While technologies for determining the potency and structural identity of macromolecules have developed significantly over recent years, the relationship between potency and structure is still an issue. Ligand-binding assays (LBA) may respond equally to related molecules that have the same epitope, but differ in their activity/potency. This is especially important in human following dosing, where the metabolites may significantly contribute to biological activity, but may (or may not) be quantified by the analytical technique. The ICH S6 (R1) guidance suggests that the consequential metabolism of proteins and peptides is via expected routes and implies only a little more investigation may be needed Citation[1]; however, a pragmatic approach to understanding their metabolism – especially modified proteins – in relation to their pharmacokinetics/pharmacodynamics is essential, while not turning the development program into a major research program.
Scope & definition
The objective of this article is to raise discussion as to how far we can go pragmatically in defining the selectivity of LBAs with current technologies, and how far should we go if technology was not limiting? To further extend the debate, how do we weave potency/activity (pharmacodynamics) issues into the discussion? This is not intended as a definitive review, but is designed to raise issues that might be developed in a future special issue dedicated to this topic.
What are biologics? One of the many alternative names, which we shall use here, is ‘biotherapeutics’. This encompasses any biologically active macromolecule, ranging from a recombinant form of an endogenous biochemical (e.g., recombinant human erythropoietin) to macromolecules, which may have been chemically modified, usually to increase their potency and/or half-life (e.g., fusion proteins). This still uses recombinant technologies, but produces an entity consisting of two or more different proteins (e.g., albiglutide, which is a fusion of human GLP-1 to human albumin). At the other extreme are pegylated products, where PEG is covalently attached to the protein of interest (e.g., interferon [PEGASYS®] and GCSF [Neulasta®]) in order to modify its half-life, activity and/or immunogenic response.
Other biotherapeutics, such as monoclonal antibodies, excluding vaccines, form probably the biggest therapeutic grouping (e.g., Avastin®, Herceptin®, Humira, Remicade® and Rituxan®), which are, for the most part, metabolically stable and typically have similar half-lives of approximately 20 days, are not part of this discussion. However, commonly held precepts are there to be questioned, and the work of Huang et al.Citation[2] and Chen et al.Citation[3] suggests that in vivo modifications (e.g., deamidation and varied glycosylation patterns) may contribute to significant changes in stability/activity.
For the sake of simplicity, this article will discuss the issues surrounding the specificity of assays used to quantify recombinant proteins and modified naturally occurring proteins in biological matrices, mainly plasma or serum (as opposed to peptides, which may be quantified intact by chromatographic techniques).
Protein breakdown products may be a consequence of cleavage or modification of individual amino acids, or modification of post-translational changes. These breakdown products have been variously designated as ‘metabolites‘, ‘catabolites’ or ‘degradants’. In this paper, whatever their fate, they will be termed ‘metabolites’.
Selectivity
Characterizing biotherapeutics is a complex process; heterogeneity brought about by the ‘manufacturing’ process is an added complication. For this reason the term ‘well-characterized biologicals’ used by the US FDA’s Center for Biologics Evaluation and Research (CBER) was coined in the mid-1990s. Although still widely used, it has been superseded by the term ‘specified biologicals‘; this reflects the increased ability to characterize these molecules in more detail due to improvements in instrument technologies. Once the molecule has been specified, it is then possible to develop a specific assay for that molecule; however, even this terminology presents a problem. The United States Pharmacopeia definition of specificity refers to the extent to which a method can determine a particular analyte in mixtures or matrices without interferences from other components: specificity is the ultimate aspect of selectivity. The usage of ‘specificity’ suggests that no component other than the analyte contributes to the result. Hardly any method is that specific and, in general, the term should be avoided.
We can only tend to perfection! Indeed, in 1999 Selby, writing about the selectivity of LBAs in measuring endogenous analytes, asked “How do you know what you are measuring?” Citation[4], a question he discusses in some detail and with great equivocation. Lee compared the differences in validating analytical methods for the measurement of biotherapeutics and biomarkers using ligand-based assays Citation[5]. While there are many differences between these two groups, there is much to be learnt from interactions at the analytical level, especially the interaction between the target ligand, the endogenous analyte and the biotherapeutic.
How much do you understand about the selectivity of the antibody? What are the endogenous analytes that may bind to it? How is the assumed analyte metabolized/degraded and, last but not least, how does activity/potency (or perhaps more appropriately in the pharmacokinetic context, pharmacodynamics) relate to molecular integrity? In recent years there has been a plethora of publications related to the issue of better identifying/characterizing the active moiety. Most recently, Ezan et al. have reviewed the use of LC–MS to accurately quantify therapeutic proteins and antibodies Citation[6].
“International Biological Standards developed by WHO are the world’s primary calibrants against which biological medicines and candidate biological medicines are assayed. International Biological Standards enable the potency of different manufacturers’ products to be directly comparable in terms of potency,” said Peter Phillips (National Institute for Biological Standards and Control [NIBSC]) in 1997 Citation[7]. While these comments are directed at the raw material/primary standard, the relationship between pharmacokinetics, metabolism and pharmacodynamics emphasizes this issue.
How can we correlate a selective LBA with the in vivo activity of the analyte? What are the factors influencing selectivity? Both De Silva Citation[8] and Lee and Ma Citation[9] have discussed the role of possible interferants, such as matrix effects, which can be a function of known and unknown diseased states, and binding proteins, both endogenous (pre-dosing), which can cross-react with the assay, and neutralizing antibodies formed post-dosing. While it may be possible to minimize these effects through dilution, or in some cases via some form of sample clean-up, the major issue of cross-reactivity with catabolites is how they relate to potency. While historically it was assumed that they would have little inherent biological activity, this has not always been found to be the case. This complex relationship between structure and activity (potency) is exemplified by erythropoietin and related molecules, as discussed by Yuen et al. in relation to the production process where differences in charge, variable sialic acid content and number of N-linked sialic acid chains and total sialic acid content are observed Citation[10]. The greater the sialic acid content, the greater the biological activity and half-life, but the lower receptor binding. A further confounding factor was that the in vivo biological activity appears to be the inverse of in vitro activity, based on the differing N-glycan content of the molecule. These findings are for pure materials; it can be much more complex for circulating biotherapeutics where metabolism/degradation (catabolism) takes place. Egrie et al.Citation[11] and Catlin et al.Citation[12] have considered this in more detail using biological fluids and similar methodologies, and they confirm the complexity of deconvoluting these issues. These issues are particularly important during the discovery/screening stage, where an understanding of the relationship between potency and pharmacokinetics of the drug and/or metabolite is pivotal in lead candidate selection.
Metabolism
For small molecules, identifying metabolites was a major driver of the drug development process where metabolites could be more or less toxic and/or potent than the parent and, therefore, prime candidates for a drug in their own right. So, how relevant is metabolism to biotherapeutics?
This analogy for macromolecules, if carried to its ultimate conclusion, could result in the elucidation of a wide variety of metabolites (more accurately ‘catabolites‘, as they are usually breakdown products) and result in considerable delays to the development process. Indeed, the focus of research should be on the activity of the circulating moieties. As long ago as 1992, Moore and Wroblewski expressed some thoughts on metabolism, which are probably more analogous to the developmental approach for small molecules Citation[13]: “A better understanding of protein degradation will likely lead to the design of modified molecules or appropriate adjuncts for protein ‘hormone’ therapy… If toxicological findings are insignificant, metabolism studies will not be critical to the development of protein drugs”. This is an important consideration when deciding the extent to which the fate of these molecules should be pursued. However, the situation is further complicated by their question “what is the significance of metabolism at the preclinical stage to metabolism in humans”. They continue: “Findings of metabolism can be relevant to the pharmacological activity of the drug … therefore application of protein metabolism to the discovery process should be an exciting and interesting approach for the future”. Indeed, since that time, technologies have developed to such an extent that detailed characterization develops daily. But should we pursue identification and characterization of metabolites just because we can, while their in vivo biological activity (potency) may be the more important parameter?
More recently, Hall et al. have discussed issues of metabolites and the limitations of LBA for developing pharmocokinetic profiles Citation[14]. The impact of these thoughts are illustrated by some examples. Atrial naturetic factor is a 28-amino acid residue peptide, synthesized in the heart from a 126-amino acid precursor, and is the 99–126-amino acid residue of the precursor. It is inactivated by endopeptidases and no biologically active metabolites are known to exist Citation[9]. However, the truncation of some proteins can influence their potency; for example, leukotactin-1, a novel chemokine involved in atherogenesis, is degraded by serial deletion of amino acids at the N-terminus, with 20-amino acid deletion there is no change in activity, but when the 24-amino acid is deleted, activity is increased 100-fold Citation[15]. Furthermore, while deletion of 27- and 28-amino acids had no further effect, all agonistic activity is lost on deletion of 29-amino acids. While these are endogenous molecules and the in vivo half-lives of the metabolites are difficult to ascertain, they do send out warnings of how small differences may influence activity. This is especially true for fusion proteins and PEGylated proteins, where considerable changes in one part of the molecule may or may not influence the activity of the drug, but could influence the half-life while still being quantified by the analytical technique of choice. How much effort should be put into tracking down ‘metabolites‘?
Potency
While it is often assumed the activity of the assay lies with the intact product, this is not always the case. Nevertheless, for the raw product, potency is a pivotal parameter. Venkat Mukku of the United States Pharmacopeia believes, in terms of the ‘pure test compound‘, the potency assay is the single most important quality attribute of a biological product Citation[16]. In his 2009 presentation he also discussed the relevance of potency assays based on whole animal through to antigen–antibody binding in terms of their relevance to the classical analysis and suitability for lot release. Thus, whole animal assays may have greater clinical significance than for lot release, although it is now more common to use cell-based potency assays rather than whole animal.
Characterizing biotherapeutics
Most biological molecules are defined by international units as developed by WHO, which are related to the biological activity (i.e., potency). How does this relate to SI traceability, the gold standard for analytical standards? The number of classical chemistry techniques used to characterize these molecules are multifarious, with a range of chromatographic and MS techniques (either separately or hyphenated) for the determination of primary structure. Determination of secondary and tertiary structures is more complex, even in the pure form.
There are a multitude of organizations working on this issue, including but not limited to: WHO, NIBSC, Consultative Committee for Amount of Substance–Metrology in Chemistry (CCQM), Working Group on Bioanalysis (BAWG), International Federation of Clinical Chemistry and Laboratory Medicine (IFCC), Laboratory of the Government Chemist (LGC), National Institute of Standards and Technology (NIST), and last but not least, the European Directorate for the Quality Medicines and Healthcare – European Union Biological Standardization Programme. These efforts cover a wide variety of scenarios from biotherapeutics as raw materials through to biomarkers and biotherapeutics in biological fluids.
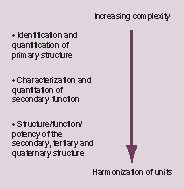
The BAWG section of the CCQM has established a calibration and measurement capability-driven route map with the ultimate objective of harmonizing the SI traceable component with the molecular changes that impact on potency. This has been fully discussed by Gray in a 2007 presentation Citation[17] and is summarized in .
How relevant are these relationships to the biotherapeutic in biological fluid? The development of some form of multivariate structure–activity index that can be correlated with activity/potency may provide a solution to the problem of relating potency to activity. This might be achieved through the use of chip-based technologies, similar to those proposed by Ekins Citation[18]. This could take into account metabolites/degradants, although the deconvolution could be complex unless there is a 1:1 structure–activity relationship between the metabolite degradant and parent. Nevertheless, the use of multivariate indices is gaining acceptance, and if coupled with some orthogonal analytical technologies it could significantly enhance the selectivity of the conclusions.
A small step in this direction has taken place with the development of the Optim™ 100, which uses multi-analysis techniques that can compare information on closely related compounds (albeit for pure compounds at this stage). If an ex vivo device could be developed (likely chip-based), it may be possible to tie together multiple structural and potency components into one assay. While this may be a dream at this stage, technology is advancing such that it may become a reality. In the meantime, we must develop a pragmatic approach to fully elucidating in vivo degradants, the specified test article and potency, so life-saving drugs can get to market quickly and safely. The use of immunocapture offers one approach, as detailed in the paper by Kumar et al. with respect to parathyroid hormone Citation[19]. Although the addition of a nonspecific immunological reagent to an assay might be deemed to be counterintuitive, this is not borne out by the data, and in fact seems to be complementary in improving selectivity. In most cases, these molecules are not measured intact, but following digestion prior to LC–MS; therefore, the digestion process must not add any artifacts and should be evaluated accordingly. This can be achieved by ensuring our analytical techniques are fit for purpose and not implemented purely because the technology is available.
Conclusion
The conundrum lies in the fact that no one technique can satisfactorily characterize the active moiety with respect to both its activity/potency and its physicochemical structure. While use of post-hydrolysis chromatographic technologies have been used to characterize one or more signature peptides specific to the protein therapeutic, it provides no information regarding its tertiary structure and concomitant activity. How does it relate to truncated or metabolized products? How does the activity of what may be a myriad of products circulating in the body relate to plasma concentration of the biotherapeutic, whether it be determined by ligand-based or chromatographic assays (although it should be recognized that the latter approach is still lacking in sensitivity for high-potency biotherapeutics)? It remains to be determined which strategy is best-suited to measuring these complex molecules in the body using current technologies, and what the gold standard should be in future. At present, many scientists involved in biotherapeutics use both quality-control and biofluid assays; hopefully this will continue to develop synergies, unlike in small-molecule development where divergences have developed into rigid philosophical and technological silos.
This article is not intended to provide answers, but hopefully it will stimulate discussion through the pages of Bioanalysis as to where we go next from both a theoretical and practical perspective.
Financial & competing interests disclosure
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Bibliography
- Addendum to ICH S6, Preclinical Safety Evaluation of Biotechnology: Preclinical Safety Evaluation of Biotechnology Products, Derived Pharmaceutics . S6(R1) (2009).
- Huang L , LuJ, WroblewskiVJ, BealsJH, RigginRM. In vivo deamidation characterization of monoclonal antibodies by LC–MS/MS. Anal. Chem. 77, 1432–1439 (2005).
- Chen X , LiuYD, FlynnGC. The effect of Fe Glycan forms on human IgGZ antibody clearance in humans. Glycolobiology19(3), 240–249 (2009).
- Selby C . Interference in immunoassay. Ann. Clin. Biochem. 36, 704–721 (1999).
- Lee JW . Method validation and application of protein biomarkers; basic similarities and differences from biotherapeutics. Bioanalysis1(8), 1461–1474 (2009).
- Ezan E , DuboisM, BeckerF. Bioanalysis of recombinant proteins and antibodies by mass spectrometry. Analyst134(5), 825–834 (2009).
- Phillips P . The preparation of International Biological Standards. J. Anal. Chem. 360(3/4), 473–475 (1998).
- De Silva B , SmithW, WeinerRet al. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm. Res. 20(11), 1885–1900 (2003).
- Lee J , MaH. Specificity and selectivity evaluations of ligand binding assay of protein therapeutics against concomitant drugs and related endogenous protein. AAPS J. 9(2), E164–E170 (2007).
- Yuen C-T , StorringPL, TipladyRJet al. Relationships between the N-glycan structures and biological activities of recombinant human erythropoietins produced using different culture conditions and purification procedures. Br. J. Haematol. 121, 511–526 (2003).
- Egrie JC , BrowneJR. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol. Dial Transplant. 16, 3–13 (2001).
- Catlin DH , BreidbachA, ElliottS, GlaspyJ. Comparison of the isoelectric focussing pattern of darbepoetin α, recombinant human erythropoietin and endogenous erythropoietin from human plasma. Clin. Chem. 48, 2057–2059 (2002).
- Moore JA , WroblewskiVJ. Pharmacokinetics and Metabolism of Protein Hormones. In:Protein Pharmacokinetics and Metabolism. Ferraiolo BL, Mohler MA, Glo CA (Eds). Plenum Press (1992).
- Hall M , LeeJ, OrtizRet al. Ligand binding – mass spectrometric methods to investigate biotransformation of protein therapeutics and impact on ligand binding assays. Poster presented at: 2009 AAPS National Biotechnology Conference. Seattle, Washington, DC, USA, 21–24 June 2007.
- Lee JK , LeeEH, YunYPet al. Truncation of NH2 – terminal amino acid residues increases potency of leukotactin-1 on CC chemokine receptors 1 and 3. J. Biol. Chem. 277(17), 14757–14763 (2002).
- Mukku V . USP bioassay standards – general guidance and product specific assays, an ipdate. Presented at: IPC-USP 8th Annual Scientific Meeting. Hyderabad, India, 11–12 February 2009.
- Gray E . (NIBSC) Challenges for international standardization and traceability biologicals. Presented at:JCTLM Symposium 11th Asia Pacific Congress for Clinical Biochemistry. Beijing, China, 16 October 2007.
- Ekins RP . Ligand assays: from electrophoresis to miniaturized microarrays. Clin. Chem. 44, 2015–2030 (1998).
- Kumar V , BarnbridgeDR, Chen L-Set al. Quantification of serum 1–84 parathyroid hormone in patients with hyperparathyroidism by immunocapture, in situ digestion liquid chromatography–tandem mass spectrometry. Clin. Chem. 56, 306–313 (2010).