
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Aim: This work aims to contribute toward development of preventive measures for the control of monkeypox (mpox) virus disease through computational design of a multiepitope vaccine. Methods: To accomplish this, we employed a robust immunoinformatics approach to design a putative chimeric vaccine candidate from 18 viral transmembrane proteins. Results: The resulting chimeric vaccine candidate is a 76.4 kDa protein containing 687 amino acids with an estimated isoelectric point of 9.39. In addition, it was predicted to adopt a stable 3D conformation that harbors discontinuous B-cell epitopes and strongly interacts with key immune receptors. Conclusion: The designed hypothetical antigen is a valuable addition to the collection of prospective vaccine candidates for future development and trials against the reemerging mpox disease.
Plain language summary
The recent outbreak of mpox virus presents a significant public health threat with no treatment available. While the use of smallpox vaccines remains the current strategy for the managing the outbreak, the approach suffers from drawbacks of adverse effects. As a consequence, our work employed computational methods to design a chimeric vaccine that targets the pathogen. We hope the putative vaccine will add to the collection of available therapeutic candidates for future trials.
Graphical Abstract
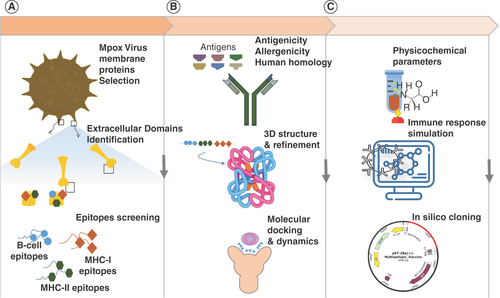
Graphical abstract illustrating the workflow of a multiepitope vaccine design against mpox virus. (A) Prediction of the transmembrane protein with identification of the extracellular domains as well as the screening of the MHC-I, MHC-II and continuous B-cell epitopes. (B) Antigenicity and allergenicity prediction with human homology analysis followed by 3D structural modeling of the linear vaccine construct, molecular docking and dynamics of the multiepitope vaccine against toll-like receptors. (C) Assessment of physicochemical properties, immune simulation of the multiepitope vaccine and in silico cloning.
The recent outbreak of the zoonotic monkeypox (mpox) virus disease has spread to about 110 endemic and nonendemic nations, presenting a global threat that requires urgent action. Since May 2022, the mpox virus has not only reemerged in endemic regions but also emerged in nonendemic regions, including Europe and North America [Citation1, Citation2]. The current mpox outbreak is global, so far recording more than 87,000 cases with 112 deaths and it is believed that the peak phase of the disease outbreak has not yet been witnessed in many countries [Citation3, Citation4].
Mpox virus, responsible for human mpox disease, belongs to the Orthopoxvirus genus, which contains some of the most complex animal viruses, including vaccinia viruses, smallpox-causing virus (Variola major) and cowpox virus [Citation5]. Mpox virus is a linear dsDNA zoonotic virus that exists in two clades: the west African strain and the Congo Basin strain, both of which are virulent, with the Congo Basin strain reported to cause more severe and fatal mpox [Citation6–8]. Since 1970, there have been several outbreaks of mpox globally, with cases ranging from mild to severe and fatal. However, reported cases have increased along with human-to-human transmission over the past three decades [Citation9, Citation10]. The fatality case rate of mpox disease (10%) sits in between the case fatality rates of V. major and Variola minor, which are 30 and 1% respectively. Mpox disease was considered rare and self-limiting, but current reports indicate otherwise [Citation1]. Moreover, the identification of the mpox virus in seminal fluid by polymerase chain reaction assays is worrying as this raises the concern that the virus could be sexually transmitted [Citation11].
The symptoms of mpox disease resemble those of smallpox, an observation on which the repurposing of smallpox vaccines against mpox virus was based, and approximately 85% effectiveness was achieved [Citation12, Citation13]. Despite the high efficacy of the smallpox vaccine (DryvaxTM, Wyeth Laboratories, PA, USA) against mpox, safety issues associated with the use of the live virus and reported side effects pose significant drawbacks to this modality. For example, severe side effects such as progressive vaccinia, postvaccinial encephalopathy and even death have been associated with the Dryvax vaccine [Citation13]. Other smallpox vaccines used to prevent mpox include the second-generation vaccine from Dryvax, ACAM2000TM, and JYNNEOSTM (Bavarian Nordic, Hellerup, Denmark), a third-generation vaccine. Like Dryvax, ACAM2000 is a live virus-based vaccine and, therefore, has the same safety concerns. In addition, reports have shown that ACAM2000 displayed similar side effects to those associated with Dryvax [Citation13, Citation14]. While JYNNEOS, a nonreplicating modified vaccinia virus Ankara, has shown similar immunogenicity to ACAM2000TM when administered in two doses, its use is limited by side effects such as swelling, itching, fatigue, muscle pain and cardiac events. LC16m8TM (KAKETSUKEN, Kumamoto, Japan) is another third-generation smallpox vaccine used against mpox, but it has been associated with lymphadenopathy and breakthrough disease. Furthermore, these third-generation vaccines generally require two doses, making them less suitable for a rapid outbreak response [Citation13]. Although several drugs have also been approved against mpox virus, vaccines remain the best arsenal for managing pandemics and eradicating diseases. This has been well demonstrated by the effectiveness of COVID-19 vaccines in restoring normalcy after more than a year of lockdown and strict social restrictions.
With the advances in genomics, bioinformatics and sequencing techniques, the omics-driven design of multiepitope vaccines (MEVs) has become a valuable and viable approach to developing vaccines against infectious diseases. A MEV represents an ensemble of immunogenic peptide-base fragments derived from proteins from infectious agents. MEVs can elicit strong immune responses while avoiding allergic reactions, thus overcoming problems associated with conventional vaccines and saving time and cost [Citation15]. A good number of MEV candidates have been designed against several pathogens, such as Trypanosoma brucei gambiense [Citation15], SARS-CoV-2 [Citation16], Coronavirus Neoromicia (NeoCoV) [Citation17] and tick-borne virus [Citation18], among others. Indeed, the remarkable result reported in clinical trials for MEVs targeting cancer and human immunodeficiency virus highlight the potential of these innovative molecular chimeras in revolutionizing the landscape of disease treatment and prevention [Citation19–21].
Learning from the COVID-19 pandemic, it is evident that zoonotic pathogens are a significant threat to global health and economy. Moreover, the numerous recurrent outbreaks of mpox disease over the years and the possibility of the existence of an animal reservoir outside Africa further increase the severity of the current and future situation. Hence, innovative therapeutic modalities are needed to contain the present situation and to avoid a resurgence of outbreaks in the future. MEV is one such therapeutic modality that will play a significant role in addressing the current needs of containing the virus as well as preparing for occurrences. In fact, several groups have exploited such ideas to propose putative MEV candidates as part of the emergency response to the mpox outbreak [Citation17, Citation22, Citation23]. Our approach herein, however, differs from the previous reports in that we screened putative epitopes from extracellularly exposed domains of transmembrane proteins from the whole mpox virus proteome. We further performed a robust vaccine structure modeling and followed the dynamic interaction of the designed MEV through molecular dynamics (MD) simulations and end-state free energy calculations (Graphical abstract). Therefore, we present our work as an addition to the collection of prospective vaccine candidates for trials against reemerging mpox disease.
Materials & methods
Prediction of transmembrane proteins
The mpox virus proteome was recovered from UniProt (accession no. UP000099051) and was submitted to DeepTMHMM [Citation24] to predict transmembrane proteins. From the predicted transmembrane proteins, extracellular domains with no fewer than 20 amino acids were collected and used for further analysis.
Prediction of MHC-I & MHC-II binding epitopes
The NetMHC-4.0 and NetMHC-II-2.3 servers were used to predict MHC-I and MHC-II binding epitopes, respectively, by inputting the extracellular domains obtained from previous predictions [Citation25, Citation26]. Strong MHC-I and MHC-II binders were selected based on a threshold of ≤0.5 percentile and ≤2.0 percentile, respectively. In both predictions, human supertype alleles were chosen for the coverage of the global population.
Prediction of continuous B-cell epitopes
Continuous B-cell epitopes in the extracellular domains were predicted using BepiPred-2.0 with a default server threshold of 0.5 [Citation27].
Prediction of antigenicity & allergenicity
The predicted epitopes (B cell, MHC-I and MHC-II) were analyzed for antigenicity and allergenicity using VaxiJen-2.0 and AllerTOP-2.0, respectively [Citation28, Citation29]. A viral threshold of 0.4 was used for the VaxiJen-2.0 prediction, while default parameters were maintained for the AllerTOP-2.0 prediction. The peptides that were antigenic and nonallergenic were then selected for further analysis.
IFN-γ-inducing epitopes
The potential of predicted antigenic and nonallergenic MHC-II binding epitopes to stimulate the production of IFN-γ were predicted on IFNepitope server by using the motif and support vector machine (SVM) hybrid methods [Citation30].
Construction of final vaccine candidate
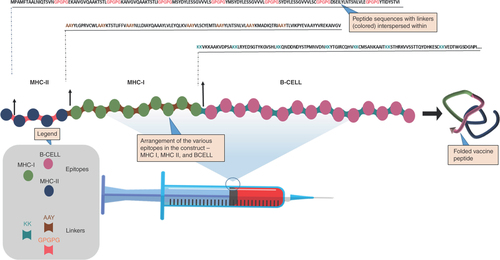
The epitopes that passed the antigenicity and allergenicity filters were assembled to obtain the final vaccine construct. To achieve this, the epitope clusters (i.e., MHC or B cell) of the epitopes were arranged in different combinations to find the most stable antigen. The arrangement presented in was the most stable and compact and was chosen as the best. The MHC-II epitopes were linked together using GPGPG linkers, while the MHC-I epitopes were linked using AAY linkers. The B-cell epitopes, on the other hand, were linked using KK linkers.
The MHC-II, MHC-1 and B-cell epitopes are joined using GPGPG linkers, AAY linkers and KK linkers, respectively.
Vaccine structure prediction & refinement
The amino acid sequence of the combined antigen (687 in total) construct was used to predict the 3D structure of the putative vaccine using RoseTTAFold [Citation31]. The antigen’s primary sequence was used as template, and the default parameters of the server were maintained. Structure refinement was done using GalaxyRefine [Citation32], and we further equilibrated the structure by subjecting it to 100 ns MD simulation using GROMACS-2021 [Citation33]. The simulation parameters are explained subsequently as a standalone method under Molecular docking & dynamics simulations section. .
Prediction of discontinuous B-cell epitopes
The formation of discontinuous B-cell epitopes in the 3D structure of the vaccine was predicted using the ElliPro server with the default minimum score of the server and a maximum distance of 0.5 and 6, respectively [Citation34].
Evaluation of physicochemical properties
Expasy ProtParam was used to characterize the physicochemical properties of the putative vaccine using the primary sequence [Citation35]. Properties evaluated included MW, charge distribution, estimated half-life in yeast and mammalian cells, and instability index, among others.
Molecular docking & dynamics simulations
Toll-like receptors 2 and 4 (TLR2 and 4) play vital roles in viral protein recognition, therefore, we docked the putative vaccine candidate with TLR2 and TLR4 to predict possible interactions with the receptors. The 3D structures of TLR2 and TLR4 were downloaded from the Protein Data Bank using their respective IDs, 6NIG and 3FXI. Docking was performed on ClusPro server with the antigen as a ligand and the immune receptors as docking receptors [Citation36]. The best docking pose in each case was then subjected to MD simulation to follow the dynamics of the interactions. To this end, three systems were prepared: MEV, MEV-TLR2 and MEV-TLR4. Each system was protonated at a pH of 6.5, solvated with TIP3P water and neutralized with 150 mM NaCl [Citation37]. The steepest descent algorithm was used to energy minimize the systems for 5000 steps. This was followed by constant temperature, constant volume (NVT) and constant temperature, constant pressure (NPT) ensembles equilibrations for 100 ps each using the Bussi velocity rescale thermostat and Parrinello–Rahman barostat for temperature and pressure coupling, respectively [Citation38, Citation39]. Following the equilibration of the systems to a pressure of 1 atm and a temperature of 298 K, a 100 ns MD run was performed for each system in the NPT ensemble. Unless otherwise told, hydrogen mass repartitioning was used to achieve a timestep of 4 fs for all simulations. The root mean square deviation (RMSD) and root mean square fluctuations as well as the radius of gyration of the systems were calculated from the trajectories information generated from the simulations. All simulations and analyses were conducted using GROMACS-2021 [Citation33], and the simulation parameters described here were used for the prior equilibration simulation of the MEV.
Binding free energy calculations
The binding free energy of the putative antigen to TLR2 and TLR4 was calculated using the molecular mechanics with the generalized Born and surface area solvation method. This method relies on the trajectory generated from explicit solvent MD simulation, where the simulation box and ions are eliminated prior to the calculation, and the solvation terms are inferred implicitly. The binding free energy (ΔGbind) is computed using the following equations (EquationEquation 1(Eq.1) , Equation2
(Eq.2) , Equation3
(Eq.3) ).
(Eq.1)
(Eq.2)
(Eq.3)
Where ΔEMM accounts for the molecular mechanics energy due to changes in the gas phase, ΔGsol represents the free energy of solvation, while TΔS accounts for the entropy due to ligand-induced conformation changes. The ΔEMM is further split into ΔEinternal, ΔEelectrostatic and ΔEvdw, representing changes in the internal, electrostatic and van der Waals energies, respectively. The polar and nonpolar contribution terms between the solute and the solvent, that is, ΔGGB and ΔGSA, constitute the solvation free energy [Citation40].
Immune simulation
The behavior of the putative antigen in mammalian hosts was predicted from an immune simulation. The simulation entailed the administration of three doses of a combination of 1000 molecules of the antigen and 100 molecules of lipopolysaccharide (LPS) to the human model at different time steps of 1, 84 and 168, where one simulation step represented 8 h. The simulation lasted 1098 steps, equivalent to 366 days, and the machine-learning algorithm C-IMMSIM was used for the simulation [Citation41].
Codon adaptation & in silico cloning
Finally, the MEV protein sequence was reverse-translated to the corresponding nucleotide sequence and adapted to Escherichia coli codons using the JAVA Codon Adaptation Tool [Citation42]. The EcoRI recognition sequence and a start codon were used to flank the 5′ end, while a stop codon and NotI recognition sequence flanked the 3′ end of the vaccine sequence. The new chimeric sequence was finally cloned in silico into pET28a(+) vector with an N-terminal hexa-His-tag.
Results
Prediction of transmembrane proteins
Given the higher probability of membrane proteins to interact with immune receptors of the host, leading to an immune response, our objective was to develop a potential vaccine by utilizing epitopes from membrane proteins of mpox virus. Accordingly, we utilized DeepTMHMM [Citation24] for the prediction of the subcellular localization of the viral proteins. The prediction returned 18 nonredundant transmembrane proteins, from which we collected 19 extracellularly exposed domains. Each domain contained a minimum of 20 amino acids (Supplementary material). This criterion was set to increase the likelihood of exposure of domains and adequately cover B-cell epitopes in subsequent prediction.
Prediction of B-cell, MHC-I & MHC-II binding epitopes
Using the selected extracellularly exposed domains, a total of 75 B-cell epitopes were predicted using BepiPred-2.0. Similarly, 30 and 37 epitopes were classified as strong binders out of the thousands of epitopes predicted by NetMHC-4.0 and NetMHC-II-2.3 for MHC-I and MHC-II binding epitopes [Citation25–27], respectively (Supplementary Data Sheet).
Selection of epitopes for vaccine construction
The VaxiJen-2.0 server was utilized to perform antigenicity analysis on the predicted B-cell- and MHC-binding epitopes. The outcome of this analysis was 45, 15 and 20 antigenic B-cell, MHC-I and MHC-II epitopes, respectively. The identified antigenic epitopes were subjected to allergenicity screening using the AllerTOP server, out of which 20 B-cell epitopes, 9 MHC-I epitopes and 7 MHC-II epitopes were predicted as nonallergenic and subsequently chosen for downstream analyses ().
Table 1. Antigenic, nonallergenic and nontoxic epitopes selected for the multiepitope vaccine construction.
IFN-γ epitopes prediction
In viral infections, IFN-γ plays a vital role in the elimination of infection, hence, the ability of the seven selected MHC-II epitopes to induce the production of IFN-γ were predicted using IFNepitope server [Citation30]. The analysis returned one epitope with a positive score, suggesting that it could trigger the desired outcome (Supplementary Table 1).
Construction of the MEV
The epitopes of the chimeric vaccine were reshuffled interclassically to obtain the most stable and compact structure, while the intraclass epitope arrangement was random and kept constant during the reshuffling. The arrangement, MHC-II∼MHC-I∼B-CELL, was found to be the best, characterized by a compact structure with Ramachandran outliers of 2.1% (Supplementary Figure 1). The MHC-II epitopes in this configuration were connected with GPGPG linkers, while the MHC-I epitopes were joined by AAY, and the B-cell epitopes linked by KK linkers (). The selection of these linkers was based on prior research demonstrating their successful application in constructing stable MEVs [Citation43].
Prediction & refinement of 3D structures
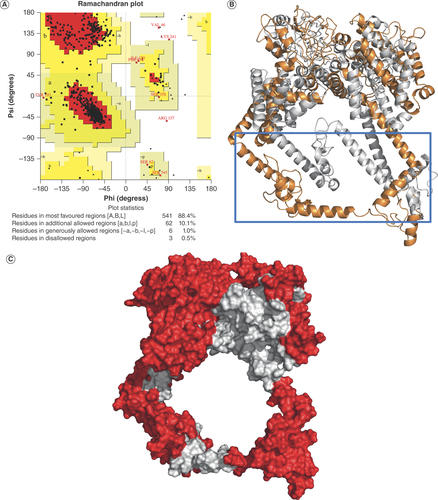
As described earlier, different arrangements of epitopes were subjected to 3D structure prediction using RoseTTAFold [Citation31], and the outcome was evaluated using a Ramachandran plot. The best structure, predicted using the order MHC-II∼MHC-I∼B-CELL, returned a Ramachandran outliers value of 2.1% (Supplementary Figure 1A). This value, however, fell outside the acceptable range of ≤0.5%. Therefore, we refined the structure using the GalaxyRefine sever and recalculated the Ramachandran outliers. Surprisingly, the value worsened to 2.6% (Supplementary Figure 1B). To address this, we equilibrated the predicted structure through a 100 ns MD simulation and extracted the structural coordinates from the last frame of the simulation trajectory. This was used to recalculate the percentage of Ramachandran outliers, which was found to be an acceptable value of 0.5% (A). Superimposition of the MEV structure after GalaxyRefine refinement with the one extracted after MD simulation reveals critical changes that the antigen underwent during the 100 ns equilibration. Specifically, the structure relaxed to form an extended conformation by opening up three alpha helices (highlighted in a blue box, B). This accounted for the improvement in the value of the Ramachandran outliers and also underscored the potential of MD simulation in studying protein folding.
(A) Ramachandran plot of the molecular dynamics (MD) simulation equilibrated structure of the designed multiepitope vaccine (MEV). (B) Superimposed structures of MEV before (in gray) and after (orange) MD simulation equilibration. The structural changes that occurred during MD simulation are highlighted in a blue box. (C) The surface representation of the equilibrated structure of MEV showing discontinuous B-cell epitopes (in red).
The chimeric vaccine adopted a conformation that promoted the formation of discontinuous B-cell epitopes
In addition to the predicted linear B-cell epitopes, the formation of discontinuous B-cell epitopes is equally desired, as the 3D conformation of the vaccine may bury some of the linear epitopes and prevent their exposure to the host’s immune receptors. Thus, we used the ElliPro server [Citation34] to predict if our vaccine’s structure forms such discontinuous B-cell epitopes. Interestingly, our prediction revealed well-exposed discontinuous B-cell epitopes that cover most of the vaccine surface and represent more than half of the antigen’s total residues (C). This indicates that the MEV has a high chance of eliciting a humoral response.
The predicted characteristics of the MEV indicate its safety, antigenicity & favorable physicochemical properties
To reevaluate the safety and the predicted antigenicity of MEV, the whole construct primary sequence was resubmitted to AllerTOP-2.0 and VaxiJen-2.0 for prediction of allergenicity and antigenicity, respectively [Citation28, Citation29]. The MEV was characterized as a nonallergen and probable antigen with a VaxiJen-2.0 score of 0.5807. A BLAST search of the MEV against UniProt’s Homo sapien reference genome was performed to further ascertain its safety. Our search returned a putative short-chain dehydrogenase with UniProt ID A0A8I5KQB5 as the closest human homolog of our MEV sequences with an identity of 35.1%. In addition to the identity falling within an acceptable range, the homologous amino acids are widely dispersed in the dehydrogenase sequence with the highest amino acid stretch of five around the GPGPG linker region, further highlighting the antigen’s safety.
Additionally, the physicochemical properties of the MEV were also evaluated. The antigen’s predicted molecular weight is 76.4 kDa, theoretical isoelectric point is 9.39 and instability index is 29.33, showing that it is stable. The vaccine has 110 and 73 positively and negatively charged residues, respectively. In addition, the predicted half-life in both yeast and mammalian cells was estimated to be 20 h. Taken together, we can conclude that the putative MEV is presumably safe and exhibits the properties of a stable antigen.
Molecular docking & MD simulation predicted the interaction of the designed vaccine with key immune receptors
TLRs are membrane-spanning receptors that are essential for pathogen recognition and the activation of innate immunity. Among these receptors, TLR2 and TLR4 are especially important in triggering an immune response against DNA viruses, including the Poxviridae virus family, to which the mpox virus belongs [Citation44, Citation45]. Therefore, to study the possible interaction of our MEV with these immune receptors, we docked the MEV against both TLR2 and TLR4 using ClusPro-2.0 [Citation36]. This docking algorithm generates 28 different clusters of binding orientation with no defined binding energies. Hence, we selected, in each case, a conformation that showed maximum occupancy into the receptor cavity. We then probed these docking states using 100 ns MD simulations to allow for better sampling of the binding conformation and a more robust evaluation of the interactions.
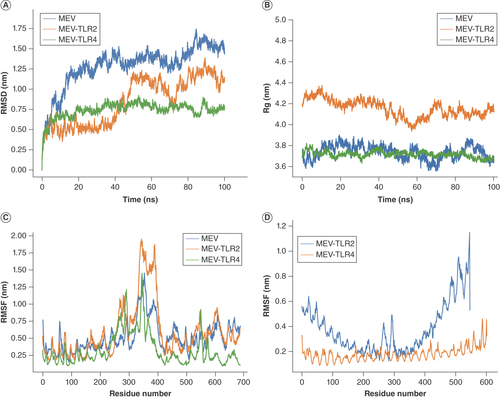
The stability of the simulation systems was first evaluated by following the RMSD of the receptors’ backbone atoms and the MEV as a function of time [Citation46]. MEV-TLR4 exhibited the most stable RMSD, stabilizing as early as the first 10 ns, while the MEV system stabilized around 50 ns with slight fluctuations toward the end (A). On the contrary, the RMSD of the MEV-TLR2 system fluctuated throughout the simulation, implying that the system had not attained equilibrium (A). This observation is also consistent with the root mean square fluctuation of MEV-TLR2, showing high flexibility around residue 300–400 of MEV (C). Notwithstanding, this region constituted majorly the helices described earlier that transformed from compact to open conformation during the initial equilibration. Therefore, the observed flexibility could be attributed to this structural transformation. Similarly, the radius of gyration revealed that the compactness and therefore the global conformation of the MEV was maintained during the simulation (B), indicating stability of the designed putative vaccine. Clustering the trajectories revealed 206 and 66 clusters for MEV-TLR2 and MEV-TLR4 systems, respectively. The centroid of the largest cluster was chosen as a reference for the interaction model of both systems. In these models, TLR4 binds more strongly to the MEV than TLR2. TLR4 encircled a large part of MEV, while TLR2 barely formed contacts with two helices of MEV (A & C). In agreement with this, MEV interaction with TLR4 included 7 salt bridges, 21 hydrogen bonds and 180 nonbonded contacts with TLR4, as opposed to the 6, 10 and 116 salt bridges, hydrogen bonds and nonbonded contacts with TLR2 (B & D). Atomic details of these interactions are presented in Supplementary Figure 2. Although the MEV interacts more robustly with TLR4 than TLR2, its interaction with TLR2 is equally good and involves strong forces like hydrogen bonds. Taken together, we can conclude that there are stable interactions between the MEV, and the vital immune receptors as predicted by the MD simulation.
(A) The RMSD of the simulation system. (B) Rg of the simulation systems. (C) The RMSF of MEV residues in apo state and bound with TLR2 and TLR4. (D) The RMSF of TLR2 and TLR4 residues bound to the designed MEV.
MEV: Multiepitope vaccine; Rg: Radius of gyration; RMSD: Root mean square deviation; RMSF: Root mean square fluctuation; TLR: Toll-like receptor.
(A) Interaction of the MEV with TLR2 in 3D. (B) Overview of the types of chemical interaction between the MEV and TLR2. (C) Interaction of the MEV with TLR4 in 3D. (D) Overview of the types of chemical interactions between the MEV and TLR4.
MEV: Multiepitope vaccine; TLR: Toll-like receptor.
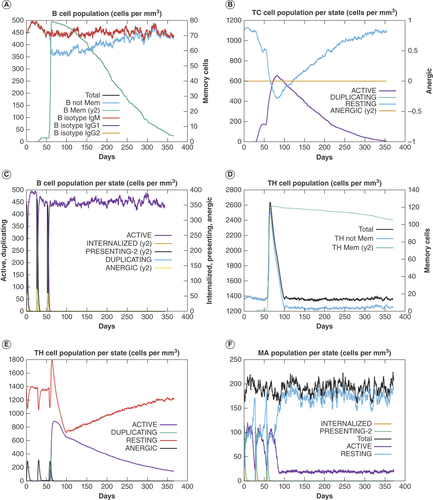
(A) B-cell population. (B) Population of cytotoxic T cells per state. (C) Population of B cells per state. (D) Helper T-cell population. (E) Population of helper T cells per state. (F) Macrophages population per state.
The predicted binding free energy correlates with the predicted interaction models
To quantitatively define the strength of the interaction of our MEV with TLR2 and TLR4, we used MM/GBSA end-state free energy calculation tool [Citation40] to estimate the interaction binding free energies of our systems from the MD simulation trajectories. The average binding energy of the MEV against TLR2 was predicted to be -66.34 kcal/mol, higher than -96.93 kcal/mol for TLR4 (). This suggests stronger binding of the MEV with TLR4 than TLR4, further substantiating the predicted interaction models. Nevertheless, the putative vaccine interacts very well with the two receptors.
Table 2. Calculation of MM/GBSA binding free energy (kcal/mol) of multiepitope vaccine against immune receptors.
Immune simulation predicts persistent cellular & humoral immunity against the putative antigen
To understand the potential immune reaction that our MEV could stimulate, we performed a year-long immune response simulation on a human model using the C-IMMSIM algorithm [Citation41]. Three doses of the antigen with LPS as adjuvant were administered in silico on day 0, day 28 and day 56, and the immune response was monitored until day 366. The simulation predicted the activation and proliferation of immunoglobulins-producing B cells as well as memory B cells with population and activity that persisted throughout the period of simulation (A & C). Similarly, the proliferation of cytotoxic and helper T cells was recorded, with a stable population of memory helper T cells persisting for 1 year (B, D & E). Furthermore, high and stable total macrophage activity was observed from the first dose to day 366 of the simulation (F). Together, these data suggest the stimulation of lasting and memorable immune responses against the putative vaccine.
Codon adaptation & in silico cloning
Finally, the corresponding nucleotide sequence of the designed MEV was obtained through the reverse translation of its amino acid sequence. The codons of the reverse-translated sequence were optimized and adapted for expression in E. coli. The adapted codon returned a GC content of 48%, which is acceptable considering the optimal range of 30–70%. To present MEV in a ready-to-use expression vector, the adapted sequence was subcloned in silico into pET28(+) vector using the multiple cloning sites. The MEV sequence was flanked by an EcoRI recognition sequence and a start codon on the 5′ end and a stop codon followed by a NotI recognition sequence on the 3′ end. The new chimeric sequence contained an N-terminal His-thrombin-tag to facilitate purification (Supplementary Figure 3).
Discussion
Mpox, a zoonotic disease of substantial public health significance, is reemerging as a notable orthopoxvirus infection in humans following the successful eradication of smallpox. Mpox virus and smallpox virus share similar clinical manifestations, and this was the basis for repurposing smallpox vaccines against mpox virus [Citation47]. Even though this approach has proven to be effective to some degree, issues with the usage of a live virus and the well-documented side effects of the smallpox vaccines limit the exploitation of this strategy [Citation13, Citation14].
On the contrary, subunit vaccines, designed based on antigenic molecules of a pathogen, are generally safer and devoid of most risks associated with live attenuated vaccines [Citation48]. The immunoinformatic approach to vaccine design is a powerful computational method used to construct subunit vaccine candidates from a cocktail of antigenic epitopes. The approach is cost-effective and timesaving and has proven to be successful in predicting highly probable antigenic epitopes to target many diseases [Citation49]. The power of MEV design is further illuminated by the potential of the approach to screen the pathogen’s proteome to identify immunogenic epitopes that elicit a precise and focused immune response, minimizing the risk of reversible pathogenesis [Citation48]. In contrast to the conventional recombinant vaccine technology that utilizes entire proteins, MEV triggers immune responses by targeting short immunogenic epitopes. An approach like this circumvents excess antigenic load and limits (or perhaps eliminates) allergenic reactions within the host [Citation50]. One more advantage of MEVs over traditional and single-epitope vaccines is the recognition of multiple MHC-I and MHC-II epitopes by T-cell receptors from various subsets of T cells. The field of MEV design is rapidly emerging and has already gained significant importance. Vaccines developed using this approach have demonstrated effectiveness in inducing protective immunity in vivo and have also progressed to phase I clinical trials [Citation51].
Capitalizing on this, we employed such an approach to design a prophylactic MEV against the mpox virus from the viral surface proteins. Surface proteins were targeted for design because of their exposure and therefore a higher chance of interacting with host molecules. Our initial prediction resulted in 18 nonredundant transmembrane proteins from which we collected 19 extracellularly exposed domains. Interestingly, these proteins are conserved in other orthopoxviruses including horsepox virus, smallpox virus, cowpox virus, vaccinia virus and camelpox virus, implying that vaccine designed from these proteins could cover a broad spectrum of diseases [Citation52]. We further predicted MHC-I, MHC-II and B-cell epitopes from the selected domains and joined them using linkers. Accordingly, MHC-I epitopes were linked by AAY linkers, MHC-II by GPGPG and B cell by KK linkers. The incorporation of linkers in the construct enhances its solubility, facilitates the creation of discontinuous B-cell epitopes and ensures the accessibility of the adjacent domains [Citation53, Citation54]. The order of epitopes arrangements in the vaccine construct was determined by reshuffling the epitope classes until a structure of reasonable quality was obtained. The sequence described in yielded the best structure with Ramachandran outliers of 2.1%. The structure was further refined and equilibrated over 100 ns MD simulation until an acceptable Ramachandran outlier value of ≤0.5% was achieved. Interestingly, the quality of our equilibrated structure outperformed all previously reported mpox MEVs [Citation17, Citation22, Citation23].
Thereafter, the equilibrated primary structure was used to assess the physicochemical properties of the designed construct. Predicting these properties is vital for future molecular and structural characterization of the antigen. The vaccine construct was determined to have a molecular weight of 76.4 kDa and a theoretical isoelectric point of 9.39, consistent with the MEV reported by Shantier et al. [Citation23]. Proteins with an instability index below 40 are anticipated to exhibit stability, whereas values exceeding this threshold indicate protein instability [Citation35]. Hence, our vaccine was predicted to be stable, as it exhibited an instability index of 29.33 [Citation35], and a predicted half-life of 20 h for both yeast and mammalian cells, in agreement with the mpox MEVs reported by Aziz et al. [Citation17]. The antigen was further classified as nonallergen by AllerTOP-2.0. Having established these, interactions of the designed antigen with TLR2 and TLR4 were assessed using molecular docking and MD simulations. TLR2 and TLR4 are important pattern recognition receptors that are involved in pathogen sensing by interacting with pathogen-associated molecular patterns. Their role in recognition of viral antigens has been well-documented [Citation55]. In a study reviewed by Totura et al. [Citation56], it was demonstrated that mice lacking TLR4 were more susceptible to infection compared with wild-type mice. Given the importance of these receptors, it is vital that the putative vaccine construct is recognized by human TLR2 and TLR4. This being the case, the MEV was docked against the two receptors and simulated for 100 ns each. By clustering the MD simulation trajectories and end-states free energy calculations, a strategy we deployed elsewhere [Citation57, Citation58], we predicted extensive interactions between the antigen and both immune receptors. These interactions entailed multiple hydrogen bonds, salt bridges and more than a hundred nonbonded contacts in each case. Nevertheless, the interaction of the antigen with TLR4 appeared to be stronger with about 11 more hydrogen bonds, an observation that was very well recapitulated by the lower predicted binding energy of the antigen to TLR4 than to TLR2. Importantly, the predicted energies of the MEV against TLR4 and TLR2 is superior to the mpoxMEV design from partial proteome [Citation59], indicating a more robust binding and potentially an increased likelihood of triggering a response.
We also simulated the immune reaction to our putative vaccine construct using an in silico technique. Three doses of the antigen (on days 0, 28 and 56) were injected (at day 0, 28 and 56) alongside LPS adjuvant to the human model using the C-IMMSIM algorithm. A direct interaction between TLR4 and LPS plays an enormous role in the activation of TLR4 [Citation60]. The simulation lasted for a year, predicting strong humoral and cellular immune responses, with a persistent population of both memory B cells and T cells that survived for the period of the simulation. While predictive, this finding suggests that the designed antigen can induce the desired response. To facilitate future molecular characterization of the designed MEV, we obtained its cDNA by reverse translating its amino acid sequence. Furthermore, the codons were optimized and adapted for expression in E. coli. We finally subcloned the optimized construct into the pET28a(+) expression vector with an N-terminal His-tag and presented it as a ready-to-use construct for future studies.
Conclusion
In conclusion, our present study proposes a putative chimeric vaccine designed through a rigorous immunoinformatics framework that is superior to conventional vaccine design approaches. MEVs do not only induce robust immune responses while preventing allergic reactions but also prove to be a time- and cost-efficient alternative. Although the study is predictive and requires experimental validation, our MEV is an addition to the collection of putative antigens ready for future vaccine trials. Thus, we hope that our work will stimulate more efforts toward developing vaccines against reemerging mpox virus. Notwithstanding, the predictive nature of this study necessitates the in vitro and in vivo validation of the efficacy and safety of the designed MEV.
Highlights
The recent zoonotic mpox virus outbreak has spread to about 110 endemic and nonendemic nations.
Immunoinformatics was employed to design a stable 76.4 kDa chimeric vaccine to target the zoonotic mpox virus.
Immune simulation of the multiepitope vaccine predicts strong immune response induction and lasting memory.
This work contributed to the mpox virus control through computational design of peptides vaccine.
The resulting chimeric vaccine candidate is a 76.4 kDa protein that is predicted to adopt a stable 3D conformation.
The designed hypothetical antigen represents a valuable addition to the collection of prospective vaccine candidates and holds promise for future development and trials aimed at combating the reemerging threat of mpox disease.
Author contributions
EO Balogun conceived the basic idea. EO Balogun, G Chang, AU Danazumi, SI Gital and LBS Dibba contributed to the design of the study. AU Danazumi, SI Gital and LBS Dibba performed the analysis and prepared the initial draft. GI Joseph, OA Adepoju and B Ibrahim reviewed the critical analysis and helped in draft preparation. EO Balogun and G Chang provided overall supervision of the study, interpreted the data and reviewed the manuscript critically. All authors contributed to the manuscript revision, read and approved the final manuscript.
Financial disclosure
The authors have no financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Writing disclosure
No writing assistance was utilized in the production of this manuscript.
Acknowledgments
The authors also acknowledge the support of the University of Groningen Peregrine for providing access to computational resources
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.future-science.com/doi/suppl/10.4155/fdd-2023-0013
References
- European Centre for Disease Prevention and Control. Risk assessment: monkeypox multi-country outbreak. (2022). www.ecdc.europa.eu/en/publications-data/risk-assessment-monkeypox-multi-country-outbreak
- AdepojuOA, AfinowiOA, TauheedAMet al. Multisectoral perspectives on global warming and vector-borne diseases: a focus on southern Europe. Curr. Trop. Med. Rep. 10(2) (2023).
- WHO. Mpox (monkeypox). (2023). www.who.int/news-room/fact-sheets/detail/monkeypox
- CDC. Technical report 4: multi-national mpox outbreak, United States, 2022. (2023). www.cdc.gov/poxvirus/mpox/cases-data/technical-report/report-4.html
- ManesNP, EstepRD, MottazHMet al. Comparative proteomics of human monkeypox and vaccinia intracellular mature and extracellular enveloped virions. J. Proteome Res. 7(3), 960–968 (2008).
- KugelmanJR, JohnstonSC, MulembakaniPMet al. Genomic variability of monkeypox virus among humans, Democratic Republic of the Congo. Emerg. Infect. Dis. 20(2), 232–239 (2014).
- Yinka-OgunleyeA, ArunaO, DalhatMet al. Outbreak of human monkeypox in Nigeria in 2017–18: a clinical and epidemiological report. Lancet Infect. Dis. 19(8), 872–879 (2019).
- BeerEM, BhargaviRao V. A systematic review of the epidemiology of human monkeypox outbreaks and implications for outbreak strategy. PLoS Negl. Trop. Dis. 13(10), e0007791 (2019).
- KisaluNK, MokiliJL. Toward understanding the outcomes of monkeypox infection in human pregnancy. J. Infect. Dis. 216(7), 795–797 (2017).
- NguyenPY, AjisegiriWS, CostantinoV, ChughtaiAA, MacIntyreCR. Reemergence of human monkeypox and declining population immunity in the context of urbanization, Nigeria, 2017–2020. Emerg. Infect. Dis. 27(4), 1007–1014 (2021).
- LapaD, CarlettiF, MazzottaVet al. Monkeypox virus isolation from a semen sample collected in the early phase of infection in a patient with prolonged seminal viral shedding. Lancet Infect. Dis. 22(9), 1267–1269 (2022).
- FinePEM, JezekZ, GrabB, DixonH. The transmission potential of monkeypox virus in human populations. Int. J. Epidemiol. 17(3), 643–650 (1988).
- PolandGA, KennedyRB, ToshPK. Prevention of monkeypox with vaccines: a rapid review. Lancet Infect. Dis. 22(12), e349–e358 (2022).
- PetersenE, KanteleA, KoopmansMet al. Human monkeypox: epidemiologic and clinical characteristics, diagnosis, and prevention. Infect. Dis. Clin. North Am. 33(4), 1027–1043 (2019).
- DanazumiAU, IliyasuGital S, IdrisS, BSDibba L, BalogunEO, GórnaMW. Immunoinformatic design of a putative multi-epitope vaccine candidate against Trypanosoma brucei gambiense. Comput. Struct. Biotechnol. J. 20 (2022).
- KarT, NarsariaU, BasakSet al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 10(1), 1–24 (2020).
- AzizS, AlmajhdiFN, WaqasMet al. Contriving multi-epitope vaccine ensemble for monkeypox disease using an immunoinformatics approach. Front. Immunol. 13, 6148 (2022).
- ImranMA, IslamMR, SahaA, FerdouseeS, MishuMA, GhoshA. Development of multi-epitope based subunit vaccine against Crimean–Congo hemorrhagic fever virus using reverse vaccinology approach. Int. J. Pept. Res. Ther. 28(4), 1–20 (2022).
- GrossS, LennerzV, GalleraniEet al. Short peptide vaccine induces CD4+ T helper cells in patients with different solid cancers. Cancer Immunol. Res. 4(1), 18–25 (2016).
- LennerzV, GrossS, GalleraniEet al. Immunologic response to the survivin-derived multi-epitope vaccine EMD640744 in patients with advanced solid tumors. Cancer Immunol. Immunother. 63(4), 381–394 (2014).
- AkbariE, SeyedinkhorasaniM, BolhassaniA. Conserved multiepitope vaccine constructs: a potent HIV-1 therapeutic vaccine in clinical trials. Braz. J. Infect. Dis. 27(3), doi:10.1016/j.bjid.2023.102774. (2023) ( Online).
- AimanS, AlhamhoomY, AliFet al. Multi-epitope chimeric vaccine design against emerging monkeypox virus via reverse vaccinology techniques – a bioinformatics and immunoinformatics approach. Front. Immunol. 13, 4645 (2022).
- ShantierSW, MustafaMI, AbdelmoneimAH, FadlHA, ElbagerSG, MakhawiAM. Novel multi epitope-based vaccine against monkeypox virus: vaccinomic approach. Sci. Rep. 12(1), 1–17 (2022).
- HallgrenJ, TsirigosKD, ArmenterosJJAet al. DeepTMHMM. (2021). https://biolib.com/DTU/DeepTMHMM/
- AndreattaM, NielsenM. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32(4), 511–517 (2016).
- JensenKK, AndreattaM, MarcatiliPet al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154(3), 394–406 (2018).
- JespersenMC, PetersB, NielsenM, MarcatiliP. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 45(W1), W24–W29 (2017).
- DoytchinovaIA, FlowerDR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8(1), 1–7 (2007).
- DimitrovI, FlowerDR, DoytchinovaI. AllerTOP – a server for in silico prediction of allergens. BMC Bioinformatics 14(Suppl. 6), S4 (2013).
- DhandaSK, VirP, RaghavaGPS. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 8(1), 1–15 (2013).
- BaekM, DiMaioF, AnishchenkoIet al. Accurate prediction of protein structures and interactions using a 3-track neural network. Science 373(6557), 871 (2021).
- KoJ, ParkH, HeoL, SeokC. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 40(W1), W294–W297 (2012).
- Lindahl, Abraham, Hess, Spoelvan der. GROMACS 2021 manual. (2021). https://zenodo.org/record/4457591
- PonomarenkoJ, BuiHH, LiWet al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 9(1), 1–8 (2008).
- GasteigerE, HooglandC, GattikerAet al. Protein identification and analysis tools on the Expasy server. In: The Proteomics Protocols Handbook. Humana, NJ, USA, 571–607 (2005). https://link.springer.com/protocol/10.1385/1-59259-890-0:571
- KozakovD, HallDR, XiaBet al. The ClusPro web server for protein–protein docking. Nat. Protoc. 12(2), 255 (2017).
- PriceDJ, BrooksCL. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 121(20), 10096 (2004).
- ParrinelloM, RahmanA. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52(12), 7182 (1998).
- BussiG, DonadioD, ParrinelloM. Canonical sampling through velocity rescaling. J. Chem. Phys. 126(1) (2007).
- WangE, SunH, WangJet al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 119(16), 9478–9508 (2019).
- RapinN, LundO, BernaschiM, CastiglioneF. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLOS ONE 5(4), 9862 (2010).
- GroteA, HillerK, ScheerMet al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33(Suppl. 2), W526–W531 (2005).
- Tahirul Qamar M, RehmanA, TusleemKet al. Designing of a next generation multiepitope based vaccine (MEV) against SARS-CoV-2: immunoinformatics and in silico approaches. PLOS ONE 15(12), e0244176 (2020).
- CartyM, BowieAG. Recent insights into the role of Toll-like receptors in viral infection. Clin. Exp. Immunol. 161(3), 397 (2010).
- OtuA, EbensoB, WalleyJ, BarcelóJM, OchuCL. Global human monkeypox outbreak: atypical presentation demanding urgent public health action. Lancet Microbe 3(8), e554–e555 (2022).
- DanazumiAU, UmarHI. You must be flexible enough to be trained, Mr. Dynamics simulator. Mol. Divers. 1–3, doi:10.1007/s11030-023-10689-5 (2023) ( Online).
- SklenovskáN, Van RanstM. Emergence of monkeypox as the most important orthopoxvirus infection in humans. Front. Public Health. 6, 241 (2018).
- ZhangL. Multi-epitope vaccines: a promising strategy against tumors and viral infections. Cell. Mol. Immunol. 15(2), 182–184 (2017).
- TostaSF de O, PassosMS, KatoRet al. Multi-epitope based vaccine against yellow fever virus applying immunoinformatics approaches. J. Biomol. Struct. Dyn. 39(1), 219–235 (2021).
- ChauhanV, RungtaT, GoyalK, SinghMP. Designing a multi-epitope based vaccine to combat Kaposi sarcoma utilizing immunoinformatics approach. Sci. Rep. 9(1), doi:10.1038/s41598-019-39299-8 (2019) ( Online).
- ToledoH, BalyA, CastroOet al. A phase I clinical trial of a multi-epitope polypeptide TAB9 combined with Montanide ISA 720 adjuvant in non-HIV-1 infected human volunteers. Vaccine 19(30), 4328–4336 (2001).
- MolteniC, ForniD, CaglianiR, MozziA, ClericiM, SironiM. Evolution of the orthopoxvirus core genome. Virus Res. 323, 198975 (2023).
- WriggersW, ChakravartyS, JenningsPA. Control of protein functional dynamics by peptide linkers. Biopolymers 80(6), 736–746 (2005).
- ChenX, ZaroJL, ShenWC. Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev. 65(10), 1357–1369 (2013).
- SartoriusR, TrovatoM, MancoR, D’ApiceL, DeBerardinis P. Exploiting viral sensing mediated by Toll-like receptors to design innovative vaccines. NPJ Vaccines 6(1), 1–15 (2021).
- ToturaAL, WhitmoreA, AgnihothramSet al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio 6(3), 1–14 (2015).
- AdamuRM, IbrahimB, IbrahimMA, BalogunEO. Identification of megacerotonic acid and a quinazoline derivative from Universal Natural Product Database as potential inhibitors of Trypanosoma brucei brucei alternative oxidase: molecular docking, molecular dynamic simulation and MM/PBSA analysis. J. Biomol. Struct. Dyn. 41(1), 45–54 (2023).
- DanazumiAU, BalogunEO. Microsecond-long simulation reveals the molecular mechanism for the dual inhibition of falcipain-2 and falcipain-3 by antimalarial lead compounds. Front. Mol. Biosci. 9, doi:10.3389/fmolb.2022.1070080 (2022) ( Online).
- TanC, ZhuF, PanP, WuA, LiC. Development of multi-epitope vaccines against the monkeypox virus based on envelope proteins using immunoinformatics approaches. Front. Immunol. 14, 1112816 (2023).
- PhongsisayV, IizasaE, HaraH, YoshidaH. Evidence for TLR4 and FcRγ-CARD9 activation by cholera toxin B subunit and its direct bindings to TREM2 and LMIR5 receptors. Mol. Immunol. 66(2), 463–471 (2015).