
Abstract
Activating mutations in the three human RAS genes, KRAS, NRAS and HRAS, are among the most common oncogenic drivers in human cancers. Covalent KRASG12C inhibitors, which bind to the switch II pocket in the ‘off state’ of KRAS, represent the first direct KRAS drugs that entered human clinical trials. However, the remaining 85% of non-KRASG12C-driven cancers remain undrugged as do NRAS and HRAS and no drugs targeting the ‘on state’ have been discovered so far. The switch I/II pocket is a second pocket for which the nanomolar inhibitor BI-2852 has been discovered. Here, we elucidate inhibitor binding modes in KRAS, NRAS and HRAS on and off and discuss future strategies to drug all RAS isoforms with this one pocket.
Keywords: :
The three human RAS genes KRAS, NRAS and HRAS encode for four highly homologous isoforms (NRAS, HRAS, KRAS4A and KRAS4B). These RAS gene products are all small GTPases that control the level of numerous cell signaling processes implicated in cell growth regulation [Citation1]. Activating mutations in RAS, most commonly at codons 12, 13 and 61, dramatically increase cell signaling and are among the most common oncogenic drivers in human cancers [Citation2]. KRAS is the most frequently mutated oncogene predominantly mutated at positions G12 and G13, with mutation rates of 86–96% in pancreatic cancers [Citation3], 40–54% in colorectal cancers [Citation4] and 27–39% in lung adenocarcinomas [Citation5]. NRAS mutations are predominantly found in melanoma and hematological malignancies [Citation6,Citation7], while for HRAS, mutations in salivary gland and urinary tract cancers [Citation8,Citation9] are the most common. In addition to cancer, germline RAS mutations have been observed in RASopathies. Rare mutations in NRAS at codons 50 and 60 [Citation10] and KRAS at positions 14, 58 and 153 [Citation11] have been reported for Noonan syndrome. A KRAS P34R alteration has been observed for a patient with cardiofaciocutaneous syndrome [Citation11] whereas Costello syndrome is driven by oncogenic HRAS mutations at positions 12 and 13 [Citation12]. The members of the RAS protein family cycle through two different conformational states defined by differential binding to nucleotides which regulate the signaling. In the ‘off’ state (RASOff), RAS proteins are bound to the nucleotide GDP, while in the ‘on’ state (RASOn) they are bound to the nucleotide GTP [Citation13]. Activation from the GDP bound KRASOff to the GTP (or nonhydrolysable analogs) bound KRASOn form is catalyzed by GEF such as SOS1 and SOS2 [Citation14]. The γ-phosphate of GTP keeps two flexible loop regions, switch I and switch II [Citation15] in a compact conformation that induces signal transduction through RAS binding to downstream effectors (e.g., CRAF, PI3K and RALGDS) and further activates GEF catalyzed nucleotide exchange through allosteric GTPase binding to GEFs (e.g., SOS). Hydrolysis of the γ-phosphate of GTP produces KRASOff and causes a conformational change in the switch regions, leading to the formation of an inactive state which is unable to bind to effector molecules [Citation16,Citation17]. RAS itself has intrinsic, but weak GTPase activity that is catalyzed by GTPase activating proteins leading to its inactivation again. As RAS proteins functionally interact with GEFs, GTPase activating proteins and a number of effector proteins they possess a high degree of intrinsic plasticity that enable the formation and adaptation of binding pockets and interfaces to specifically adapt to these interaction partners (Supplementary Figure 1). Single point mutations in RAS proteins alter the specific dynamic of the protein and hence differentially influence various steps of the cycle, leading to increased equilibrium levels of KRASOn and ultimately, via downstream effector pathways, to amplified cell growth.
Pros & cons of RAS’ two pockets
The RAS family of proteins has been notoriously recalcitrant to drug discovery efforts since their discovery as oncogenes in the early 1980s [Citation18] until the recent discovery of two potentially druggable pockets on RAS (A & B). The Shokat group discovered a pocket on RAS positioned above the switch II loop (SII-pocket) using covalently linked small molecules [Citation17,Citation19]. The SII-pocket is closed in the active form of KRAS and can only be observed in inactive KRAS. Covalent inhibitors binding to the SII-pocket have successfully reached human clinical trials [Citation20,Citation21] but adaptive resistance mechanisms have already been elucidated [Citation22]. Drugs targeting KRASOff are unlikely to be successful on KRAS mutants such as KRASG12R which do not cycle through the inactive form [Citation23].
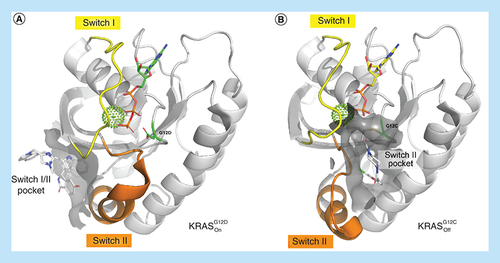
Overview of the overall structure of KRAS highlighting the switch I (yellow) and switch II regions (orange). The respective switch pockets are shown with surface representation around the ligand-binding sites. (A) Ribbon plot of KRASG12D in the active conformation. The switch I/II region is highlighted around BI-2852 (PDB ID: 6GJ8). (B) Ribbon plot of KRASG12C in the inactive conformation with an acrylamide ligand covalently bound to amino acid G12C in the switch II pocket (PDB ID: 4M22).
PDB: Protein Data Bank.
Another pocket that has been characterized is situated between the switch I and II regions (SI/II-pocket) of RAS, and is occupied by up to three distinct water molecules as seen in several apo RASOn crystal structures (PDB-Codes, e.g., 6QUU for KRASG12D, 3OIW for HRASG12V and 5UHV for NRASwt) (B) with water #1 being the most conserved and bound in all three RAS forms. In contrast, this is not observed in apo KRASOff protein crystal structures (e.g., 4LPK for KRASwt or 6ZIO for NRASwt; A). The SI/II-pocket is small and shallow having a volume and surface area of 12 Å3 and 36 Å2, respectively. Fragments binding to the SI/II-pocket were discovered by the Fesik group at Vanderbilt [Citation24] and researchers at Genentech [Citation25]. Recently, we published the first nanomolar chemical probe, BI-2852, which binds to the SI/II-pocket of all three RAS isoforms in both the on and off states [Citation26].
Surface representation of KRASOff (grey, PDB ID: 4LPK) and KRASOn (blue, PDB ID: 6QUU) with binding site crystallographic waters (cut-off radius 5 Å, waters are colored by their RISM free energy calculated with flare (water ‘happiness’): yellow (indifferent waters): -1.5 < ΔG < 1.5; red (unhappy water): ΔG >1.5 (#1: 2.16, #2: 0.49, #3: 2.68; all units in kcal/mol) around the switch I/II binding site [Citation27]. The three most conserved waters are highlighted with numbers. There are no very ‘happy’ waters with highly negative ΔG values.
PDB: Protein Data Bank; RISM: Reference interaction site model.
![Figure 2. Switch I/II pocket of KRAS.Surface representation of KRASOff (grey, PDB ID: 4LPK) and KRASOn (blue, PDB ID: 6QUU) with binding site crystallographic waters (cut-off radius 5 Å, waters are colored by their RISM free energy calculated with flare (water ‘happiness’): yellow (indifferent waters): -1.5 < ΔG < 1.5; red (unhappy water): ΔG >1.5 (#1: 2.16, #2: 0.49, #3: 2.68; all units in kcal/mol) around the switch I/II binding site [Citation27]. The three most conserved waters are highlighted with numbers. There are no very ‘happy’ waters with highly negative ΔG values.PDB: Protein Data Bank; RISM: Reference interaction site model.](/cms/asset/490620a8-d352-45a2-b3d1-87952c7966f4/ifmc_a_12361812_f0003.jpg)
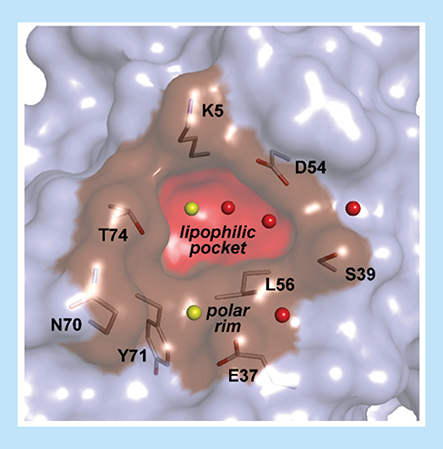
The SI/II-pocket presents opportunities that are not accessible to the SII-pocket; develop drugs for each of the three isoforms KRAS, HRAS and NRAS as the region of the SI/II pocket is fully conserved (Supplementary Figure 2); target the on state of RAS to overcome adaptive resistance to KRASOff drugs; and target the on state of RAS to address non-cycling RAS mutants. However, the SI/II-pocket also poses significant challenges. Sparring at least one wild-type RAS isoform is an expected requirement for a RAS drug, as triple RAS knockout mice embryos are not viable but can be rescued by reintroduction of an HRAS transgene [Citation28]. Given that KRAS, HRAS and NRAS have identical sequences at the SI/II-pocket (Supplementary Figure 2), achieving sufficient RAS isoform selectivity poses a formidable hurdle. Also, the SI/II-pocket is a very small lipophilic pocket mainly formed by the key amino acids K5, V7, D54, L56, Y71 and T74. The lipophilic core is surrounded by a shallow polar rim () which will require precision design in order to achieve the potency required by drug molecules. In this work, we provide x-ray crystal structures elucidating in detail how ligands bind to the SI/II-pocket in KRAS, NRAS and HRAS in both the on and off states. The establishment of robust cocrystallization systems [Citation26] and high-throughput soaking systems [Citation29] has allowed us to generate a high coverage of relevant RAS crystal structures and thus gain insights into designing more potent and specific SI/II-pocket inhibitors or proteolysis targeting chimeras (PROTACs) for the three RAS family of proteins. The high throughput crystallization system also allowed us to develop our so called ‘x-ray first’ approach where we crystallized every newly synthesized compound in the active KRASG12D form before proceeding toward biophysical or biochemical affinity testing. Based on the binding mode we selected the interesting molecules for further measurements to neglect the typical affinity biased optimization strategies that often lead to wrong conclusions with respect to binding interactions.
SI/II-pocket with the relevant crystallographic water molecules and amino acids in the small lipophilic pocket and the shallow polar rim surrounding the small cavity.
Indoles: a privileged scaffold for the SI/II-pocket
Similarly to Maurer et al. and Sun et al., we also identified indole starting points with millimolar affinity, using our fragment-based screening approaches (). The high propensity for indoles to bind to the SI/II-pocket is determined by two structural key elements. First, the bicyclic aromatic indole core forms CH–π interactions with the side-chains of K5 and L56 (C). Second, the indole NH forms an H-bond with the carbonyl of D54, selecting the in position over the out position. Both positions have also been observed in the apo KRASOn structure. The privileged binding of indoles to the SI/II-pocket is also explained by the apo structure of KRASOff, where Tyr71 from switch II occupies the SI/II-pocket also forming H-bonds to D54 and CH–π interactions with K5 and L56 (B). Upon indole binding Tyr71 is flipped toward the switch II pocket, turning the S2 helix by about 35° and adopting a conformation similar to that observed in the apo KRASOn structure.
Published indole starting points in the switch I/II of KRASG12D with respective NMR HSQC binding affinities. (A) Maurer et al. (4DST) [Citation25], (B) Sun et al. (4EPV) [Citation24], (C) Kessler et al. with compound 13 (6ZLI) [Citation26].
HSQC: Heteronuclear single quantum coherence; NMR: Nuclear magnetic resonance.
![Figure 4. Indoles as privileged scaffold.Published indole starting points in the switch I/II of KRASG12D with respective NMR HSQC binding affinities. (A) Maurer et al. (4DST) [Citation25], (B) Sun et al. (4EPV) [Citation24], (C) Kessler et al. with compound 13 (6ZLI) [Citation26].HSQC: Heteronuclear single quantum coherence; NMR: Nuclear magnetic resonance.](/cms/asset/5e1fa414-cfd1-4400-9094-39def60a131e/ifmc_a_12361812_f0005.jpg)
(A) Key compounds during optimization of BI-2852 with respective KD values in active KRASG12D. (B) Shows the apo KRASwt structure (4LPK), (C–E) shows key residues in the switch I/II pocket of active KRASG12D with 13 (6ZLI), 18 (6GJ6) and BI-2852 (6GJ8). Important CH–π interactions and H-bonds are marked with dotted lines.
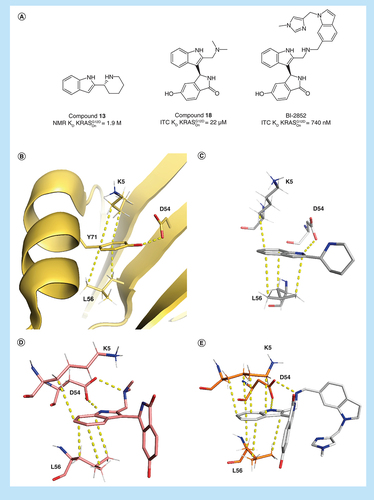
The indole-binding elements are maintained as the compound potency is optimized from the starting fragments, leading to compound 18 from Kessler et al. and BI-2852 itself (A–E). Of note is the altered position of K5 in the structure of BI-2852 which forms four CH–π interactions with the ligand in contrast to only one CH–π interaction with the fragments and 18 (E). The 2,4-dichloroindole (4DST) shows a flipped binding mode maintaining the CH–π interactions with L56 without the H-bond to D54, explaining its very weak potency. The additional charge–charge interaction formed between D54 and the amino group of compound 13 with an NMR KD of 1.9 mM is an important additional interaction.
Selectivity between active KRAS, HRAS & NRAS
Despite the amino acid sequences of KRAS, HRAS and NRAS being identical in the region of the SI/II-pocket, surprisingly moderate selectivity between RAS isoforms in the active state has been observed for BI-2852 and compound 18. All interactions established by the original fragments were maintained by BI-2852 and 18 across all three RAS isoforms, namely the CH–π interactions between the indole moiety and K5 and L56 and the H-bond and charge–charge interactions between D54.
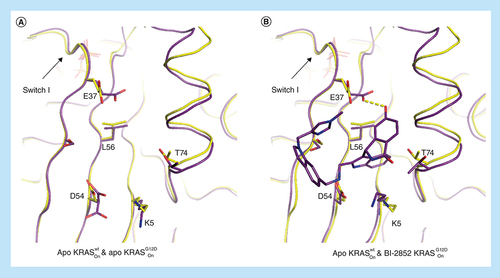
While isothermal titration calorimetry (ITC) data clearly demonstrate that BI-2852 binds all three RAS isoforms, intriguingly it binds tenfold more strongly to active KRASG12D versus KRASwt (740 nM vs 7.5 μM). We have been unsuccessful in obtaining crystal structures of BI-2852 in the active form of wild-type KRAS. However, an overlay of apo phosphoaminophosphonic acid guanylate ester (GNP)-bound KRASwt (6GOD) and apo phosphomethylphosphonic acid guanylate ester (GCP)-bound KRASG12D (6QUU) structures shows a subtle difference in the switch I conformation (A) leading to E37 adopting a position in wild-type KRAS which is unable to form an H-bond to the phenyl group and clashes with the imidazole of BI-2852 (B). This could explain the observed weaker activity for wild-type KRAS.
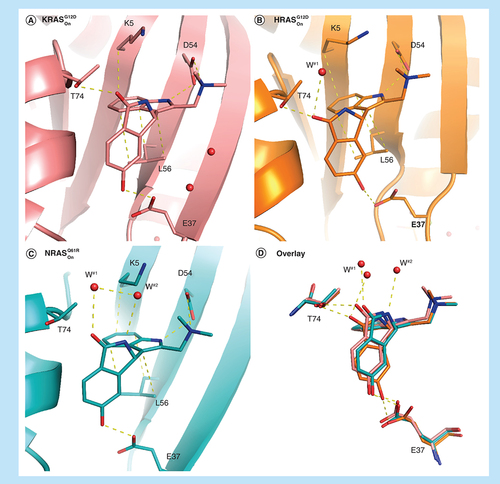
Intriguingly, the less decorated compound 18 displays a different selectivity profile compared with BI-2852, and binds most strongly to active HRASwt (6.7 μM), seven-times more weakly to active KRASwt (49 μM), and exhibits intermediate binding affinity to active KRASG12D (22 μM) and active NRASwt (33 μM). In order to elucidate the structural source of this selectivity profile we crystalized compound 18 into the active forms of KRAS, HRAS and NRAS utilizing crystallization systems of the strongly stabilizing oncogenic mutants KRASG12D, HRASG12D and NRASQ61R, respectively. In addition to the indole interactions, 18 forms two additional H-bonds with T74 and E37 in the active KRASG12D x-ray crystal structure (A) binding with a KD of 22 μM and accounting for the approximate 50-fold increase affinity from the original fragment hits. In active HRASG12D compound 18 forms all the interactions as observed in active KRASG12D but the iso-indolinone rotates slightly to stabilize a water molecule (water#1) that is not observed in KRAS, most likely leading to the observed threefold increased KD of 6.7 μM for active HRASwt. To accommodate the rotation of 18, E37 moves slightly away maintaining the H-bond to 18 but from the alternate carbonyl oxygen (B). Binding of 18 to active GTP loaded NRASQ61R (C), the weakest complex of the three isoforms, completely loses the H-bond to T74, maintains bonding to water#1 as in the GCP-bound HRASG12D structure, maintains the H-bond to E37 as in the GCP-bound KRASG12D structure, and finally forms an H-bond to a newly observed second water via the iso-indolinone NH. Of note is the GTP loaded nucleotide state of NRASQ61R without further need for a nucleotide exchange demonstrating the strong prevalence of KRASOn for that very specific mutation. These subtle differences in binding modes highlight both the potential for potency and selectivity optimization in the SI/II-pocket but also the precision of design required to achieve it. For design, the intrinsic plasticity of RAS proteins mentioned above (Supplementary Figure 1) constitutes both boon and bane. Here, the opening of the SI/II-pocket results in the binding of energetically unfavorable water molecules in the newly formed pocket, offering the replacement by other binding partners that is energetically favorable. This underpins the importance of ligand binding to water molecules, and more generally the potential of stringent incorporation of solvation analysis in compound design [Citation30].
Overlay of protein crystal structures of different KRAS variants in the active apo and ligand bound form. (A) Overlay of apo active KRASwt (yellow, 6GOD) and apo KRASG12D (purple, 6QUU) with E37 in a conformation that does not accommodate binding of BI-2852 (B) (active KRASG12D in purple, 6GJ8).
Comparison of the binding mode of 18 in active RAS mutant forms. (A) Active KRASG12D (6GJ6), (B) active HRASG12D (6ZJ0), (C) active NRASQ61R (6ZIZ) and (D) overlay of all three forms. Important waters in the binding pockets are highlighted with labels.
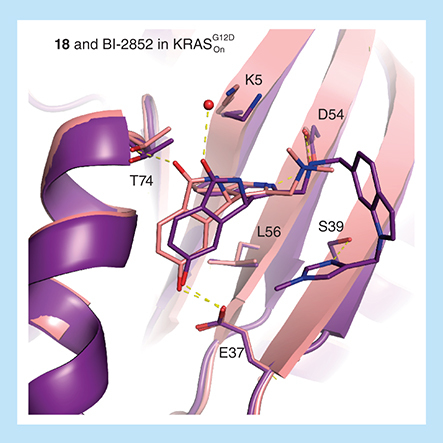
Precision of binding modes required to gain selectivity for one RAS isoform over another also needs to be maintained as molecules are grown to increase potency. This was not completely achieved growing from compound 18 to BI-2852 whereby the H-bond to T74 was completely lost for KRASG12D () similarly as observed in the compound 18 NRASQ61R complex.
Overlay of active KRASG12D with 18 (6GJ6) and BI-2852 (6GJ8) demonstrating a conserved binding mode except for the lost H-bond of T74 with isoindolinone–oxygen when bound to BI-2852.
Differences between the active & inactive states
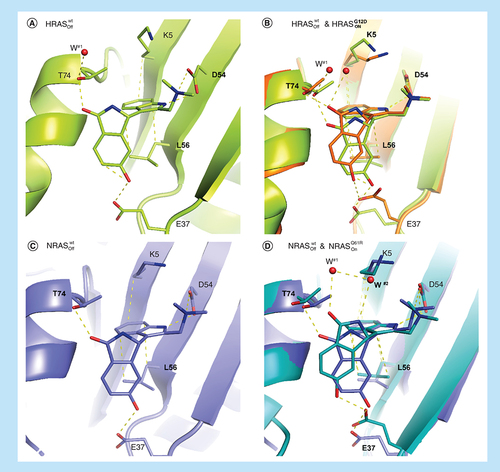
In order to spare one of the wild-type isoforms of RAS, binding to both the active and inactive forms needs to be avoided. Especially as inhibition of the RASOff form is expected to have a stronger effect on wild type versus oncogenic mutant due to the former cycling faster through the GDP-bound state. Although 18 binds most strongly to the active form of HRASG12D it displays eightfold weaker binding to inactive HRASwt (6.7 vs 54 μM). In order to understand this difference we determined the x-ray crystal structure of 18 and inactive HRASwt. There is a significant movement in switch I on going from the active to inactive form of HRAS resulting in a 2.5–3 Å displacement of E37. Despite this movement 18 still maintains the E37 H-bond albeit with the alternative carbonyl oxygen (A & B). The consequence of this is that 18 can no longer H-bond to water #1 which shifts slightly and forms an H-bond with T74 instead. Also, L56 in the off-state forms only two CH–π interactions with the indole of 18 versus the three interactions in the on-state. The loss of these two interactions accounts for the loss in binding affinity for inactive HRASwt. Compound 18 binds with the same affinity (ITC KD = 33 μM) to the active and inactive states of NRASwt. In the binding mode of 18 in inactive NRASwt (C) the iso-indolinone is again rotated to accommodate the E37 movement on going from the active to inactive conformation but all interactions are maintained in both structures (A), again in line with the observed affinities.
Comparison of the binding modes of 18 to (A) inactive HRASwt (6ZI3) and (B) inactive HRASwt (6ZI3) overlayed with active HRASG12D (6ZJ0) showing the adapted binding mode in the switch I/II pocket. (C) Shows the binding mode of 18 in inactive NRASwt (6ZL3) and (D) the overlay of inactive NRASwt (6ZL3) with active NRASQ61R (6ZIZ), again showing an adapted binding mode with E37 moving 2.5–3 Å.
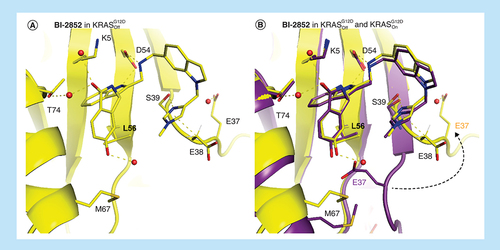
18 binds to all forms of KRAS, active and inactive KRASG12D and active and inactive KRASwt, with approximately equivalent ITC KD values of 22, 29, 49 and 15 μM, respectively. As we were unable to obtain x-ray crystal structures beyond GCP–KRASG12D (A) we can only assume that the binding modes are the same. It is however important to obtain crystal structures for all forms when optimizing SI/II-pocket drugs as surprises can occur. Such is the case for BI-2852 binding to active KRASG12D and inactive KRASG12D which also have very similar binding affinities, 0.74 and 2 μM, respectively, and could be assumed to have the same binding modes. While the ligand BI-2852 overlays in the active and inactive states of KRASG12D, due to a large movement of the switch I loop in the GDP structure, E37 moves away from the SI/II-pocket losing the phenolic H-bond to BI-2852 which is replaced by water. Interestingly, a new CH–π interaction between the methylene of D38 and the π system of the N-methyl imidazole group is formed regaining the lost affinity from the missing H-bond to E37. ().
BI-2852 bound to (A) inactive KRASG12D (6ZL5) and (B) overlaid with BI-2852 in active KRASG12D (6GJ8).
Future perspective
Closely tracking compound binding modes when attempting to drug the RAS SI/II-pocket is crucial if structure–activity relationships are to be interpreted correctly. Subtle differences in binding mode across the RAS isoforms can lead to significant differences in ligand-binding affinities. Also compounds with similar binding affinities can adopt alternate binding modes. Ideally, crystal structures need to be obtained for all compounds synthesized which has now become possible with the advances in protein crystallography and given sufficiently robust and high throughput crystallization systems. For the optimization of BI-2852, we applied an approach that we refer to as ‘x-ray first’ whereby newly synthesized compounds were first co-crystallized in the active form of KRASG12D and based on the x-ray crystallographic data compounds were then chosen for affinity measurement. ‘x-ray first’ avoids the compound selection bias which occurs with the classical compound profiling approach where affinity measurements are performed first and used to then select compounds for further profiling such as x-ray crystallography. An x-ray first approach will move drug design away from structure–activity relationships toward interaction–activity relationships which will be required to design molecules with the precision needed to drug challenging pockets such as the SI/II-pocket.
We have developed an approach that aggregates data from 3D structural ensembles into relevant hotspot information that can directly inform compound design and we could also demonstrate that non-obvious hidden opportunities can be found [Citation31]. Given the distinct plasticity and dynamic nature of RAS proteins it is evident that a thorough understanding of RAS structures must be complemented by molecular dynamics approaches as a theoretical foundation. A number of papers, see for example, the work by Pantsar [Citation32] and Gorfe et al. [Citation33], have been published on this topic. However, bridging the gap between such analyses and their swift and direct application in drug discovery remains a challenge, and is outside the scope of this paper. Our future efforts are committed toward vanishing this gap and seamlessly incorporating complex theory-driven approaches into our compound design processes.
It is interesting to note that all the subtly different binding modes for compound 18 arise from a rotation of the iso-indolinone with the privileged indole scaffold remain fixed in all structures. Structural rigidification of the bond between the indole and iso-indolinone resulting in natural product-like structures similar to the approach we took with mdm2 [Citation34] should lead to an improvement of both potency and selectivity. It should be noted that all the observed SI/II-pocket interactions, including those with bound water molecules, have not yet been successfully addressed with one molecule. This highlights the potential for significant improvement beyond the current ligands but also the precision design challenge that this pocket poses.
The observation of protein–ligand interactions by x-ray crystallography is clearly a powerful tool for drug design. However, the strength of individual interactions can only be inferred and not quantified. CH–π interactions between drug molecules and their target proteins, while often overlooked, are crucial determinants for their affinity. Indeed, CH–π interactions are the reason that indoles are a privileged structure for the SI/II-pocket. We have recently reported a new methodology, Pi by NMR [Citation35], which can gauge the strength of individual CH–π interactions. We plan to extent the utility of this methodology and apply it to the SI/II-pocket which could identify scaffolds binding more tightly to the SI/II-pocket than indoles.
The challenge of drugging the SI/II-pocket has been significantly reduced with the advent of PROTACs which can substantially improve potency and selectivity of inhibitors [Citation36]. Also, the first KRAS PROTACs utilizing covalent ligands binding to the SII-pocket have recently been described [Citation37]. The insights discussed here can be equally applied to the optimization of SI/II-pocket binders and SI/II-pocket PROTACs. The SI/II-pocket clearly offers numerous opportunities to provide future medicines for RAS driven diseases particularly where an RASOn drug is needed and for HRAS and NRAS driven disease for which no other options currently exist.
Overview
The three human RAS GTPases control numerous cell-signaling processes in cancer and RASopathies.
Druggability of RAS
The RAS family has two known pockets than can be drugged:
The switch II pocket in inactive RAS has led to covalent inhibitors, now in clinical trials, for KRASG12C;
The switch I/II pocket offers the possibility to develop inhibitors for all three RAS isoforms in both their active and inactive states. Attaining sufficient drug potency and selectivity in the switch I/II pocket is a significant challenge which will require high levels of structural information for drug design.
Indoles are a privileged scaffold of the SI/II pocket
Two structural key elements of indoles are responsible for this preference:
The aromatic indole core forms CH–π interactions with the side chains of L56 and K5;
The indole N–H forms an H-bond with the carbonyl of D54 leading to an ‘Asp in’ conformation.
Selectivity between KRAS, HRAS & NRAS is achievable
Biophysical and biochemical selectivity can be structurally explained with slightly differing binding modes of compound 18 and BI-2852 in inactive and active RAS forms.
Future perspective
Tracking compound binding modes by obtaining x-ray crystal structures for all compounds synthesized is critical when attempting to drug pockets as challenging as the SI/II pocket on RAS.
Recent advances in 3D data analysis and molecular dynamics further help to understand the plasticity of RAS.
Proteolysis targeting chimeras offer further possibilities to obtain sufficient potency and selectivity for RAS SI/II-pocket drugs.
New modalities like proteolysis targeting chimeras are on the way that offer possibilities for more selective drugs for such difficult targets.
Author contributions
D Kessler, J Böttcher and G Fischer designed the experiments, which were performed by S Döbel, M Hinkel, B Müllauer and A Weiss-Puxbaum. Data analysis were performed by D Kessler, J Böttcher, G Fischer and A Bergner performed the pocket plasticity and water analysis. D Kessler and DB McConnell drafted and edited the manuscript.
Supplemental Text 1
Download Zip (2.1 MB)Acknowledgments
The authors would like to thank M Gmachl, A Mantoulidis, L Martin, M Mayer, A Gollner, B Wolkerstorfer and A Zöphel for their fundamental work in the project; I Vorwahlner, M Pearson, J Quant and N Kraut for their support, the opnMe.com team for providing the BI-2852 on the collaboration platform and A Waterson, J Phan and SW Fesik at Vanderbilt University, TN for the fruitful collaboration to target KRAS. We thank the staff at beamline X06SA at the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland, for support and the Expose GmbH for crystallographic data collection.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at:www.tandfonline.com/doi/full/10.2217/epi-2016-0184
Financial & competing interests disclosure
This work was supported by the Austrian Forschungsförderungsgesellschaft (FFG) (grant nos. 854341, 861507, 867897 and 874517) (‘Basisprogramme’). All authors are currently employees of Boehringer Ingelheim. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Colicelli J . Human RAS superfamily proteins and related GTPases. Sci. STKE2004(250), Re13 (2004).
- Prior IA , LewisPD , MattosC. A Comprehensive survey of Ras mutations in cancer. Cancer Res.72(10), 2457–2467 (2012).
- Biankin AV , WaddellN , KassahnKSet al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature491(7424), 399–405 (2012).
- Neumann J , Zeindl-EberhartE , KirchnerT , JungA. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol. Res. Pract.205(12), 858–862 (2009).
- Network CGAR . Comprehensive molecular profiling of lung adenocarcinoma. Nature511(7511), 543–550 (2014).
- Colombino M , CaponeM , LissiaAet al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J. Clin. Oncol.30(20), 2522–2529 (2012).
- Bacher U , HaferlachT , SchochC , KernW , SchnittgerS. Implications of NRAS mutations in AML: a study of 2502 patients. Blood107(10), 3847–3853 (2006).
- Yoo J , RobinsonRA. H-RAS gene mutations in salivary gland mucoepidermoid carcinomas. Cancer88(3), 518–523 (2000).
- Jebar AH , HurstCD , TomlinsonDC , JohnstonC , TaylorCF , KnowlesMA. FGFR3 and RAS gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene24(33), 5218–5225 (2005).
- Cirstea IC , KutscheK , DvorskyRet al. A restricted spectrum of NRAS mutations causes Noonan Syndrome. Nat. Genet.42(1), 27–29 (2010).
- Schubbert S , ZenkerM , RoweSLet al. Germline KRAS mutations cause Noonan Syndrome. Nat. Genet.38(3), 331–336 (2006).
- Aoki Y , NiihoriT , KawameHet al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat. Genet.37(10), 1038–1040 (2005).
- Wittinghofer A , PalEF. The structure of RAS protein: a model for a universal molecular switch. Trends Biochem. Sci.16, 382–387 (1991).
- Liceras-Boillos P , García-NavasR , Ginel-PicardoAet al. SOS1 disruption impairs cellular proliferation and viability through an increase in mitochondrial oxidative stress in primary MEFs. Oncogene35(50), 6389–6402 (2016).
- Milburn MV , TongL , deVosAMet al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science247(4945), 939–945 (1990).
- Vetter IR , WittinghoferA. The guanine nucleotide-binding switch in three dimensions. Science294(5545), 1299–1304 (2001).
- Ostrem JM , ShokatKM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discov.15(11), 771 (2016).
- Karnoub AE , WeinbergRA. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol9(7), 517–531 (2008).
- Ostrem JM , PetersU , SosML , WellsJA , ShokatKM. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature503(7477), 548–551 (2013).
- Canon J , RexK , SaikiAYet al. The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature575(7781), 217–223 (2019).
- McCormick F . Sticking it to KRAS: covalent inhibitors enter the clinic. Cancer Cell37(1), 3–4 (2020).
- Xue JY , ZhaoY , AronowitzJet al. Rapid non-uniform adaptation to conformation-specific KRAS (G12C) inhibition. Nature577(7790), 421–425 (2020).
- Hunter JC , ManandharA , CarrascoMA , GurbaniD , GondiS , WestoverKD. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res.13(9), 1325–1335 (2015).
- Sun Q , BurkeJP , PhanJet al. Discovery of small molecules that bind to K-Ras and inhibit SOS-mediated activation. Angew. Chem. Int.51(25), 6140–6143 (2012).
- Maurer T , GarrentonLS , OhAet al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl Acad. Sci. USA109(14), 5299–5304 (2012).
- Kessler D , GmachlM , MantoulidisAet al. Drugging an undruggable pocket on KRAS. Proc. Natl Acad. Sci. USA116(32), 15823–15829 (2019).
- Cheeseright T , MackeyM , RoseS , VinterA. Molecular field extrema as descriptors of biological activity: definition and validation. J. Chem. Inf. Model46(2), 665–676 (2006).
- Nakamura K , IchiseH , NakaoKet al. Partial functional overlap of the three RAS genes in mouse embryonic development. Oncogene27(21), 2961 (2008).
- Bergner A , CockcroftX , FischerGet al. KRAS binders hidden in nature. Chem. Eur. J.25(52), 12037–12041 (2019).
- Klebe G . Applying thermodynamic profiling in lead finding and optimization. Nat. Rev. Drug Discov.14(2), 95–110 (2015).
- Schmalhorst PS , BergnerA. A grid map based approach to identify nonobvious ligand design opportunities in 3D protein structure ensembles. J. Chem. Inf. Model60(4), 2178–2188 (2020).
- Pantsar T . The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J.18, 189–198 (2020).
- Gorfe AA , GrantBJ , McCammonJA. Mapping the nucleotide and isoform-dependent structural and dynamical features of RAS proteins. Structure16(6), 885–896 (2008).
- Gollner A , WeinstablH , FuchsJEet al. Targeted synthesis of complex spiro [3H-indole-3,2′-pyrrolidin]-2(1H)-ones by intramolecular cyclization of azomethine ylides: highly potent MDM2–p53 inhibitors. ChemMedChem14(1), 88–93 (2019).
- Platzer G , MayerM , BeierAet al. PI by NMR: probing CH–π interactions in protein-ligand complexes by NMR. Angew. Chem. Int. Ed.
- Farnaby W , KoeglM , RoyMJet al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol.15(7), 672–680 (2019).
- Zeng M , XiongY , SafaeeNet al. Exploring targeted degradation strategy for oncogenic KRASG12C. Cell Chem. Biol27(1), 19–31e16 (2020).