeutics, Accuracy/LLOQ for Flow Cytometry (Part 2 – Recommendations on Biomarkers/CDx Assays Development & Validation, Cytometry Validation & Innovation, Biotherapeutics PK LBA Regulated Bioanalysis, Critical Reagents & Positive Controls Generation). Bioanalysis.14(10), 627–692 (2022).
- Loo L , HarrisS, MiltonMet al. 2021 White Paper on Recent Issues in Bioanalysis: TAb/NAb, Viral Vector CDx, Shedding Assays; CRISPR/Cas9 & CAR-T Immunogenicity; PCR & Vaccine Assay Performance; ADA Assay Comparability & Cut Point Appropriateness (Part 3 – Recommendations on Gene Therapy, Cell Therapy, Vaccine Assays; Immunogenicity of Biotherapeutics and Novel Modalities; Integrated Summary of Immunogenicity Harmonization). Bioanalysis.14(11), 737–793 (2022).
- Kaur S , AlleySC, SzapacsMet al. 2021 White Paper on Recent Issues in Bioanalysis: Mass Spec of Proteins, Extracellular Vesicles, CRISPR, Chiral Assays, Oligos; Nanomedicines Bioanalysis; ICH M10 Section 7.1; Non-Liquid & Rare Matrices; Regulatory Inputs (Part 1A – Recommendations on Endogenous Compounds, Small Molecules, Complex Methods, Regulated Mass Spec of Large Molecules, Small Molecule, PoC & Part 1B – Regulatory Agencies' Inputs on Bioanalysis, Biomarkers, Immunogenicity, Gene & C. Bioanalysis.14(9), 505–580 (2022).
- Suk JS , XuQ, KimN, HanesJ, EnsignLM. PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv Drug Deliv Rev.99 (2016).
- U.S. Department of Health and Human Services Food and Drug Administration Center for Biologics Evaluation and Research . Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products Draft Guidance for Industry. https://www.fda.gov/media/156896/download (2022).
- Gorovits B , KorenE. Immunogenicity of Chimeric Antigen Receptor T-Cell Therapeutics. BioDrugs.33(3), (2019).
- Potthoff B , McBlaneF, SpindeldreherS, SickertD. A Cell-Based Immunogenicity Assay to Detect Antibodies Against Chimeric Antigen Receptor Expressed by Tisagenlecleucel. J Immunol Methods.476 (2020).
- Jawa V , JoubertMK, ZhangQet al. Evaluating Immunogenicity Risk Due to Host Cell Protein Impurities in Antibody-Based Biotherapeutics. AAPS Journal.18(6), (2016).
- Joubert MK , HokomM, EakinCet al. Highly Aggregated Antibody Therapeutics can Enhance the in vitro Innate and Late-Stage T-cell Immune Responses. Journal of Biological Chemistry.287(30), (2012).
- U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) . Guidance for Industry Immunogenicity Assessment for Therapeutic Protein Products. https://www.fda.gov/media/85017/download (2014).
- U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) . Immunogenicity Information in Human Prescription Therapeutic Protein and Select Drug Product Labeling – Content and Format Guidance for Industry. https://www.fda.gov/media/155871/download (2022).
- U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) . Immunogenicity Testing of Therapeutic Protein Products — Developing and Validating Assays for Anti-Drug Antibody Detection Guidance for Industry. https://www.fda.gov/media/119788/download (2019).
- U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) . Clinical Pharmacology Considerations for the Development of Oligonucleotide Therapeutics Guidance for Industry. https://www.fda.gov/media/159414/download.
- Rogers H , AdeniyiO, RamamoorthyA, BaileyS, PacanowskiM. Clinical Pharmacology Studies Supporting Oligonucleotide Therapy Development: An Assessment of Therapies Approved and in Development Between 2012 and 2018. Clin Transl Sci.14(2), (2021).
- Bano N , EhlingerC, YangT-Y, SwansonM, AllenS. Considerations in the Immunogenicity Assessment Strategy for Oligonucleotide Therapeutics (ONTs). AAPS Journal.24(5), (2022).
- Henry SP , ArfvidssonC, ArringtonJet al. Assessment of the Immunogenicity Potential for Oligonucleotide-Based Drugs. Nucleic Acid Ther.32(5), (2022).
- Yu RZ , WangY, NorrisDAet al. Immunogenicity Assessment of Inotersen, a 2-O-(2-Methoxyethyl) Antisense Oligonucleotide in Animals and Humans: Effect on Pharmacokinetics, Pharmacodynamics, and Safety. Nucleic Acid Ther.30(5), (2020).
- Piccoli S , MehtaD, VitalitiAet al. 2019 White Paper on Recent Issues in Bioanalysis: FDA Immunogenicity Guidance, Gene Therapy, Critical Reagents, Biomarkers and Flow Cytometry Validation (Part 3-Recommendations on 2019 FDA Immunogenicity Guidance, Gene Therapy Bioanalytical Challenges, St. Bioanalysis. (2019).
- INTERNATIONAL COUNCIL FOR HARMONISATION OF TECHNICAL REQUIREMENTS FOR PHARMACEUTICALS FOR HUMAN USE . Bioanalytical Method Validation and Study Sample Analysis M10. https://database.ich.org/sites/default/files/M10_Guideline_Step4_2022_0524.pdf (2022).
- Validation BM . Guidance for Industry Bioanalytical Method Validation Guidance for Industry Bioanalytical Method Validation. Fda [ Internet]. (May), 1–22 (2018). Available from: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
- Giannoukos G , CiullaDM, MarcoEet al. UDiTaS™, A Genome Editing Detection Method for Indels and Genome Rearrangements. BMC Genomics.19(1), (2018).
- Tsai SQ , ZhengZ, NguyenNTet al. GUIDE-seq Enables Genome-Wide Profiling of Off-Target Cleavage by CRISPR-Cas Nucleases. Nat Biotechnol.33(2), 187–197 (2015).
- Cameron P , FullerCK, DonohouePDet al. Mapping the Genomic Landscape of CRISPR–Cas9 Cleavage. Nat Methods.14(6), 600–606 (2017).
- Bae S , ParkJ, KimJS. Cas-OFFinder: A Fast and Versatile Algorithm That Searches for Potential Off-Target Sites of Cas9 RNA-Guided Endonucleases. Bioinformatics.30(10), (2014).
- Chaudhari HG , PentermanJ, WhittonHJet al. Evaluation of Homology-Independent CRISPR-Cas9 Off-Target Assessment Methods. CRISPR Journal.3(6), (2020).
- Zhang Y , BlomquistTM, KuskoRet al. Deep Oncopanel Sequencing Reveals Within Block Position-Dependent Quality Degradation in FFPE Processed Samples. Genome Biol.23(1), (2022).
- Liu Z , RobertsR, MercerTR, XuJ, SedlazeckFJ, TongW. Towards Accurate and Reliable Resolution of Structural Variants for Clinical Diagnosis. Genome Biol.23(1), (2022).
- Liu Z , RobertsR, MercerTR, XuJ, SedlazeckFJ, TongW. Author Correction: Towards Accurate and Reliable Resolution of Structural Variants for Clinical Diagnosis (Genome Biology, (2022), 23, 1, (68), 10.1186/s13059-022-02636-8). Genome Biol23(1), (2022).
- Mercer TR , XuJ, MasonCE, TongW. The Sequencing Quality Control 2 Study: Establishing Community Standards for Sequencing in Precision Medicine. Genome Biol.22(1), (2021).
- Sequencing Benchmarked. Nat Biotechnol.39(9), (2021).
- Lawler SE , SperanzaMC, ChoCF, ChioccaEA. Oncolytic Viruses in Cancer Treatment a Review. JAMA Oncol.3(6), (2017).
- Santos Apolonio J , Limade Souza Gonçalves V, CordeiroSantos MLet al. Oncolytic Virus Therapy in Cancer: A Current Review. World J Virol.10(5), (2021).
- Russell SJ , PengKW, BellJC. Oncolytic Virotherapy. Nat Biotechnol.30(7), (2012).
- U.S. Department of Health and Human Services Food and Drug Administration Center for Biologics Evaluation and Research . Design and Analysis of Shedding Studies for Virus or Bacteria-Based Gene Therapy and Oncolytic Products Guidance for Industry. https://www.fda.gov/files/vaccines%2C%20blood%20%26%20biologics/published/Design-and-Analysis-of-Shedding-Studies-for-Virus-or-Bacteria-Based-Gene-Therapy-and-Oncolytic-Products--Guidance-for-Industry.pdf (2015).
- Wissel M , PoirierM, SatterwhiteCet al. Recommendations on qPCR/ddPCR Assay Validation by GCC. Bioanalysis.14(12), 853–863 (2022).
- Sugimoto H , ChenS, MinembeJPet al. Insights on Droplet Digital PCR–Based Cellular Kinetics and Biodistribution Assay Support for CAR-T Cell Therapy. AAPS Journal.23(2), (2021).
- Yamamoto S , MatsumotoS ichi, GotoAet al. Quantitative PCR Methodology with a Volume-Based Unit for the Sophisticated Cellular Kinetic Evaluation of Chimeric Antigen Receptor T Cells. Sci Rep.10(1), (2020).
- Wu B , SternheimN, AgarwalPet al. Evaluation of Atezolizumab Immunogenicity: Clinical Pharmacology (part 1). Clin Transl Sci.15(1), 130–140 (2022).
- Guinn D , MadabushiR, WangYM, ZinehI, MaxfieldK. Elucidating the Impact of Immunogenicity Assessment Postapproval: A Targeted Analysis of Immunogenicity Postmarketing Requirements and Commitments. Clin Pharmacol Ther.109(3), (2021).
- Rossert J , CasadevallN, EckardtK-U. Anti-Erythropoietin Antibodies and Pure Red Cell Aplasia. Journal of the American Society of Nephrology.15(2), 398–406 (2004).
- Bartelds GM . Development of Antidrug Antibodies Against Adalimumab and Association With Disease Activity and Treatment Failure During Long-term Follow-up. JAMA.305(14), 1460 (2011).
- Swanson S . Immunogenicity of Therapeutic Proteins. Translational Medicine Optimizing Preclinical Safety Evaluation of Biopharmaceuticals.CavagnaroJ, CosenzaM ( Eds). CRC Press (2021).
- Garcês S , DemengeotJ, Benito-GarciaE. The Immunogenicity of Anti-TNF Therapy in Immune-Mediated Inflammatory Diseases: A Systematic Review of the Literature with a Meta-Analysis. Ann Rheum Dis.72(12), 1947–1955 (2013).
- Garay RP , El-GewelyR, ArmstrongJK, GarrattyG, RichetteP. Antibodies Against Polyethylene Glycol in Healthy Subjects and in Patients Treated with PEG-Conjugated Agents. Expert Opin Drug Deliv.9(11), 1319–1323 (2012).
- Yang Q , LaiSK. Anti-PEG immunity: Emergence, Characteristics, and Unaddressed Questions. Wiley Interdiscip Rev Nanomed Nanobiotechnol.7(5), 655–677 (2015).
- Larimore K , NguyenT, BadilloBet al. Depletion of Interfering IgG and IgM is Critical to Determine the Role of IgE in Pegvaliase-Associated Hypersensitivity. J Immunol Methods.468, 20–28 (2019).
- Verhoef JJF , CarpenterJF, AnchordoquyTJ, SchellekensH. Potential Induction of anti-PEG Antibodies and Complement Activation Toward PEGylated Therapeutics. Drug Discov Today.19(12), 1945–1952 (2014).
- Joyce A , SheaC, YouZ, GorovitsB, LepsyC. Determination of Anti-drug Antibody Affinity in Clinical Study Samples Provides a Tool for Evaluation of Immune Response Maturation. AAPS Journal.24(6), (2022).
- Stevens FJ , BobrovnikSA. Deconvolution of Antibody Affinities and Concentrations by Non-Linear Regression Analysis of Competitive ELISA Data. J Immunol Methods.328(1–2), 53–58 (2007).
- Karle AC , WrobelMB, KoepkeSet al. Anti-Brolucizumab Immune Response as One Prerequisite for Rare Retinal Vasculitis/Retinal Vascular Occlusion Adverse Events. Sci Transl Med.15(681), (2023).
- Kearns JD , WassmannP, OlgacUet al. A Root Cause Analysis to Identify the Mechanistic Drivers of Immunogenicity Against the Anti-VEGF Biotherapeutic Brolucizumab. Sci Transl Med.15(681), (2023).
- Yasuno K , Hamamura-YasunoE, NishimiyaDet al. Host Cell Proteins Induce Inflammation and Immunogenicity as Adjuvants in an Integrated Analysis of in vivo and in vitro Assay Systems. J Pharmacol Toxicol Methods.103 (2020).
- Carpenter JF , RandolphTW, JiskootWet al. Overlooking Subvisible Particles in Therapeutic Protein Products: Gaps That May Compromise Product Quality. J Pharm Sci.98(4), (2009).
- Gjølberg TT , LodeHE, MeloGBet al. A Silicone Oil-Free Syringe Tailored for Intravitreal Injection of Biologics. Frontiers in Ophthalmology.2 (2022).
- Clinical and Laboratory Standards Institute . H62 Validation of Assays Performed by Flow Cytometry, 1st Edition. https://clsi.org/standards/products/hematology/documents/h62/ (2021).
- Ducret A , AckaertC, BessaJet al. Assay Format Diversity in Pre-Clinical Immunogenicity Risk Assessment: Toward a Possible Harmonization of Antigenicity Assays. MAbs.14(1), (2022).
- Morcos PN , LiJ, HosseiniI, LiC. Quantitative Clinical Pharmacology of T-Cell Engaging Bispecifics: Current Perspectives and Opportunities. Clin Transl Sci.14(1), 75–85 (2021).
- INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE . ICH Harmonised Tripartite Guideline Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals S6(R1). https://database.ich.org/sites/default/files/S6_R1_Guideline_0.pdf (2011).