
Abstract
Plasma protein binding (PPB) is an important parameter for a drug’s efficacy and safety that needs to be investigated during each drug-development program. Even though regulatory guidance exists to study the extent of PPB before initiating clinical studies, there are no detailed instructions on how to perform and validate such studies. To explore how PPB studies involving bioanalysis are currently executed in the industry, the European Bioanalysis Forum (EBF) has conducted three surveys among their member companies: PPB studies in drug discovery (Part I); in vitro PPB studies in drug development (Part II); and in vivo PPB studies in drug development. This paper reflects the outcome of the three surveys, which, together with the team discussions, formed the basis of the EBF recommendation. The EBF recommends a tiered approach to the design of PPB studies and the bioanalysis of PPB samples: ‘PPB screening’ experiments in (early) drug discovery versus qualified/validated procedures in drug development.
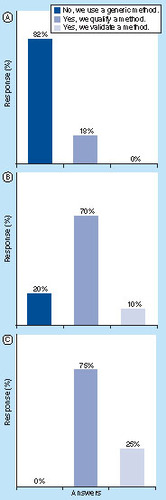
(A) Survey I: PPB in drug-discovery phase. (B) Survey II: in vitro PPB in drug-development phase. (C) Survey III: in vivo PPB in drug-development phase.
PPB: Plasma protein binding.
Drug molecules in animals and humans are either bound to proteins and lipids or are freely available. This article focuses on analysis of protein binding of small molecules in plasma Citation[1–3]. The extent of protein binding may impact the efficacy and toxicology of a drug as it is generally believed that the free drug concentration drives the therapeutic outcome Citation[4]. Interspecies differences in plasma protein binding (PPB) can lead to a reduced or increased drug-safety margin Citation[4]. Several regulatory authorities recommend the determination of PPB in animal and human plasma before commencing Phase I trials and to use it as supporting data set when evaluating drug–drug interactions Citation[5–7]. Therefore, PPB is considered to be an important parameter throughout an ongoing drug-development program.
In drug discovery, the free fraction obtained from in vitro PPB experiments is frequently used in combination with intrinsic clearance to guide structural design and prioritize compounds for downstream in vivo experiments. So far, there is no clear guidance on how to conduct these studies and how to interpret the results. Still, it is generally recognized that the protein binding of a drug has a major influence on PK parameters, such as volume of distribution and clearance Citation[8]. High protein binding is usually associated with high lipophilicity and low free-drug concentration in the systemic circulation Citation[9]. A correct implementation of PPB and interpretation of respective data is therefore critical and essential for further drug optimization.
Disease states such as renal Citation[10] or hepatic Citation[11] impairment, or pregnancy can also alter protein binding and, consequently, the therapeutic efficacy of a drug Citation[12]. Protein binding can play a major role in drug–drug interactions when different drugs target the same enzyme/receptor Citation[13–15]. Compared with PPB of the drug, the protein binding of specific metabolites can change significantly and, as a result, contribute to efficacy and/or adverse side effects Citation[16,17].
There are a number of commonly used methods to separate the bound and unbound fraction in plasma samples: these include ultrafiltration, ultracentrifugation and equilibrium dialysis Citation[18–20]. Equilibrium dialysis is the most commonly used method. However, if a drug or a metabolite is not stable in plasma for 3–5 h, this technique cannot be used and ultrafiltration might be a better alternative. Another critical aspect in performing PPB experiments is adsorption. Adsorption at membranes and on the surfaces of devices might significantly impact the measurement. Therefore, the mass balance in the PPB experiments should be evaluated carefully. Ultracentrifugation requires long centrifugation times. This technique is rather expensive and is not commonly used.
Although many papers have been published regarding PPB studies, including the techniques to separate the protein-bound drug from the unbound drug, information regarding bioanalysis of radiolabeled as well as nonlabeled compounds in PPB samples is very limited. PPB samples containing nonlabeled compounds are often analyzed using LC in combination with mass spectrometric detection (LC–MS/MS) Citation[21] or, alternatively, with radiolabeled compounds in combination with liquid scintillation counting/combustion and HPLC purity profiles. Moreover, there is no detailed guidance about the level of bioanalytical method validation for samples originating from PPB studies in drug discovery or drug development and laboratories often use a variety of methods ranging from basic screening to qualified and fully validated methods Citation[22].
To obtain information about current practice regarding PPB studies, including bioanalysis within the European Bioanalysis Forum (EBF), three surveys were conducted:
(I) PPB studies in drug discovery;
(II) In vitro PPB studies in drug development;
(III) In vivo PPB studies in drug development.
This article proposes recommendations as a result of the outcome of the three surveys and the subsequent discussions in the team.
Part I: PPB studies in drug discovery
Results of the survey
Overview
In drug discovery, PPB studies are conducted by drug metabolism PK (DMPK) scientists (70% of responders), bioanalytical scientists or in cooperation between DMPK and bioanalytical scientists. They are usually performed for PK/PD evaluation, for compound selection and for allometric scaling. Typically, plasma from different species, such as humans, rats, dogs, mice, rabbits, monkeys and guinea pigs, is investigated. Generally, PPB is investigated using an in vitro setting (i.e., spike compound to blank plasma), however, some members also perform PPB from in vivo studies (i.e., plasma from dosed animals). In drug discovery, the majority of the EBF member companies do not have standard operating procedures (SOPs) in place, either for separating bound and unbound fractions or for the bioanalysis of the free drug.
Techniques used to assess PPB
In drug discovery, equilibrium dialysis, ultrafiltration, ultracentrifugation and Transil™ partitioning are all used for separation of bound and unbound drugs. Although preliminary experiments, such as investigation of nonspecific binding, drug stability in plasma and buffer, in silico evaluation and assessment of the time to reach equilibrium (equilibrium dialysis) were reported, a majority of EBF member companies do not perform such experiments at the drug-discovery phase. In general, PPB studies are conducted in triplicate at one standard drug concentration ranging from approximately 0.3 to 10 µM. Some companies perform more elaborate experiments, using two or three different drug concentration levels with one to five replicates at each level. In very early drug discovery ‘PPB screening’ is conducted by approximately 50% of the responders. A minority of the responders evaluate the mass balance/recovery using an acceptance criterion of 80%.
Equilibrium dialysis is the most popular technique for separation. Most companies (82%) use a rapid equilibrium dialysis (RED) device Citation[23]. PBS is the standard dialysis buffer. Fewer than 10% of the responders correct for the change of volume induced by equilibrium dialysis. Equilibrium dialysis membranes are checked for leakages by 27% of the responders. The incubation time for equilibrium dialysis ranges from 2 to 7 h. Approximately 50% of the responders assess nonspecific binding; either in the plasma compartment, the buffer compartment or in both compartments. Nonspecific binding of the drug to the membrane is mostly investigated at low and medium drug levels, although some also include a high level as well. Hardly any companies had an acceptance criterion for nonspecific binding.
If ultrafiltration is applied, most companies use regenerated cellulose membranes with a cut-off value of 10 kDa. Nonspecific binding of the drug to the membrane is mostly investigated at similar drug levels as those used in dialysis experiments. Plasma samples spiked with drug are mostly incubated at 37°C for 30 min. After incubation plasma samples are centrifuged in the ultrafiltration device at 37°C.
Level of qualification of bioanalytical method used
Most use nonlabeled drug material for their PPB studies. Bioanalysis is generally performed using qualified or screening methods Citation[22]; 82% of the responders use generic analytical methods (18% qualified methods). The majority of responders (64%) quantify the drugs in plasma and ultrafiltrate/dialysate using a calibration curve; alternatively, relative quantification, the comparison of peak responses in the sample before and after PPB, is also practiced in several companies. QC samples are prepared by approximately 50% of the responders. Calibration and QC samples are prepared in plasma, buffer (equilibrium dialysis), ultrafiltrate (ultrafiltration) or buffer/plasma (50/50 v/v).
Prestudy method qualification is performed by approximately 50% of the responders by demonstrating linearity, accuracy and precision of the analytical method; stability, selectivity, dilution and matrix effect are investigated by only a few companies. During the analysis of PPB samples, 60–70% of the responders include calibration and QC samples. Only a few responders have batch acceptance criteria for calibration samples, using either the acceptance criteria used in regulated bioanalysis (15% [20% at LLOQ]), or widened acceptance criteria (25% at all levels). A similar strategy is followed for the QC samples.
Reporting of the results
The report usually includes a short description of the separation experiment (equipment, materials and conditions), a description of the bioanalytical method, the results of the preliminary experiments, the unbound fraction (average and individual results) at all concentration levels (most companies do not correct for nonspecific binding when calculating the unbound fraction), and the average and individual nonspecific binding results.
Best practice & recommendations for in vitro PPB in drug discovery
Selection of the technique for PPB assessment
Equilibrium dialysis is recommended as the first choice for assessment of PPB in drug discovery as this technique can be used for nearly all small-molecule compounds. In RED, cycle times are limited to approximately 4 h, thus offering advantages for increased sample throughput. For compounds with limited stability, ultrafiltration might be the method of choice.
Important aspects to include in PPB assessment
It is recommended to perform PPB experiments at least in duplicate. Fresh or frozen pooled plasma may be used. Equilibrium dialysis should be performed at 37°C. For RED, the dialysis time of approximately 4 h should be applied (if not optimized in a preliminary experiment). A nonspecific binding test of the drug during the PPB experiment (acceptance criterion ≤30%) as well as a stability test of the drug in plasma and buffer at 37°C (4 h) can be performed as preliminary experiments; however, instead of these tests, the mass balance (recovery) of the drug can be evaluated at the end of the PPB study.
Generally, it is not required to perform a prestudy qualification of the bioanalytical method during this phase. For the analysis of study samples after dialysis it is recommended to perform limited in-study qualification by including a limited number of calibration samples. Although not critical at this stage, QC samples may also be included. For efficiency and to enhance simplicity of the analysis, the PPB samples should be matrix-matched (e.g., buffer added to the plasma and plasma added to the buffer samples, at a ratio of 50:50 v/v). In a screening setup, relative quantification can also be applied but the linearity of the analyte response should be investigated and confirmed.
Reporting of the results
Raw data of the analysis should be available upon request. In some cases, for instance when the data are used in later stages of drug development, it may add value to write a bioanalytical report that includes the key features of the analytical method used.
Part II: in vitro PPB studies in drug development
Results of the survey
Overview
In drug development, in vitro PPB studies are conducted by DMPK scientists (radiolabeled compounds), by bioanalytical scientists (nonlabeled compounds) or in cooperation (DMPK and bioanalytical scientists). In vitro PPB studies are performed for PK/PD evaluation or in support of a safety evaluation. Although PPB data is often available from the drug-discovery phase, PPB studies are extended in the drug-development phase to investigate protein binding at more concentrations and in more species. The majority of the EBF member companies have SOPs in place for PPB analysis.
To identify the main binding protein or because of regulatory requirement, most companies assess in vitro PPB studies for specific proteins, such as human serum albumin and α1-acid-glycoprotein. Typically, plasma from different species such as humans, rats, dogs, mice, rabbits, monkeys and minipigs is investigated (strains and species dependent on the species used in the toxicity studies). In addition to mixed gender, individual plasma batches (male and female) are normally investigated. In a later stage, plasma from different disease states can be included (e.g., liver and kidney impairment). Most EBF companies (83%) have quality criteria for the plasma, such as pH, storage temperature, anticoagulant, expiry date, treatment of animals during blood sampling and total protein. Fresh plasma is used as well. The majority (70%) measures the pH in plasma before use. If the pH is not within the lower limit of 7.2–7.4 and the upper limit of 7.4–7.8 the pH is adjusted.
Techniques used to assess PPB
Equilibrium dialysis is the commonly used technique for PPB studies of radiolabeled compounds as well as nonlabeled compounds. Other important techniques used are ultrafiltration and ultracentrifugation. To a lesser extent, distribution methods (e.g., Transil partitioning), erythrocyte partitioning and gel filtration are used. More than 90% of the companies perform preliminary experiments prior to the actual PPB study, such as the determination of the equilibrium time (equilibrium dialysis), nonspecific binding, drug stability in plasma, buffer and ultrafiltrate, solubility of the drug and in silico evaluation.
The majority of responders (62%) select a PPB technique based on compound characteristics (; Answers to survey II: in vitro plasma protein binding). For highly lipophilic drugs, equilibrium dialysis is selected by most companies. For highly hydrophilic drugs, both equilibrium dialysis and ultrafiltration are used. Not many companies have experience with PPB studies of biologicals (ultracentrifugation). For labile compounds ultrafiltration is selected. For sticky compounds ultracentrifugation is the most used technique.
Approximately 40% of the responders select a PPB technique based on anticipated protein binding (; Answers to survey II: in vitro plasma protein binding): equilibrium dialysis is mainly selected for anticipated high (85–98%) and very high (>98%) protein binding, whereas ultrafiltration is the method of choice (for two-thirds of responders) for compounds with anticipated medium to low (<85%) protein binding. Only 15% of the responders use a positive control compound to control the experimental phase, that is, warfarin for high and very high protein binding, bupivacaine for high protein binding, and atenolol and ketamine for medium to low protein binding.
Most companies (62%) do not have standard concentration levels for PPB studies. In general, for nonlabeled and radiolabeled compounds PPB studies are performed at three drug concentration levels (three replicates) with some using more or fewer replicates/concentration levels. Just before the PPB experiment the blank plasma is spiked with drug (dissolved in water or organic solvent). To prevent protein denaturation and subsequent disturbance of binding, the maximum accepted percentage of solvent in plasma is limited.
Equilibrium dialysis is used for PPB studies by all EBF companies. In addition to the conventional equilibrium dialysis devices, RED devices are used by 54% of the responders. The buffer compartment is filled with PBS. Other buffers used are Sörensen’s phosphate buffer and dextran (to compensate for osmotic volume shift). Approximately 50% of the responders correct for the change of volume induced by equilibrium dialysis. If nonspecific binding is measured, some measure it in both the plasma and buffer compartments while others only measure it in the buffer compartment. The acceptance criterion for nonspecific binding varies from 10 to 30%. Some companies do not have an acceptance criterion for nonspecific binding; it is considered not critical as long as the recovery is at least 85%. If the acceptance criterion is not met another PPB technique is selected, the results are corrected or additives are used to minimize nonspecific binding. The incubation time for equilibrium dialysis is determined as part of the preliminary experiments (83% of responders). Some companies (23%) use a protective atmosphere (CO2) for equilibrium dialysis. Equilibrium dialysis membranes are checked for leakage by 23% of the responders.
Another important PPB technique within the EBF is ultrafiltration. The ultrafiltration membranes consist of (regenerated) cellulose with a cut-off value of 5, 10 or 30 kDa. Before centrifugation, the plasma sample spiked with drug is incubated at approximately 37°C (by 78% of the responders) for 10, 15 or 30 min. Subsequently, centrifugation is performed at approximately 37°C (67% of responders). 44% of the companies have specified the allowed minimum and maximum filtrate volume percentage from the total sample volume: the minimum is 10–15% and the maximum is 15–30%. The majority of responders (90%) carry out a nonspecific binding test, mostly at low and medium drug-concentration levels, but also at high levels. The acceptance criterion for nonspecific binding varies from 10 to 30%. Some companies do not have an acceptance criterion for nonspecific binding; as for equilibrium dialysis, it is considered not critical as long as the recovery is at least 85%.
Most companies (90%) indicated that if a radiolabeled compound is available for PPB studies they prefer to use the radiolabeled compound instead of the nonlabeled compound.
Level of validation of bioanalytical method used
For the bioanalysis of nonlabeled compounds generic methods are used (20%), methods are qualified (70%) or validated (10%). The drugs in the samples originating from the PPB studies are quantified using absolute quantification (calibration curve; 78% of responders) or relative quantification (comparison of peak response in the sample before and after PPB; 22% of responders). Calibration and QC samples are prepared in plasma, buffer (equilibrium dialysis), ultrafiltrate (ultrafiltration) or in mixed plasma/buffer (50/50 v/v). For companies that perform prestudy method qualification/validation, at least the linearity, accuracy and precision are determined; other parameters such as the selectivity, stability, matrix effect, carryover, dilution and recovery are not included by all. The batch acceptance criteria for calibration and QC samples are ±15% (±20% at LLOQ level) for the majority of responders. Others increase the criteria to ±20 or 25% at all levels.
For the bioanalysis of radiolabeled compounds originating from PPB studies, the most used technique is liquid scintillation counting. In addition, radio-HPLC and combustion combined with liquid scintillation counting are used. The minimum radiochemical purity of the test compound is 90–99%. The stability of radiolabeled compounds in plasma and in the protein-free fraction is checked using spiked samples and radio-HPLC. However, the stability is not checked by all companies if the nonlabeled compound is stable. Prestudy qualification of the analytical method is performed by 50% of the responders. Radiolabeled compounds are measured by individual counting (80% of responders); only 20% use a calibration curve. QC samples are used by only 30% of the responders. If acceptance criteria for calibration and QC are used they are ±20% at all levels or ±15% (20% at LLOQ level).
Reporting of the results
It is common practice for in vitro PPB results in drug development to be presented in a formal report that includes a short description of the PPB experiment (equipment, materials and conditions), description of the bioanalytical work, results of the preliminary experiments, the unbound fraction (average and individual results) at all concentration levels (nonspecific binding not taken into account while calculating the unbound fraction by most companies), and the average (and individual) nonspecific binding results.
Best practice & recommendations for in vitro PPB in drug development
Selection of the technique for PPB assessment
Even though regulatory guidance exists that requires PPB to be investigated prior to the initiation of clinical studies there are no instructions on how to perform such studies Citation[5–7]. The technical aspects of the different techniques used to separate the free fraction are not the primary scope of this paper, but it must be stressed that different techniques, all with their own specific limitations, may give different results. The decision of which technique to use must be carefully evaluated based on the physical and chemical characteristics of the compound, and the experience gained in drug discovery.
Important aspects to include in PPB assessment
As PPB studies are not considered to be formal safety studies there is no need to claim GLP, but it is advisable to have a study protocol and a study report in place, and perform the study in accordance to the SOPs of the test facility.
It is recommended that in vitro PPB studies are performed in human plasma, the animal species used in toxicology and pharmacology, and with human serum albumin and α1-acid glycoprotein before clinical dosing begins. We recommend using at least three concentration levels (at least three replicates at each level) encompassing the concentrations in animal toxicity studies and the anticipated concentrations in human.
Preliminary experiments (at least stability at 37°C and nonspecific binding) should be performed before the actual PPB experiments are done, to be confident that the most suitable technique is selected. The recommended acceptance criterion for nonspecific binding is ≤30%. Depending on the selected technique more specific preliminary experiments might be needed as well, for example, time to reach the equilibrium and recovery in case of dialysis or stratification during the ultracentrifugation.
Positive control compounds with well-known PPB characteristics (e.g., warfarin for high-bound compounds and atenolol for low-bound compounds) should be included into the study to give further confidence on the performance of the selected technique. Criteria for the acceptance of the positive control compound results should be based on the literature and the historical data generated at the test facility.
In vitro PPB can be studied with nonlabeled and radiolabeled compounds. However, for high-bound compounds, the purity of the radiolabeled compound should be carefully considered. The purity level of the radiotracer should be at least in the range of expected PPB, that is, a radiochemical purity of at least 99% is recommended for a PPB of 99%. Additionally, HPLC purity profiles of plasma and especially buffer compartments are recommended. A nonlabeled compound combined with a LC–MS/MS based PPB assay should be considered, especially for very high protein binders (PPB ≥99%).
Bioanalytical aspects
The recommendation for in vitro PPB study samples is to analyze matrix-matched samples (i.e., buffer added to plasma samples and plasma to the protein-free samples at equal ratio) to avoid the need to develop and qualify bioanalytical methods for each and every matrix. Matrix-matching the study samples immediately after the PPB experiment is performed is especially beneficial for sticky compounds that tend to adsorb to storage containers in aqueous solutions.
To analyze the study samples from in vitro PPB studies it is recommended that qualified bioanalytical methods are used, that is, a bioanalytical method with an appropriate level of scientific validation. Then when the method is applied to study samples, it generates accurate concentration data that allows good decision making Citation[22]. The use of a stable labeled IS is also recommended.
The recommended method qualification (either prestudy or in-study) consists of the following parameters:
Calibration standards; at least six levels, prepared freshly every day (if long-term stability has not been proven) in plasma of one species (human) and matrix-matched before the sample preparation. Acceptance criteria: at least six calibration points, accuracy of the back-calculated concentrations within ±20% (LLOQ ±25%) of the nominal concentrations;
Blank samples with and without IS to be included into each run. The response at the retention time of the analyte should be ≤20% of the response at the LLOQ (lowest calibration sample);
QC samples prepared at low-, medium- and high-concentration levels in plasma of each species and matrix-matched before they are frozen. One precision and accuracy batch; n = 4–6 at each QC level, prepared in plasma of each species. The accuracy of the mean concentration at each concentration level should be within ±20% of the nominal concentration. The precision of the mean concentration should be <20% at each concentration level;
Carryover; matrix-matched blank sample after the highest calibration standard. If it is seen and is not avoidable, care should be taken to minimize it, for example, by the inclusion of additional blanks;
Stability experiments (low and high QC samples, n = 4–6): freeze–thaw (two cycles), short-term stability at room temperature and short-term stability at 37°C for the intended storage time. The freeze–thaw stability test and the short-term stability test at room temperature should be performed with matrix-matched QC samples, whereas the short-term stability test at 37°C should be conducted using plasma QC samples. The mean concentration at each level should be within ±20% of the nominal concentration. There is no need to repeat the stability experiments if the stability data is already available.
Reporting of the results
For in vitro PPB studies in drug development, we recommend a short report is written. The following information on bioanalytics (either in a separate bioanalytical report or included into the PPB study report) should be reported: detailed experimental description of the PPB experiment, complete experimental description of the bioanalytics, the results of the preliminary experiments, the bioanalytical method qualification results (prestudy and in-study), the bioanalytical results, and the individual and mean PPB results (bound and free fraction).
Part III: in vivo PPB studies in drug development
Results of the survey
Overview
In vivo PPB studies in drug development are performed for PK/PD evaluation in human plasma samples. The plasma samples originate from healthy subjects or from special populations (e.g., renal and hepatic patients). The specific plasma protein (human serum albumin, α1-acid-glycoprotein) to which the test compound is bound is often known in advance from in vitro PPB studies in earlier phases of drug development. Equilibrium dialysis and ultrafiltration, as well as ultracentrifugation and distribution methods (Transil partitioning) are used to determine the free fraction of the drug. In vivo PPB studies are not only performed for the parent drug but also for major pharmaceutically active metabolites. Inactive major metabolites are investigated on a case-by-case basis.
Techniques used to assess PPB
Selection of PPB techniques used follows similar practices as described in Part II of this article (; Answers to survey III: in vivo plasma protein binding). Companies do not have standard procedures for the selected number of PK time points; the number of time points varied from one (at anticipated Tmax), two (Cmax and post-Cmax), four (Cmax and three other time points depending on compound PK profile) to seven (predose and six time points covering Cmax and Cmin). Most companies do not include predose human plasma samples in in vivo PPB studies. The number of replicates per time point is one, two or three.
A few companies have quality criteria for the plasma sample, such as pH, treatment of subject during blood sampling (e.g., storage of plasma in freezer within 1 h after sampling), total plasma protein, human serum albumin and α1-acid-glycoprotein, and the anticoagulant. The QC samples are prepared by spiking human plasma with the test compound at three concentration levels (low, medium and high) over the anticipated pharmacological concentration or at one concentration level.
Equilibrium dialysis (including RED) is the most used PPB technique for in vivo PPB studies in drug development. The experimental details are similar to those described in Part II. Again, similar to the assessment of in vitro PPB, ultrafiltration is also used for in vivo assessment of PPB, using grossly the same analytical parameters. Typically, 1–2 ml of incurred plasma is added to the ultrafiltration device.
Level of validation of bioanalytical method used
The level of bioanalytical method qualification or validation is similar to the one used for in vitro assessment in drug development (see Part II; ).
Reporting of results
The report of an in vivo PPB study in drug development is similar to the one used for in vitro assessment in drug development (see Part II).
Best practice & recommendations for in vivo PPB in drug development
In principle, best practices and recommendations given in Part II for in vitro PPB in drug development also apply for in vivo PPB in drug development. However, some additional aspects have to be considered.
In vivo plasma samples show higher variation due to different matrix effects Citation[24–33]. Common causes of matrix effects are formulation excipients, co-medication and food. Additional controls are needed to check the quality of the assay. The use of stable isotope labeled IS is recommended as an elegant way to circumvent varying matrix interferences.
Metabolites, especially Phase II conjugates, such as acylglucuronides of the parent drug in in vivo samples, might cause interferences either due to instability in plasma/buffer and back-conversion to the parent drug, or due to ion-source fragmentation to the parent drug. The latter issue can be circumvented by applying adequate chromatography that separates these metabolites from the parent drug. A possible stability issue of Phase II conjugates during the PPB experiment and/or clean-up of samples should be evaluated in close collaboration with metabolism experts because stability of, for example, some acyl- and N-glucuronides strongly depends on compound structure and pH environment applied Citation[34,35].
In vivo plasma samples may be subject to GLP/GCP regulations (e.g., informed consent is required for samples from clinical trials).
Conclusions & EBF recommendation
In conclusion, the EBF recommends a tiered approach to the design of PPB studies and the bioanalysis of PPB samples:
Drug discovery phase
No study plan;
No need to do prestudy qualification;
No need for positive control compound;
Use one concentration (in duplicate);
Use generic analytical method (based on principles of screening or qualified assay) or limited prestudy method qualification (calibration samples);
In-study qualification: limited calibration samples included with a priori defined broadened acceptance criteria (25% on calibration accuracy), QC samples optional;
Appropriate IS recommended;
Raw data available. Short summary report if requested.
Drug development phase (in vitro or in vivo PPB studies)
Study plan recommended (no need to claim GLP);
Preliminary experiments can be performed before the actual PPB study, for example, to document nonspecific binding or other key parameters (ad hoc, based on prior knowledge or compound class characteristics);
Positive control compound optional;
Use at least three drug concentration levels (at least three replicates at each level);
Use qualified assay with prestudy or in-study method qualification: to document calibration, accuracy, precision, specificity, carryover and stability;
In-study and a priori defined acceptance criteria on calibration and QC (broadened to 20%; 25% at LLOQ);
Use of stable labeled IS recommended;
Study report includes all experimental details and results.
Based on the outcome of the three surveys, it can be concluded that equilibrium dialysis (RED) is selected for most PPB studies throughout drug-development programs (drug discovery and drug development). Depending on compound characteristics and/or anticipated protein binding other PPB techniques can be selected as well.
Table 1. Plasma protein binding techniques selected based on compound characteristics.
Table 2. Plasma protein binding techniques selected based on anticipated protein binding: very high protein binding (>98%), high protein binding (85–98%) and medium to low protein binding (<85%).
Binding of a drug to plasma proteins.
Test to investigate if an analytical method is working according to predefined acceptance criteria for calibration, QC and so on.
Determination of the free fraction of a drug in plasma.
Appropriate level of qualification/validation of a bioanalytical method at each stage of the drug-development process.
Disclaimer
The views expressed in this paper are the ones of the European Bioanalysis Forum and do not necessarily represent the views of its individual member companies.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Fichtl B , NiecieckiA, WalterK. Tissue binding versus plasma binding of drugs: general principles and pharmacokinetic consequences. Adv. Drug Res. 20, 118–166 (1991).
- Levitt DG . PKQuest: capillary permeability limitation and plasma protein binding – application to humen inulin, dicloxacillin and ceftriaxone pharmacokinetics. BMC Clin. Pharmacol. 2, 7 (2002).
- Bohnert T , Gan L-S. Plasma protein binding: from discovery to development. J. Pharm. Sci. 102(9), 2953–2994 (2013).
- Trainor GL . The importance of plasma protein binding in drug discovery. Expert Opin. Drug Discov. 2(1), 51–64 (2007).
- European Medicines Agency. ICH Guideline M3(R2) on Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorisation for Pharmaceuticals. EMA/CPMP/ICH/286/1995. European Medicines Agency, London, UK (2009).
- European Medicines Agency, Committee for Human Medicinal Products. Guideline on the Investigation of Drug Interactions, Final. CPMP/EWP/560/95/Rev. 1 Corr.*. European Medicines Agency, London, UK (2012).
- US Department of Health and Human Services, US FDA, Center for Drug Evaluation and Research Guidance for Industry, Drug Interaction Studies – Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. Draft Guidance . FDA, Rockville, MD, USA (2012).
- Proost JH , WierdaJMKH, MeijerDKF. An extended pharmacokinetic/pharmacodynamic model describing quantitatively the influence of plasma protein binding, tissue binding and receptor binding on the potency and time course of action of drugs. J. Pharmacokinet. Biopharm. 24(1), 45–77 (1996).
- Bekersky I , FieldingRM, DresslerDEet al. Plasma protein binding of amphotericin B and pharmacokinetics of bound versus unbound amphotericin B after administration of intravenous liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate. Antimicrob. Agents Chemother. 46(3), 834–840 (2002).
- European Medicines Agency, Committee for Medicinal Products for Human Use. Note for Guidance on the Evaluation of the Pharmacokinetics of Medicinal Products in Patients with Impaired Renal Function, CHMP/EWP/225/02. European Medicines Agency, London, UK (2004).
- European Medicines Agency, Committee for Medicinal Products for Human Use. Guideline on the Evaluation of the Pharmacokinetics of Medicinal Products in Patients with Impaired Hepatic Function, CPMP/EWP/2339/02. European Medicines Agency, London, UK (2005).
- Benet LZ , OieS, SchwartzJB. Design and optimization of dosage regimens; pharmacokinetic data. In: Goodman and Gilman’s The Pharmacological Basis of Therapeutics (9th Edition). McGraw-Hill, NY, USA, 1707–1711 (1996).
- Mullangi R , SrinivasNR. Clopidogrel: review of bioanalytical methods, pharmacokinetics/pharmacodynamics, and update on recent trends in drug-drug interaction studies. Biomed. Chromatogr. 23(1), 26–41 (2008).
- Sousa M , PozniakA, BoffitoM. Pharmacokinetics and pharmacodynamics of drug interactions involving rifampicin, rifabutin and antimalarial drugs. J. Antimicrob. Chemother. 62(5), 872–878 (2008).
- Wada M , IkedaR, NakashimaK. Microdialysis in drug-drug interactions. In: Applications of Microdialysis in Pharmaceutical Science. John Wiley & Sons, Inc., NJ, USA, 465–507 (2011).
- Zhao Z , XueF, ZhangLet al. The pharmacokinetics of nitazoxanide active metabolite (tizoxanide) in goats and its protein binding ability in vitro. J. Vet. Pharmacol. Ther. 33(2), 147–153 (2010).
- Drayer DE , LorenzoB, WernsSet al. Plasma levels, protein binding, and elimination data of lidocaine and active metabolites in cardiac patients of various ages. Clin. Pharmacol. Ther. 34(1), 14–22 (1983).
- Bowers WF , FultonS, ThompsonJ. Ultrafiltration vs. equilibrium dialysis for determination of free fraction. Clin. Pharmacokinet. 9(1), 49–60 (1984).
- Lee KJ , MowerR, HollenberckTet al. Modulation of nonspecific binding in ultrafiltration protein binding studies. Pharm. Res. 20(7), 1015–1021 (2003).
- Zhang F , XueJ, ShaoJet al. Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov. Today17(9–10), 475–485 (2012).
- Musteata FM . Monitoring free drug concentrations: challenges. Bioanalysis3(15), 1753–1768 (2011).
- Timmerman P , KallMA, GordonBet al. Best practices in a tiered approach to metabolite quantification: views and recommendations of the European Bioanalysis Forum. Bioanalysis2(7), 1185–1194 (2010).
- Rapid Equilibrium Dialysis (RED) Device. www.piercenet.com/product/rapid-equilibrium-dialysis-red
- Mei H , HsiehY, NardoCet al. Investigation of matrix effects in bioanalytical high-performance liquid chromatography/tandem mass spectrometric assays: application to drug discovery. Rapid Commun. Mass Spectrom. 17(1), 97–103 (2003).
- Matuszewski BK , ConstanzerML, Chavez-EngCM. Matrix effect in quantitative LC–MS/MS analyses of biological fluids: a method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal. Chem. 70(5), 882–889 (1998).
- Matuszewski BK , ConstanzerML, Chavez-EngCM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC–MS/MS. Anal. Chem. 75(13), 3019–3030 (2003).
- Müller C , SchäferP, StörtzelMet al. Ion suppression effects in liquid chromatography–electrospray-ionisation transport-region collision induced dissociation mass spectrometry with different serum extraction methods for systematic toxicological analysis with mass spectra libraries. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 773(1), 47–52 (2002).
- Choi BK , HerculesDM, GusevAI. Effect of liquid chromatography separation of complex matrices on liquid chromatography–tandem mass spectrometry signal suppression. J. Chromatogr. A907(1–2), 337–342 (2001).
- Bonfiglio R , KingRC, OlahTVet al. The effects of sample preparation methods on the variability of the electrospray ionization response for model drug compounds. Rapid Commun. Mass Spectrom. 13(12), 1175–1185 (1999).
- Larger PJ , BredaM, FraierDet al. Ion-suppression effects in liquid chromatography–tandem mass spectrometry due to a formulation agent, a case study in drug discovery bioanalysis. J. Pharm. Biomed. Anal. 39(1–2), 206–216 (2005).
- Xu X , MeiH, WangSet al. A study of common discovery dosing formulation components and their potential for causing time-dependent matrix effects in high-performance liquid chromatography tandem mass spectrometry assays. Rapid Commun. Mass Spectrom. 19(18), 2643–2650 (2005).
- Schuhmacher J , ZimmerD, TescheFet al. Matrix effects during analysis of plasma samples by electrospray and atmospheric pressure chemical ionization mass spectrometry: practical approaches to their elimination. Rapid Commun. Mass Spectrom. 17(17), 1950–1957 (2003).
- Tong XS , WangJ, ZhengSet al. Effect of signal interference from dosing excipients on pharmacokinetic screening of drug candidates by liquid chromatography–mass spectrometry. Anal. Chem. 74(24), 6305–6313 (2002).
- Dell D . Labile metabolites. Chromatographia59(2 Suppl.), 139–148 (2004).
- Regan SL , MaggsJL, HammondTGet al. Acyl glucuronides: the good, the bad and the ugly. Biopharm. Drug Dispos. 31(7), 367–395 (2010).