
Abstract
Here, we describe our action plan for hit identification (APHID) that guides the process of hit triage, with elimination of less tractable hits and retention of more tractable hits. We exemplify the process with reference to our high-throughput screening (HTS) campaign against the enzyme, KAT6A, that resulted in successful identification of a tractable hit. We hope that APHID could serve as a useful, concise and digestible guide for those involved in HTS and hit triage, especially those that are relatively new to this exciting and continually evolving technology.
High-throughput screening (HTS) has become among the most successful sources of first-in-class US FDA-approved drugs [Citation1]. However, such stellar outcomes can only be realized after intensive medicinal chemistry optimization, and this endeavor in turn can only be successful if optimizable screening hits are identified in the first place. In the context of target-based HTS, primary hits are typically confirmed by repeating the primary assay on cherry-picked actives and obtaining a dose–response curve. However, even here, it is common that only a minority of compounds among the pool of confirmed primary screening hits are actually tractable against that specific target [Citation2]. The term tractable here refers to a compound that yields to a rational structure–activity relationship (SAR) and optimization, whereas an intractable compound – for what can be a very large number of different reasons as outlined below – does not. The process of identifying such compounds can be called hit triage. Far from being passive, this is a very active process and is key to setting up successful target-based drug discovery.
At the Australian Translational Medicinal Chemistry Facility (ATMCF) [Citation3], we have implemented an action plan for hit identification (APHID) that is useful to guide the process of hit triage. As shown in , a confirmed screening hit should be able to withstand considerable interrogation before it reveals itself to be an optimizable hit.
APHID: Action plan for hit identification; HTS: High-throughput screening; PAINS: Pan-assay interference compounds; SAR: Structure–activity relationship.
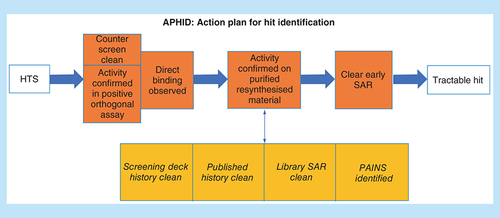
This figure describes an approximate order, and in general one would expect counter screens and positive orthogonal assays to be followed in quick succession by direct-binding assays, resynthesis and purification, then SAR by analogue and focused SAR. At any stage, analysis of screening deck and published hit history, library SAR and pan-assay interference compounds (PAINs) analysis could be undertaken in parallel. However this routine need not be implemented intransigently and may change depending upon the context of the program. For example, assay order is influenced by the capacity of the constituent assays, their accuracy and in particular their false-positive rate in cases where large numbers or large costs are involved.
Here, we discuss APHID with reference to a particular target-based HTS campaign for the discovery of inhibitors of the histone acetyltransferase (HAT), KAT6A.
Lysine acetyltransferases (KATs) acetylate the side chain amino group of specific lysine residues in proteins and alter the molecular recognition properties of the newly acetylated protein in so doing. All KATs use acetyl-coenzyme A (Ac-CoA) as the primary acetylation source. A significant subset of KATs acts on histones within cell nuclei. The net effect of action by such HATs is to neutralize the positive charge on those specific histone lysine side chains, removing electrostatic attractive forces with DNA phosphate groups, causing chromatin to adopt a more open conformation and allowing regional access to DNA by regulatory proteins, thereby promoting transcription.
Aberrant KAT function has been widely implicated in many disease states [Citation4] but in particular cancer [Citation5]. There has consequently been significant effort expended in order to discover KAT inhibitors but in general only PAINs [Citation6–8] and associated promiscuous compounds have been unearthed [Citation9,Citation10].
Of the KAT superfamily, HATs comprise a dominant class, and of HATs, MYST HATs – HATs that contain a MYST domain – are a dominant family, the other significant two being GNAT and PCB/p300 [Citation5]. MYST HATs are the largest of the three and represent about a third of the HATs in the human genome [Citation5]. Within the class of Type A KATs, MYST HATs comprise KAT5 (Tip60), KAT6A (MOZ/MYST3), KAT6B (MORF/QKF/MYST4), KAT7 (HBO1/MYST2) and KAT 8 (MOF/MYST1) [Citation11].
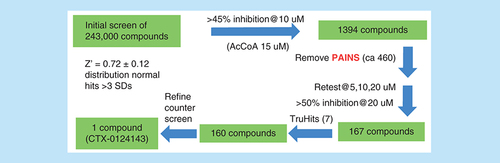
We have a focus on KAT6A. The action of KAT6A in histone acetylation results in stimulating transcription of target genes maintaining cells in the cell cycle, including genes that repress INK4A/ARF-induced senescence [Citation12]. We were interested in developing small-molecule inhibitors of KAT6A in order to induce cellular senescence. There is significant recent interest in this cellular response for the treatment of certain cancers [Citation13]. We developed a miniaturized assay biased toward detection of Ac-CoA-competitive KAT6A inhibitors that used AlphaScreen™ technology. This technology represents a robust and widely used platform for undertaking HTS. We selected two of our HTS diversity libraries that we call Stage 1 and Stage 5 as described elsewhere [Citation14] and undertook HTS of the combined 240,000 compounds, only to determine that identified hits were predominantly readout modulators (as we have defined elsewhere) [Citation15]. After extensive triage, schematically shown below, ultimately only a single hit remained, called CTX-0124143 (). Certain details of this triage have been previously reported [Citation16], but not in the context of our more recently developed APHID, as now described.
AcCoA: Acetyl coenzyme A; PAINS: Pan assay interference compounds; SD: Standard deviation.
Counter screen clean
A well-designed counter screen is one of the most useful ways to remove uninteresting active compounds, and considerable thought may be required for the best counter screen design. Assuming that the counter screen is based on similar technology used for the initial HTS, it is likely to be able to prosecute large numbers of compounds. This is important because a 1% hit rate in an HTS of multiple-millions of compounds can yield tens of thousands of hits and so a well-designed counter screen may be one of the most important early triage parameters to establish. AlphaScreen assays often utilize the TrueHits counter screen, where the bound interface of interest is replaced by a biotin–avidin interaction. In this way, any compounds that interfere with the signaling technology, such as singlet oxygen quenching, will be detected through maintenance of a positive signal in the counter screen. In our case this was useful, but we determined that there were also a large number of compounds that gave a positive readout by interfering with the interaction between the anti-acetylysine antibody and the acetylated histone peptide; that is, they were effectively Ac-Lys mimetics. Use of a counter screen that was identical in all facets to the primary assay but contained no KAT6A, and utilizing pre-acetylated histone peptide, successfully weeded out these compounds, as we have described in more detail elsewhere [Citation16]. CTX-0124143 was not active in these AlphaScreen counter screen assays [Citation16]. Reference to our recently described classification [Citation15] points to such compounds as being readout modulators falling under the subcategory of interference with the coupled assay step, the specific classification tree being: Readout modulators → Interference with the assay's specific or general technology → Interference with coupled assay step. Other readout modulators can interfere through fluorescence absorption or quenching, chelation interference in anchoring interaction such as hexaHis–nickel, reactivity with signal chemistry through singlet oxygen quenching, metal reaction-chelation, thiol reactivity and redox cycling. Assay readout can also modulated via target interaction but in a nonproductive sense through similar as well as expanded mechanisms, such as hydrophobicity-driven burial, colloidal interaction and denaturation. Appropriately designed counter screens can identify these interference compounds [Citation15].
Activity confirmed in positive orthogonal assay
If a compound is confirmed to be active in an assay that is designed to test the same mechanism of action, but which is set up differently and uses different technology, the chances are greatly increased that the compound in question is on-target and not interfering in assay signaling. Such positive orthogonal assays may be much lower in throughput, but in other cases might be higher throughput, depending upon the technology involved. For example, a positive orthogonal assay for a large set of AlphaScreen primary assay screening hits could usefully be based on a higher throughput platform such as fluorescent polarization. Although APHID as described is directed toward target-based screening, an example in the context of a phenotypic screening hit involving a luciferase-mediated readout would be the recommended use of orthogonal Luc pairs [Citation17]. For our purposes, in addition to AlphaScreen technology utilized for primary screening, CTX-0124143 was confirmed as active in a radiometric assay [Citation16], which in this case could be considered to be a low-throughput positive orthogonal assay. No other compound that passed the counter screen was active in this assay.
Direct binding observed
For a target-based screen, it is obviously an imperative to confirm target engagement, but the importance of this undertaking was recognized relatively late after the establishment of HTS for drug discovery. Today, biophysical interrogation of target engagement is increasingly seen as being an early task to impose and its utility in deprioritization of less useful compounds soon after HTS has been convincingly discussed [Citation18]. Beyond confirmation of direct binding, subsequent elucidation of a specific or nonspecific binding mode, and in the case of the former, relevant location, adds considerable value to a program and is enabled through the use of technologies such as probe displacement assays and nuclear magnetic resonance spectroscopic approaches. An impressive array of sophisticated biophysical methods approaches for confirmation of direct binding has been discussed in detail [Citation19]. In academic settings, it is recognized that access to best-practice biophysical interrogation may be limited by the resources available. Surface plasmon resonance is a useful technique that readily reveals whether binding is irreversible, but there are alternatives such as jump dilution assays and several others that can be applied for such purposes [Citation20]. When a compound identified through HTS covalently modifies a protein, even if it does so with 1:1 stoichiometry in an active site or binding interface, our view is that the risk of optimization failure is generally too high to warrant progression, although it could be a useful structural biology tool [Citation8]. In our case, surface plasmon resonance kinetic analysis of CTX-0124143 confirmed direct and reversible binding to KAT6A at a level congruent with biochemical assay data [Citation16]. Of particular relevance to this section, we note that an excellent treatise on hit triage, biophysical interrogation, and the importance of interdisciplinary collaboration between chemists and biologists has been published and is strongly recommended for further reading [Citation21].
Activity confirmed on purified, resynthesized material
Resynthesis and purification of confirmed hits removed significant numbers of compounds that lost bioactivity [Citation16], but the activity of resynthesized CTX-0124143 was confirmed in the AlphaScreen assay that had been utilized for the primary screen, returning an IC50 value of 0.97 μM.
Screening deck history clean
Substructure searches retrieved 12 N-(benzoyl)-N’-(phenylsulfonyl)hydrazine (ArSO2NHNHC(=O)Ar) analogues in the Stage 1 Walter and Eliza Hall Institute (WEHI) HTS library of 93,000 compounds. This core is not a PAINs moiety, but nonetheless all 12 analogues were subjected to additional analysis for PAINs behavior, with assignment of a screening enrichment value calculated by a process previously described [Citation7,Citation8] As shown in , only one analogue had registered as a hit in only one of the six HTS campaigns used to originally define PAINs. This is a very clean profile and the low enrichment value of 0% is indicative of a compound class that is not obviously prone to assay interference.
Table 1. Propensity for N-(benzoyl)-N’-(phenylsulfonyl)hydrazine (ArSO2NHNHC(=O)Ar) analogues (12 in total) to register as hits in six early high-throughput screening campaigns.
HTS hit CTX-0124143 itself was discovered from screening the Stage 5 (CRC-CTX) library of 153,000 compounds. This compound and nine other N-(benzoyl)-N’-(phenylsulfonyl)hydrazine (ArSO2NHNHC(=O)Ar) analogues have been tested as part of nine HTS campaigns, involving different targets and different assay technologies (1 CellLux, 4 AlphaScreen, 3 fluorescence polarization, 1 luminescence). Only CTX-0124143 and a close analogue (CTX-0123508) displayed any activity and this was only in the KAT6A assay.
Literature history clean
As depicted below, a substructure search of the SciFinder database undertaken on November 1st (2016) revealed a large number of commercially available N-(benzoyl)-N’-(phenylsulfonyl)hydrazines (1484). Of these, only 51 compounds were associated with references for biological activity and this corresponded to 24 references ().

Of the 24 references, only one related to a screening hit with reported biological activity. This compound was unearthed as a weak inhibitor of the cytosolic form of human branched-chain amino acid transferase [Citation22].
An important point that needs to be made is that a rare or new compound or chemotype may be associated with very little literature and may be assumed to be benign, but this is not necessarily the case, because it simply may not have been studied. In such cases, compelling evidence has been provided by Nissink and Blackburn that physicochemical properties can be usefully predictive, with high lipophilicity and solubility less than 10 μM being strongly implicated in directing frequent hitter behavior [Citation23].
Library SAR clean
The KAT6A activity for the total of 22 N-(benzoyl)-N’-(phenylsulfonyl)hydrazine analogs from the Stage 1 and Stage 5 HTS libraries was retrieved and is reported in .
Table 2. Structure–activity relationship for the target (here KAT6A) from the high-throughput screening deck and the structure of CTX-0124143.
As can be seen, the SAR for these compounds is sharply negative. Considering that most of these are relatively divergent analogues, containing at least two structural differences from CTX-0124143, these data are very reassuring. Perhaps even more reassuring is that the three compounds WEHI-0101079, WEHI-0076686, CTX-0123508 represent quite close analogues with all bearing the 3,4-naphthalenyl left-hand side and differing only in the right-hand side phenyl substituents yet are essentially inactive. This is reminiscent of an activity cliff and is aligned with the notion of a ligand that is inherently optimizable toward increased potency and selectivity. Having said that, on occasions where two entirely homologous compounds display inexplicably disparate bioactivities, such as a chloro to bromo replacement, caution is required and investigation of, for example, metal contamination in one of the samples would be judicious.
Of note, the closest analogue, CTX-0123508, which has only one difference with replacement of the 2-fluoro with a 2-chloro substituent, registered weak activity at the limit of the assay with an IC50 value of approximately 50% inhibition at the highest concentration tested (125 μM). We have commented elsewhere on the broader implication of such sharp negative SAR, activity cliffs and the relative ease with which whole classes of bioactive compounds may be missed if insufficient analogue representation is present in the screening deck. Further to this, our previous view that the diversity of lead-like screening space can be captured within 350,000 compounds [Citation14] necessarily needs to be broadened to 1.4 million compounds to account for such activity cliffs and the observation by Yvonne Martin that an analogue set of four compounds more than 90% similar to each other was required to maximize the chances of capturing just one with bioactivity [Citation24,Citation25]. In we list the similarity computed by comparison of molecular fingerprints, reflecting our library design that predominantly excludes the large numbers of very similar analogues (>0.90) in order to capture maximum diversity in available chemical space [Citation14].
Pan-assay interference compounds
As indicated in , as part of the hit triage we removed PAINs using our PAINs electronic filters. As all our HTS libraries after Stage 1 were prefiltered of PAINs prior to purchase, all 460 PAINs that were removed were from the Stage 1 library upon which PAINs were defined. As an exercise purely driven by curiosity, we extracted from this set the top 52 compounds that historically had been the cleanest in all HTS campaigns run to that point, and titrated them to measure an IC50 against KAT6A. We determined that 21 were inactive, 20 were active in the counter screen, but that 11 were active and apparently selective. These 11 are shown in .
Table 3. Pan-assay interference compound that registered as selective high-throughput screening hits against KAT6A.
As can be seen, entries 1–4 represent compounds recognized as bearing the quinone-like PAINs motif. We have previously discussed reasons why quinone reactivity could give rise to PAINs behavior [Citation2,Citation7,Citation8]. Entries 1–4 nicely illustrate how PAINs compounds often come with other features and in all cases extensive conjugation would likely give rise to highly colored compounds that perhaps also possess additional redox properties. It is not certain why certain 3-alkylaminoindoles such as those for entries 5 and 6 are associated with PAINs behavior but oxidative decomposition products may play a role [Citation7,Citation8]. Entry 7 contains an amidothiophene that may have interference redox activity [Citation7,Citation8], though in the case the additional succinimide is a clear electrophile. Entry 8 represents alkylidene barbiturates that are readily recognized PAINs and Michael acceptors [Citation7,Citation8]. Entry 9 represents an unusual PAIN where activity disappears on purification [Citation7,Citation8]. The same is the case for the well-known arylpyrrole carboxylic acids (entries 10 and 11) [Citation7,Citation8] that likely form anionic interference polymers [Citation26] and perhaps the same is the case for the tetrahydroquinoline carboxylic acid PAINs (entry 9).
It is instructive to look at the detail of the AlphaScreen assay used for detection of KAT6A inhibitors, this is shown in . Just as is the case for most assays, interference could arise through a variety of mechanisms, such as absorption of irradiated or emitted light, interference with the photochemistry, reaction with singlet oxygen, interference with the various interfaces represented by the acetylated histone peptide, antibody, protein A.
AcCoA: Acetyl coenzyme A.
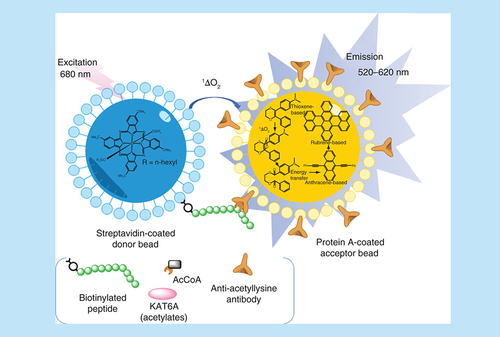
The counter screen differs in that no KAT6A enzyme is added and fully acetylated histone peptide is used. That the PAINs in are apparently selective for KAT6A inhibition, and therefore not interfering in assay technology, illustrates their subversive, misleading and distracting properties. It is possible that reactive PAINs (most obviously entries 4, 7 and 8 in ) could label KAT6A catalytic machinery or elsewhere and destroy its function. It is also possible that PAINs containing interfering contaminants such as metals or anionic polymers could selectively block or coat the KAT6A active site. We did not undertake any further investigation of these compounds and so it is not certain exactly how any interference mechanisms may be operating. Pursuit of this information may be viewed as intellectually engaging but in our view the chances are very high that it will not help advance the program. The point of this exercise is to illustrate that by removing the distraction of PAINs from further consideration in an initial pass, and then implementing APHID on the remaining preferred starting points, a project has a greater chance of successful outcomes. That said, there are occasions where a compound excluded by PAINs filters warrants further investigation. The reasons can be many, and vary from obvious to more subtle in nature, as we have discussed in detail elsewhere [Citation15]. A key danger is a PAINs compound such as that shown in (entry 8) which is a Michael acceptor. In such cases where active site covalent labeling is a plausible mechanism for signal readout, such a compound will progress through all APHID gates, but it does not mean that the compound will be optimizable. In our experience is that in all such cases SAR becomes uninterpretable and the compound nonprogressable. Structural recognition alone is the best arbiter for hit removal of these types of PAINs.
Clear early SAR
Any assay hit that has not raised any alarm bells while progressing through all stages to this point is looking in pretty good shape. Nevertheless, as we have previously emphasized [Citation27], strong SAR is the final arbiter on this matter, and for this reason we include early SAR as part of hit identification. It is unusual for adequate SAR to be available by analogue [Citation27] and design and synthesis of a focus set is generally necessary, though we make this statement with academic HTS libraries in mind. This undertaking does not need to be too onerous and in our view, fewer than a few dozen well chosen analogues are usually sufficient to satisfy this requirement, with concomitant location of a more potent analogue. From there, it is simply a matter of the effort expended to develop compounds with nanomolar levels of potency. Large corporate libraries may contain appropriate analogues to furnish early SAR. Our libraries were designed with diversity in mind to maximize initial capture of bioactivity, such that most SAR-relevant compounds would be obtained as a subsequent step from vendors after substructure searching. The result of this procedure is shown in & .
Table 4. Structure–activity relationship by analogue for the target (here KAT6A)Table Footnote†.
Shown in are substituted benzoyl derivatives. Here more meaningful SAR is directly evident. This is shown for illustrative purposes and will not be discussed in detail but in general one can observe an appropriately undulating activity profile with peaks and troughs as expected for a more defined analogue set. Note that the fully unsubstituted analogue, which is the best template on which to judge a positive or negative influence of an incoming substituent, was not commercially available. In for our search of commercially available analogues we have kept the right hand side (RHS) fluorobenzoyl and just varied the left hand side (LHS) ring. Here, too, there are signs of clear early SAR with distinct preferences and dislikes at particular positions, although none were better than the naphthyl group.
Table 5. Structure–activity relationship by analogue for the target (here KAT6A)Table Footnote†.
Finally, we explored some broader probing of the RHS aryl group and the resulting commercially available compounds are shown in . All these compounds were inactive. The point of including similarity values is to illustrate that their use in establishing a focus set for SAR purposes is very limited.
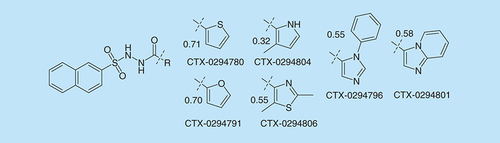
The results of these investigations were signs of clear early SAR. With focused medicinal chemistry, this became strong SAR with significant optimization towards therapeutically-relevant levels of activity, as we have recently described for CTX-0124143 [Citation28].
General comments on assay optimization
In this discussion of hit triage we have not commented on the process of prior assay optimization itself, the scope of which is outside the remit of this article. However, its importance cannot be underestimated, whether it is the addition of detergent, chelators or finding the right buffer. In one HTS campaign, for example, successful outcomes in our Bcl-XL program [Citation29] would not have been possible without the discovery that addition of α-casein to the buffer was uniquely suited to improving the signal-to-noise (S/N) ratio to useful levels by adsorbing nonspecific lipophilics. Assay precision is obviously key to successful primary HTS but also vital for subsequent SAR support. In our experience as medicinal chemists, assays optimized to reliably detect as little as a threefold difference in bioactivity, for example, an IC50 of 0.10 μM for one analogue compared with an IC50 of 0.30 μM for another analogue, even if tested at different times, can make a vast difference to SAR interpretation and the rate of progress of an optimization program.
In this article, we do not delve significantly into the influence of the target nature and the magnitude of the HTS undertaken and so it is worth noting that a screen of 1 million compounds will clearly return larger number of primary hits than a screen 20,000 compounds and generate more work at every stage of APHID. The nature of the target will also strongly shape the composition of the set of hits. A protein–protein interaction may be expected to return a cleaner set of hits that an enzyme competition assay and in the context of KAT6A, we have previously commented that part of the reason why KAT inhibitor discovery has been relatively unsuccessful to date could be the noisy nature of the hit sets returned after HTS for this enzyme class [Citation9]. However, our retrospective analyses convince us that whether a protein–protein inhibition HTS, such as our Bcl-XL/BH3 example, or an enzyme inhibition HTS, such as our KAT6A case study discussed herein, application of APHID provides for a streamlined and most efficient approach to hit triage. We also do not discuss fragment-based triage, which houses broadly similar principles but hallmarked by screening very few compounds at very high concentrations with a strong focus on biophysical interrogation.
At the other end of APHID, we have not extended our discussion to include assessment of cell-based activity because we regard this as outside the scope of hit triage in the sense described herein. Although cell-based activity can be used as part of hit triage to identify compounds with intrinsically better cell-based activity and lower serum binding, there is a strong danger that one will select for compounds with off-target activity. In contrast, target-based HTS hits with low-micromolar levels of activity are typically inactive in most cell-based assays. In our experience, optimization to less than 500 nM IC50 values is generally necessary in order to dial-in cell-based activity and in some cases such as the field of Mcl-1 antagonists [Citation30], sub nanomolar levels of target-based inhibition in vitro may be required before significant cell-based activity is observed.
Conclusion
By implementing APHID as part of the hit triage process one can be confident that subsequent medicinal chemistry optimization will have a greater chance of being successful. An added benefit is that APHID necessarily requires input from medicinal chemists, since such experience is vital for substructure searches, whereas in contrast similarity searches – that can be undertaken with little thought – are generally of far less utility for elucidation of SAR [Citation27]. Furthermore, APHID increases the objectivity of hit judgment and may allow for more constructive discussions between the chemists and biologists in the program. Finally, the removal of the distraction of less optimizable compounds allows focus on the more optimizable compounds and hence greater return for effort. The misleading KAT inhibitors reported to date fail APHID at almost every level, generally with few efforts to confirm activity on purified, resynthesized material, or in an orthogonal positive assay, not having been tested in an appropriate counter screen, not having been tested in a direct binding assay, no screening deck history assessed, no literature history assessed, no library SAR analyzed and neither any optimization nor SAR demonstrated [Citation10]. In contrast, CTX-0124143 convincingly passes APHID requirements. Indeed, APHID was established partly in response to the difficulties encountered during the hit triage process that ultimately led to successful identification of CTX-0124143. In particular, had we established an appropriate counter screen or direct-binding assay or both earlier in the program and in line with APHID, CTX-0124132 would have been identified earlier and with greater efficiency because the initial set of confirmed hits was beset with assay readout modulators and other nuisance compounds. Once CTX-0124132 was identified, medicinal chemistry optimization smoothly led to single-digit nanomolar Ac-CoA-competitive inhibitors of KAT6A that we showed bound reversibly to KAT6A at the Ac-CoA binding site, potently inhibiting KAT6A in biochemical assays, inactivating its catalytic machinery, inducing cellular senescence as a consequence and successfully treating mouse models of lymphoma. These research outcomes were published in the journal, Nature, on the 1st August 2018 [Citation31].
Future perspective
Spurred by widespread realization that today's first-in-class drugs typically have as their starting point hit discovery facilitated by HTS, technological advances in this field continue apace. Relatively newly revived entries such as DNA encoded libraries, increasingly enabled by new DNA-compatible chemistries, and that allow for the construction and bioassay of vast compound libraries that previously were imaginable comprising trillions of compounds, are causing considerable excitement [Citation32].
It is human nature to find new alternatives to a current paradigm alluring, but one should not lose sight of the fact that traditional HTS is also continuing to be refined, such that screening of as few as 100,000 compounds – or even fewer – in a well-designed compound library stands a good chance of finding optimizable starting points for many biological targets. This is complemented by the fact that today and looking forward there are increasingly sophisticated assay technologies available to prosecute almost any type of protein modulation. In particular, advances in direct binding technologies are particularly exciting and will become increasingly prevalent in HTS and hit triage. Furthermore, continuing advances in medicinal chemistry knowledge and optimization protocols allow for more efficient and smoother progression of screening hits to drug candidates that are less likely to fail in later clinical trials due to compound-related toxicity or pharmacokinetic and efficacy issues. Indeed, exemplification of these facets is becoming manifest in current FDA drug approvals.
This perspective runs counter to another that is sometimes espoused, which is that ‘HTS does not work.’ However, often when HTS is deemed to have failed, it is not that HTS technology has failed, but rather that its execution was suboptimal. Common reasons include that the screening deck is too small for the target difficulty, that the HTS library is poorly designed, that the expectations of high affinity are too great and hence the screening concentration too low, that the intricacies of assay technology and compound behavior are not fully understood, and that PAINs are unearthed as hits that may appear to be selective, may appear to have early SAR, but ultimately go nowhere, but that their subversive behavior is not fully appreciated. Interwoven among all these reasons, and irrespective of whether ‘traditional HTS’ or an alternative approach is involved such as that represented by DNA-encoded libraries, is the inevitability that one ends up with a set of active compounds where non-progressable hits contribute distracting noise to the dataset.
Every drug discovery scientist's dream is to be involved in the discovery of a first-in-class molecule. However, like a game of chess, such a successful end game requires a solid opening and where HTS is involved, well thought-out moves are all the more important. Beyond this, successful translation requires project champions for several key phases. Indeed, shortly after HTS was undertaken, this KAT6A project was judged to be too high risk from both the biology and chemistry standpoint to be continued, and only did so through considerable individual effort. This highlights an advantage of undertaking early medicinal chemistry-led drug discovery in an academic setting. In a risk-averse pharmaceutical company, with or without a champion, this project may have been terminated before enough data could be obtained to make a case. This is not just because there was uncertainty about target validation, but also the hydrazide-core raised alerts for potential toxicity and the combination might represent insurmountable risk. The same comments apply to the Bcl-XL program, where our identified hit was a singleton, with a cLogP over 5, containing a furan, a 2-aminothiazole moiety, a hydrazone and a carboxylic acid and with no cell-based activity. These would likely represent insurmountable risks in a corporate environment but we determined very rapidly there was clear early SAR and that therefore our compound would yield to optimization.
With respect to the broader field of KAT inhibitors, the future is very exciting indeed. Once considered to be undruggable, we believe that KATs other than KAT6A will yield to development of potent and selective inhibitors that bind to the Ac-CoA binding site. This situation is akin to the development of selective kinase inhibitors that bind to the ATP-binding site. Indeed, we have recently designed an analogue of WM-1119 that is selective for the related MYST HAT, KAT7, binding to its Ac-CoA site and efficacious against patient-derived primary acute myeloid leukemia cells, demonstrating for the first time the relevance of KAT7 as a disease-relevant target for acute myeloid leukemia. This work has just been published in Nature [Citation33].
In this article, we describe APHID, an action plan for hit identification, which we hope may operate as a useful guide during the all-important hit triage process, in particular to those future hit-to-lead medicinal chemists new to HTS.
High-throughput screening campaigns typically furnish sets of confirmed hits that comprise intractable hits among tractable hits.
We have outlined an action plan for hit identification (APHID) that can help guide the triage process to eliminate less tractable hits and to reveal those that are inherently more optimizable.
We hope implementation of APHID could be useful others during the process of hit triage, in particular those relatively new to high-throughput screening.
Financial & competing interests disclosure
The National Health and Medical Research Council of Australia (NHMRC) is thanked for the research support (grant numbers 1030704 and 1080146) and fellowship support for JB Baell (2012−2016 senior research fellowship number 1020411, 2017 principal research fellowship number 1117602). Acknowledged is the Australian Federal Government Education Investment Fund Super Science Initiative and the Victorian State Government, Victoria Science Agenda Investment Fund for infrastructure support and the facilities and the scientific and technical assistance of the Australian Translational Medicinal Chemistry Facility (ATMCF), Monash Institute of Pharmaceutical Sciences (MIPS). ATMCF is supported by Therapeutic Innovation Australia (TIA). TIA is supported by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Eder J , SedraniR , WiesmannC. The discovery of first-in-class drugs: origins and evolution. Nat. Rev. Drug Discov.13(8), 577–587 (2014).
- Baell JB . Redox active nuisance screening compounds and their classification. Drug Discov. Today16(17/18), 840–841 (2011).
- The Australian Translational Medicinal Chemistry Facility. https://monash.edu/atmcf
- Farria A , LiW , DentSY. KATs in cancer: functions and therapies. Oncogene34(38), 4901–4913 (2015).
- Simon RP , RobaaD , AlhalabiZ , SipplW , JungM. KATching-up on small molecule modulators of lysine acetyltransferases. J. Med. Chem.59(4), 1249–1270 (2016).
- Baell J , WaltersMA. Chemical con artists foil drug discovery. Nature513(7519), 481–483 (2014).
- Baell JB , HollowayGA. New substructure filters for removal of pan assay interference compounds (PAINs) from screening libraries and for their exclusion in bioassays. J. Med. Chem.53(7), 2719–2740 (2010).
- Baell JB . Observations on screening-based research and some concerning trends in the literature. Future Med. Chem.2(10), 1529–1546 (2010).
- Baell JB , MiaoW. Histone acetyltransferase inhibitors: where art thou?Future Med. Chem.8(13), 1525–1528 (2016).
- Dahlin JL , NelsonKM , StrasserJMet al. Assay interference and off-target liabilities of reported histone acetyltransferase inhibitors. Nat. Commun.8(1), 1527 (2017).
- Voss AK , ThomasT. Histone lysine and genomic targets of histone acetyltransferases in mammals. Bioessays40(10), e1800078 (2018).
- Sheikh BN , PhipsonB , El-SaafinFet al. MOZ (MYST3, KAT6A) inhibits senescence via the INK4A-ARF pathway. Oncogene34(47), 5807–5820 (2015).
- Sieben CJ , SturmlechnerI , vande Sluis B , van DeursenJM. Senescence two-step senescence-focused cancer therapies. Trends Cell Biol.28(9), 723–737 (2018).
- Baell JB . Broad coverage of commercially available lead-like screening space with fewer than 350,000 compounds. J. Chem. Inf. Model.53(1), 39–55 (2013).
- Baell JB , NissinkJWM. Seven year itch: pan-assay interference compounds (PAINs) in 2017 – utility and limitations. ACS Chem. Biol.13(1), 36–44 (2018).
- Falk H , ConnorT , YangHet al. An efficient high-throughput screening method for MYST family acetyltransferases, a new class of epigenetic drug targets. J. Biomol. Screen.16(10), 1196–1205 (2011).
- Auld DS , NarahariJ , HoPet al. Characterization and use of turboluc luciferase as a reporter for high-throughput assays. Biochemistry57(31), 4700–4706 (2018).
- Folmer RH . Integrating biophysics with HTS-driven drug discovery projects. Drug Discov. Today21(3), 491–498 (2016).
- Renaud JP , ChungCW , DanielsonUH , EgnerU , HennigM , HubbardRE , NarH. Biophysics in drug discovery: impact, challenges and opportunities. Nat. Rev. Drug Discov.15(10), 679–698 (2016).
- Dahlin JL , BaellJ , WaltersMA. Assay interference by chemical reactivity. In: Assay Guidance Manual [Internet]. SittampalamGS, CoussensNP, NelsonHet al.et al. ( Eds). Eli Lilly & Company and the National Center for Advancing Translational Sciences, MD, USA (2004).
- Dahlin JL , WaltersMA. The essential roles of chemistry in high-throughput screening triage. Future Med. Chem.6(11), 1265–1290 (2014).
- Hu LY , BoxerPA , KestenSRet al. The design and synthesis of human branched-chain amino acid aminotransferase inhibitors for treatment of neurodegenerative diseases. Bioorg. Med. Chem. Lett.16(9), 2337–2340 (2006).
- Nissink JWM , BlackburnS. Quantification of frequent-hitter behaviour based on historical high-throughput screening data. Future Med. Chem.6(10), 1113–1126 (2014).
- Martin YC , KofronJL , TraphagenLM. Do structurally similar molecules have similar biological activity?J. Med. Chem.45, 4350–4358 (2002).
- Muchmore SW , DebeDA , MetzJTet al. Application of belief theory to similarity data fusion for use in analog searching and lead hopping. J. Chem. Inf. Model.48, 941–948 (2008).
- Zhu W , GrohM , HaupenthalJ , HartmannRW. A detective story in drug discovery: elucidation of a screening artifact reveals polymeric carboxylic acids as potent inhibitors of RNA polymerase. Chem. Eur. J.19(26), 8397–8400 (2013).
- Baell JB . Screening-based-translation of public research encounters painful problems. ACS Med. Chem. Lett.6(3), 229–234 (2015).
- Leaver DJ , ClearyB , NguyenNet al. Discovery of benzoylsulfonohydrazides as potent inhibitors of the histone acetyltransferase KAT6A. J. Med. Chem.62(15), 7146–7159 (2019).
- Lessene G , CzabotarP , SleebsBEet al. Structure-guided design of a selective BCL-XL inhibitor. Nat. Chem. Biol.9(6), 390–397 (2013).
- Leverson JD , ZhangH , ChenJet al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis.6, e1590 (2015).
- Baell JB , LeaverDJ , HermansSJet al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature560(7717), 253–257 (2018).
- Satz AL . What do you get from DNA-encoded libraries?ACS Med. Chem. Lett.9(5), 408–410 (2018).
- MacPherson L , AnokyeJ , YeungMMet al. HBO1 is required for the maintenance of leukaemia stem cells. 577(7789), 266–270 (2020).