At the recent International Conference on Antiviral Research in San Francisco, CA, USA, 25 years of the use of azidothymidine (AZT) in the treatment of HIV/AIDS were celebrated, along with all the subsequent impressive successes in the therapy of the disease Citation[1]. This is also discussed in the editorial by DeClercq in this issue Citation[2]. Of the oral presentations, 57% dealt with the discovery and development of antivirals for HIV, hepatitis C, and herpes viruses, all of which lead to chronic infection and the need for prolonged or lifelong therapy Citation[3].
However, there was little room (8% of the talks) for emerging viruses and the large number of viruses that are obviously and rightly known as ‘neglected viruses.’ These include highly pathogenic RNA viruses with a large number of victims, such as measles virus (> 160,000 child deaths per year), rotavirus (∼500,000–600,000 child deaths every year) or dengue virus (∼50 million people infected each year; thousands, again mostly children, die from the haemorrhagic form of dengue fever).
Many of these viruses predominantly hit developing countries in tropical and subtropical regions of the world (40% of the world’s population are now at risk of contracting dengue fever), but developed countries are by no means immune to their impact. This is exemplified by West Nile virus, which was introduced from Israel and Egypt into the USA, where it apparently arrived on the East coast in 1999 and became endemic in all 48 contiguous states within 5 years (1356 cases of fever and encephalitis, with 44 deaths, in the USA in 2008). Another example is the global severe acute respiratory syndrome (SARS) epidemic, which, from its origin in Southern China spread to 29 countries within a few weeks between February and June 2003. While the death toll resulting from the epidemic was relatively low (∼800 deaths among ∼8700 people infected), the global economic cost was estimated at approximately US$59 billion. Those who witnessed the deserted streets of Beijing at the peak of the SARS outbreak in May 2003, will never forget the enormous impact on normal daily life caused by the epidemic. Chikungunya virus (CHIKV) is another example of an emerging pathogen. For decades, this virus (which causes high fever and very painful inflammation of the small joints) was associated with relatively small and infrequent outbreaks in West, East and Central Africa. However, in 2005/2006, the virus was apparently dispersed from Eastern Kenya to the islands of the Indian Ocean and subsequently to India (where it had previously made its last appearance in the early 1960s). Just as with dengue virus, CHIKV is spread by the Aedes aegypti mosquito. Following the 2006 outbreak in India, the virus underwent mutations in two surface proteins, thereby facilitating a switch of vector to Aedes albopictus, the Asian Tiger mosquito, which has gradually dispersed from Asia into the Mediterranian region, including Spain, Southern France and Italy. Moreover, the Tiger mosquito has displaced Aedes aegypti in some regions of Central Africa. This is a very worrying trend because the Tiger mosquito is an agressive feeder on humans and is gradually dispersing further afield, with the potential of spreading CHIKV much more widely. The first ever outbreak in Europe occurred in northern Italy in 2007, with more than 200 cases of chikungunya fever. This outbreak was traced back to an infected individual traveling from India to Italy. CHIKV continues to cause widespread outbreaks in India and South East Asia. During a recent visit to Pondicherry in southern India, a physician told me that on average, he sees five new chikungunya patients per day in his practice alone.
Despite the recognized emergence of these viruses and the consequent human morbidity and mortality involving millions of individuals, no antiviral agents are available with which to treat the diseases. The major factors for the apparent neglect of these and other viruses are clear: infections with these pathogens are usually self-limiting, either in the vast majority of cases, leading to clearance of the virus by the host’s immune system (often in less than 2 weeks) or, in a few cases, the death of the patient.
Consequently, infections by these viruses do not require life-long treatment and, hence, the commercial interest in these widespread diseases is limited. There are a few notable exceptions (for example, Novartis’ research program on dengue virus), but the general prospects remain gloomy. Thus, since big pharmaceutical companies are not interested, it is up to scientists in academia and small enterprises (‘micropharma’) to accept the challenge to commence activities in antiviral drug discovery, with special emphasis on neglected and emerging viruses. While this is a costly venture, some initiatives for new funding schemes are available and will be discussed.
Scientists in academia do not normally have access to the huge chemical libraries that became central to drug discovery via high-throughput screening (HTS) in the pharmaceutical industry during the 1990s. However, as has been emphasized in reviews elsewhere Citation[4,5], there is (once again) a paradigm change going on in industry at present. Rational drug-design approaches, first introduced in the 1980s, are enjoying a well -deserved comeback, with new methodology such as virtual screening and fragment-based design being added. Individual academic laboratories may still have a hard time raising the funds for implementing the entire range of technologies required for a modern preclinical drug-discovery pipeline, but consortia of complementary research groups, which may also include small or medium-sized pharmaceutical companies, can certainly cope with this. The advantages of drug discovery in academia have been discussed elsewhere Citation[6].
HIV protease is a good example of what can be at least initiated in an academic environment. The crystal structure of the enzyme was determined in 1989 by the groups of Wlodawer at the National Cancer Center in Frederick, MD, USA and TL Blundell, then at Birkbeck College, London, UK Citation[7,8], in addition to a structural study carried out by Navia at Vertex, Inc. (PA, USA) Citation[9]. It was also in academic groups that the first inhibitors of the enzyme (mostly modified peptides) were designed and co-crystallized with the target Citation[8]. The resulting structures provided the starting point for the pharmaceutical industry, which subsequently synthesized thousands of different inhibitors (PIs) and determined hundreds of crystal structures of their complexes with HIV-1 protease Citation[10]. In some cases, the compounds were derived from inhibitors that had been made in the mid-to-late 1980s against the human aspartic protease renin, which is involved in blood pressure regulation and is distantly related to HIV protease. In any case, none of the nine protease inhibitors introduced into the market starting from 1995 were discovered by HTS; thus, PIs are generally considered to be a prime example of success resulting from structure-based design.
With this encouraging example and the lack of antivirals for large groups of highly pathogenic, neglected RNA viruses in mind, many structural virologists now wish to use the 3D structures of viral enzymes determined for more than the elucidation of molecular mechanisms. So much more is possible with these results, if they are used for structure-based design of inhibitors targeting these proteins. Of course, structural virologists will either have to expand their repertoire of methods to include virtual screening, molecular modeling, fragment screening and medicinal chemistry, or team up with specialists in these fields. There is ample opportunity for medicinal chemists here, particularly if they are guided by the 3D structures of their macromolecular targets.
Let us look at two examples of neglected viruses, enteroviruses and coronaviruses, where the structure-based design of inhibitors has been successful or is well underway in academia. Although both groups of viruses have single-stranded, positive-sense RNA genomes, they are very different: the enteroviruses are small, nonenveloped icosahedral particles harboring a genome of fewer than 8000 nucleotides; and the coronaviruses are enveloped with the largest RNA genome currently known (∼30,000 bases). Nevertheless, their main proteases, called 3C and 3C-like protease, respectively, have a number of features in common, making the design of broad-spectrum inhibitors targeting both enzymes a possibility Citation[11]. In general, proteases are useful targets because they are relatively easy to assay, and the initial steps of drug design are usually based on the known cleavage specificities of the enzymes Citation[12]. The difficult step is translating peptidic leads into nonpeptidic inhibitors with the required stability and bioavailability (a novel approach to this challenge is discussed later).
Belonging to the family Picornaviridae (‘small RNA viruses’), the genus Enterovirus includes rhinoviruses, a major cause of the common cold in humans, as well as the enteroviruses. Human rhinovirus (HRV) has been and is of interest to several pharmaceutical companies (see elsewhere for a comprehensive review Citation[13]). Rupintrivir was designed by Agouron to interfere with the HRV 3C protease Citation[14–16]. Its development was halted in Phase III clinical trials owing to low efficacy in patients Citation[17]. This failure demonstrates that the demands on safety and efficacy are particularly high in case of treatments of viral infections that are not life-threatening or which cause mild infections lasting only a few days. It is not even clear whether health insurance policies would pay for these drugs, as there is no urgent medical need for them in most patients (although this is different in patients with asthma and chronic obstructive pulmonary disease).
However, there are many relatives of rhinovirus among the enteroviruses that do cause more severe and, sometimes, life-threatening disease. Coxsackieviruses often infect neonates and young children. Most infections are mild or even asymptomatic but, in some cases, patients develop severe conditions, such as encephalitis, meningitis, pancreatitis, myocarditis or acute paralysis Citation[18]. No reliable statistics exist, but it is estimated that several thousand small children succumb to infection by these viruses in Western and Central Europe every year. A causative treatment is not available. This is also true for enterovirus 71 (EV-71), which, like some members of the Coxsackievirus A family, can cause hand, foot and mouth disease (HFMD), a condition that starts with painful blisters on the extremities and the tongue and can proceed to meningoencephalitis or pulmonary edema (which may be fatal) in severe cases. In 2008 and 2009, there were two major outbreaks of HFMD in China, with more than 400,000 people infected during the first outbreak. Relatively few died from the infection but, again, those who did were young children Citation[19,20]. EV-71 is believed to have emerged only relatively recently and is now a major global health concern.
Being RNA viruses, the enteroviruses have an RNA-dependent RNA polymerase, which lacks a proofreading function. They therefore exhibit high mutation rates, which is a major driving factor for virus emergence. For this reason, it is difficult to predict which enteroviruses will be important 5 years from now. Consequently, enterovirus inhibitors with broad specificity need to be developed to ensure adequate coverage of the majority of the species in the family. Until recently, there was a lack of structural data on suitable targets for anti-enteroviral therapy, but this has changed in the past few years, thanks largely to the European Comparative Structural Genomics of Viral Enzymes Involved in Replication (VIZIER)consortium of virologists and structural biologists, who characterized more than 170 RNA viruses and determined approximately 85 crystal structures of key enzymes Citation[21,101]. This included the 3C protease of Coxsackievirus B3 [Tan et al. Manuscript in Preparation].
The 3C proteases of these viruses are useful targets for broad-spectrum inhibitors, because they are highly conserved. The amino acid sequences share 40–60% sequence identity between individual family members, and the 3D structures are highly similar. This includes the structures of the HRV-14 enzyme Citation[22], as well as the 3C proteases from Coxackievirus B3 Citation[11] [Tan et al., Manuscript in Preparation], EV-68 and EV-71 [Tan et al., Unpublished Data]. At their active site, these proteases carry a Cys–His–Glu (or Asp) catalytic triad, with the cysteine offering ample opportunity for covalent modification by nonreversible inhibitors. The common approach to this is to start with a peptide corresponding to the nonprimed half (see elsewhere for nomenclature Citation[23]) of the enzyme’s substrates, which are the cleavage sites in the viral polyprotein. Next, an electrophilic warhead is attached to the carboxylic end of the P1 residue, to react with the catalytic cysteine of the protease. Carrying an α,β-nonsaturated alkyl ester moiety, rupintrivir , as well as many of the experimental inhibitors synthesized in academic laboratories, act as Michael acceptors for the nucleophilic Cys residue Citation[14]. Elaboration of the ester group will address the S1´ and S2´ pockets of the target enzyme Citation[24,25]. Other electrophilic groups that have been used in noncompetitive inhibitors include aldehydes, isatins, homophthalimides and halomethylketones (see elsewhere for review Citation[13]). After modification of their P2 residue, these inhibitors are often not only active against the picornavirus 3C proteases, but also against coronavirus 3C-like proteases (also known as main proteases, Mpro) Citation[26,27]. The most prominent examples of such compounds have been described as inactivators of the SARS-coronavirus MproCitation[11,28,29]. In addition, benzotriazole esters have been described as efficient modifiers for the SARS-CoV enzyme Citation[30,31].
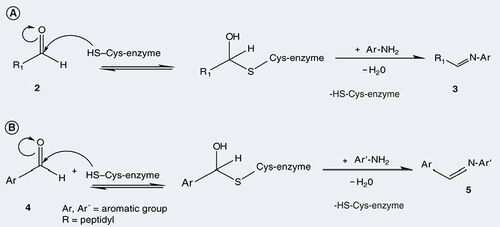
Peptidic aldehydes are of particular interest, because their binding to the catalytic center of cysteine proteases is reversible Citation[32]. This makes them useful as active site-directing probes. In a process known as ‘dynamic ligation screening’ (DLS) Citation[33], the covalent adduct between a peptide aldehyde and the target protease, in that case the SARS-CoV Mpro, has been used to screen a small focused library of nonpeptidic nucleophiles, mostly amines, for compounds that would replace the aldehyde (2) from the active site cysteine by formation of a Schiff base (3) and, subsequently an imine . This coupling reaction will only work in the presence of the target enzyme, and only with aromatic amines (Ar-NH2); the resulting mixed peptidic/nonpeptidic imine is often a moderately active inhibitor (Ki ∼50 µM). In a second step, the most efficient amine in this reaction is modified to carry an aldehyde moiety itself (4), which in turn is reacted with the catalytic cysteine of the protease, and the same library of nucleophiles is once again screened. This procedure has been used to discover nonpeptidic, competitive inhibitors (5) of the target protease in a very short time, starting from a peptidic aldehyde that was easily designed on the basis of the known subsite specificity of the protease.
While the DLS procedure works both for coronavirus 3C-like proteases and enterovirus 3C proteases, the former do harbor some special features. A total of 8 years after the first crystal structures of coronavirus Mpros were reported Citation[26,27], the enzyme still surprises us. Recently, two articles addressed the question of how the protease can liberate itself from the viral polyprotein, of which it is an integrated part (nonstructural protein 5) before autocleavage. Chen et al. showed that mutations known to destroy the Mpro dimer and, thereby, inactivate the mature enzyme, still allow auto-activation Citation[34]. Furthermore, Zhang et al. elucidated how this ‘premature’ enzyme may look in three dimensions. They presented the crystal structure of an Mpro octamer, carrying an interesting domain swap, which explained the data presented by Chen et al. in structural terms Citation[35]. These new views on a viral protease in its premature state may offer new ideas for the design of inhibitors directed at the maturation process.
By no means do I want to suggest that inhibitors of viral cysteine proteases cannot be discovered by screening of a chemical library. On the contrary, the chances of identifying competitive inhibitors that do not modify the active site covalently are much higher with this approach (or with virtual screening) than with structure-based de novo design, which will always be biased by the knowledge of the interaction between the peptidic substrate and the target enzyme. Occasionally, laboratories in academia have their own moderately sized compound libraries, or they have access to libraries established by consortia of academic institutions Citation[36]. For example, by screening a library of approximately 50,000 compounds, a nonpeptidic, small-molecule hit has been discovered as an inhibitor of the second cysteine protease of the SARS virus, the papain-like protease, and optimized to fit the 3D structure of the enzyme Citation[37].
Of course, one might ask, does it make sense to design and synthesize new SARS-CoV inhibitors, 7 years after the outbreak in 2003? Apart from a few subsequent cases that occurred in China in 2004, the virus has not been detected in humans since. My answer to this question is a definite ‘yes’. The virus is almost certainly hiding in its natural reservoir, most likely bats. Coronaviruses of both groups 1 and 2b (to which SARS-CoV belongs) have been detected in bats in Southern China as well as in Ghana, Eastern Europe and Northern Europe Citation[38–42]. In various studies, coronaviruses were detected in 5–25% of the bats investigated. Therefore, zoonotic transmission to humans in the future has to be expected, whether directly or via an intermediate carrier (such as civets in the 2003 SARS outbreak). We must not again be caught by surprise or be almost completely helpless, as we were during the first outbreak. Moreover, since 2003, several new coronaviruses have been discovered, with two of them, HCoV-NL63 and HCoV HKU1, being important human pathogens causing infection of the upper respiratory tract Citation[43,44].
As much as the work on inhibitor discovery for the SARS-CoV proteases may bear fruit for future outbreaks of the same or related viruses, the inhibitors that have been designed for enterovirus 3C proteases may also become relevant for the complete eradication of poliovirus Citation[45]. Through a very successful global vaccination program, this member of the genus Enterovirus is almost extinct, but it is proving almost impossible to eradicate the virus completely. Poliomyelitis is still endemic in Nigeria, India, Pakistan and Afghanistan Citation[102], and, at present, there is an outbreak in Tajikistan Citation[103]. Furthermore, there is evidence for recombination of the attenuated live vaccination strain with field strains of Coxsackieviruses of the A clade and for immunocompromised people shedding the vaccination strain of the virus Citation[46,47]. As a result, the WHO has launched a call for the development of antivirals directed at poliovirus Citation[48]. The good news is that on the basis of structural similarity between the target enzymes, it can be predicted that inhibitors of the 3C protease of other enteroviruses will also inactivate the poliovirus enzyme Citation[49]. Preliminary tests are being carried out at this time.
Having realized the value of their viral protein structures for antiviral drug discovery, a number of research groups from the VIZIER project Citation[101] (which ended in 2009) have teamed up with medicinal chemists and experts in small animal models of diseases caused by RNA viruses, to form a new consortium designated ‘small-molecule inhibitor leads versus neglected and emerging RNA viruses’ (SILVER), which aims at capitalizing on the structural output of VIZIER and using this large body of information for antiviral drug discovery. The goal of the project is to demonstrate proof of principle for a minimum of two antiviral compounds in a small animal model. Compounds to be developed initially will likely be directed at enteroviruses (including the emerging EV-71) and dengue virus. It is hoped that, if successful, the SILVER project will motivate the pharmaceutical industry to collaborate and perhaps develop some of the lead compounds discovered within the new project. Alternatively, another consortium, the International Consortium on Antivirals (ICAV) has been created by Canadian scientists in response to the SARS outbreak in Toronto, and has since formed a global network of scientists in academia who design and synthesize antivirals Citation[104]. The idea behind ICAV is to identify the best antiviral compounds available within the network and develop them further in preclinical and, ultimately, clinical trials. Such objectives will be staggeringly expensive, but ICAV has developed new funding schemes involving governments, nongovernmental organizations and charities, and has so far been quite successful in implementing these. The ambitious goal of the consortium is to provide access to new antivirals at a reasonable cost for the developing countries that need them most, but also to license the compounds to the pharmaceutical industry in developed countries. Clearly, both SILVER and ICAV will need the best available antiviral medicinal chemistry. Hopefully, these academic initiatives will have a positive impact on the current gloomy prospects for antiviral therapies for infections caused by neglected and emerging RNA viruses.
Acknowledgements
The author thanks Professor EA Gould (Oxford/Marseille) and Dr J Tan, Dr L Zhu and Dr Y Kusov (Lübeck) for discussion.
Financial & competing interests disclosure
The author is a member of the three not-for-profit consortia active in antiviral research that have been mentioned in the text: VIZIER, SILVER and ICAV. He has received grants from VIZIER and ICAV in support of his academic research and expects to receive such a grant from SILVER. The author also acknowledges funding for his structural virology and antiviral drug design work from DFG, the European Commission, the Sino-German Center for the Promotion of Research (Beijing), and the Schleswig-Holstein Innovation Fund. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript, apart from those disclosed.
No writing assistance was utlized in the production of this manuscript.
Additional information
Funding
Bibliography
- Broder S . The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic.Antiviral Res.85, 1–18 (2010).
- DeClercq E . Antiviral therapy: quo vadis?Future Med. Chem.2(7), 1049–1053 (2010)
- Abstracts of the 23rd International Conference on Antivirals, San Francisco, CA, 25–28 April, 2010. Antiviral Res.86, A1–A76 (2010).
- Ojima I . Medicinal chemistry at a crossroads: challenges and new possibilities.Future Med. Chem.1, 401–403 (2009).
- Raveglia LF , GiardinaGAM. Accelarating the drug-discovery process: new tools and technologies available to medicinal chemists.Future Med. Chem.1(6), 1019–1023 (2009).
- Wyatt PG . The emerging academic drug-discovery sector.Future Med. Chem.1(6), 1013–1017 (2009).
- Wlodawer A , MillerM, JaskólskiMet al. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science 245, 616–621 (1989).
- Blundell TL , LapattoR, WilderspinAFet al. The 3D structure of HIV-1 proteinase and the design of antiviral agents for the treatment of AIDS. Trends Biochem. Sci. 15, 425–430 (1990).
- Navia MA , FitzgeraldPM, McKeeverBMet al. 3D structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 337, 615–620 (1989).
- Vondrasek J , WlodawerA. HIVdb: a database of the structures of human immunodeficiency virus protease.Proteins49, 429–431 (2002).
- Lee CC , KuoCJ, KoTPet al. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J. Biol. Chem. 284, 7646–7655 (2009).
- Steuber H , HilgenfeldR. Recent advances in targeting viral proteases for the discovery of novel antivirals.Curr. Top. Med. Chem.23, 323–345 (2010).
- De Palma AM , VliegenI, De Clercq E, Neyts J. Selective inhibitors of picornavirus replication. Med. Res. Rev.28, 823–884 (2008).
- Matthews DA , DragovichPS, WebberSEet al. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl Acad. Sci. USA 96, 11000–11007 (1999).
- Johnson TO , HuaY, LuuHTet al. Structure-based design of a parallel synthetic array directed toward the discovery of irreversible inhibitors of human rhinovirus 3C protease. J. Med. Chem. 45, 2016–2023 (2002).
- Binford SL , MaldonadeF, BrothersMAet al. Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob. Agents Chemother. 49, 619–626 (2005).
- Patick AK . Rhinovirus chemotherapy.Antiviral Res.71, 391–396 (2006).
- Sawyer MH . Enterovirus infections, diagnosis and treatment.Semin. Pediatr. Infect. Dis.13, 40–47 (2002).
- Yang F , RenL, XiongZet al. Enterovirus 71 outbreak in the People’s Republic of China in 2008. J. Clin. Microbiol. 47, 2351–2352 (2009).
- Zhang Y , TanXJ, WangHYet al. An outbreak of hand, foot, and mouth disease associated with subgenotype C4 of human enterovirus 71 in Shandong, China. J. Clin. Virol. 44, 262–267 (2009).
- Coutard B , GorbalenyaAE, SnijderEJet al. The VIZIER project: preparedness against pathogenic RNA viruses. Antiviral Res. 78, 37–46 (2008).
- Matthews DA , SmithWW, FerreRAet al. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell 77, 761–777 (1994).
- Schechter I , BergerA. On the size of the active site in proteases. I. Papain.Biochem. Biophys. Res. Commun.27, 157–162 (1967).
- Lee ES , LeeWG, YunSHet al. Development of potent inhibitors of the Coxsackievirus 3C protease. Biochem. Biophys. Res. Commun. 358, 7–11 (2007).
- Kuo CJ , ShieJJ, FangJMet al. Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg. Med. Chem. 16, 7388–7398 (2008).
- Anand K , PalmGJ, MestersJR, SiddellSG, ZiebuhrJ, HilgenfeldR. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain.EMBO J.21, 3213–3224 (2002).
- Anand K , ZiebuhrJ, WadhwaniP, MestersJR, HilgenfeldR. Coronavirus main proteinase (3CLpro) structure: basis for design of antiSARS drugs. Science300, 1763–1767 (2003).
- Yang H , YangM, DingYet al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl Acad. Sci. USA 100, 13190–13195 (2003).
- Yang H , XieW, XueXet al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 3, 1742–1752 (2005).
- Wu CY , KingKY, KuoCJet al. Stable benzotriazole esters as mechanism-based inactivators of the severe acute respiratory syndrome 3CL protease. Chem. Biol. 13, 261–268 (2006).
- Verschueren KHG , PumporK, AnemüllerS, ChenS, MestersJR, HilgenfeldR. A structural view of the inactivation of the SARS-coronavirus main proteinase by benzotriazole esters.Chem. Biol.15, 597–606 (2008).
- Al Gharabli SI , ShahST, WeikSet al. An efficient method for the synthesis of peptide aldehyde libraries employed in the discovery of reversible SARS coronavirus main protease (SARS-CoV Mpro) inhibitors. ChemBioChem 7, 1048–1055 (2006).
- Schmidt MF , Isidro-LlobetA, LisurekMet al. Sensitized detection of inhibitory fragments and iterative development of nonpeptidic protease inhibitors by dynamic ligation screening. Angew. Chem. Int. Ed. Engl. 47, 3275–3278 (2008).
- Chen S , JonasF, ShenC, HilgenfeldR. Liberation of SARS-CoV main protease from the viral polyprotein: N-terminal autocleavage does not depend on the mature dimerization mode.Protein Cell,1, 59–74 (2010).
- Zhang S , ZhongN, XueFet al. 3D domain swapping as a mechanism to lock the active conformation in a super-active octamer of SARS-CoV main protease. Protein Cell 1, 371–383 (2010).
- Lisurek M , RuppB, WichardJet al. Design of chemical libraries with potentially bioactive molecules applying a maximum common substructure concept. Mol. Divers. 14, 401–408 (2010).
- Ratia K , PeganS, TakayamaJet al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl Acad. Sci. USA 105, 16119–16124 (2008).
- Lau SK , LiKS, HuangYet al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 84, 2808–2819 (2010).
- Yuan J , HonCC, LiYet al. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J. Gen. Virol. 91, 1058–1062 (2010).
- Pfefferle S , OppongS, DrexlerJFet al. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg. Infect. Dis. 15, 1377–1384 (2009).
- Rihtaric D , HostnikP, SteyerA, GromJ, ToplakI. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch. Virol.155, 507–514 (2010).
- Gloza-Rausch F , IpsenA, SeebensAet al. Detection and prevalence patterns of group I coronaviruses in bats, Northern Germany. Emerg. Infect. Dis. 14, 626–631 (2008).
- van der Hoek L , PyrcK, JebbinkMFet al. Identification of a new human coronavirus. Nature Med. 10, 368–373 (2004).
- Woo PC , LauSK, ChuCMet al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 79, 884–895 (2005).
- Collett MS , NeytsJ, ModlinJF. A case for developing antiviral drugs against polio.Antiviral Res.79, 179–187 (2008).
- Jiang P , FaaseJAJ, ToyodaH, PaulA, WimmerE, GorbalenyaAE. Evidence for emergence of diverse polioviruses from C-cluster Coxsackie A viruses and implications for global poliovirus eradication.Proc. Natl Acad. Sci. USA104, 9457–9462 (2007).
- Rakoto-Andrianarivelo M , GuillotS, IberJet al. Co-circulation and evolution of polioviruses and species C enteroviruses in a district of Madagascar. PloS Pathogens 3, 1950–1961 (2007).
- Couzin J . Report concludes polio drugs are needed after disease is eradicated.Science311, 1539 (2006).
- De Palma AM , PuerstingerG, WimmerEet al. Comparative activity of a selected series of antipicornavirus compounds against poliovirus replication in vitro. Emerg. Infect. Dis. 14, 545–551 (2008).
Websites
- VIZIER Project www.vizier-europe.org
- Global polio eradication initiative. Programme of Work 2009 and financial resource requirements 2009–2013, as of May 2009 WHO, Rotary International, CDC, Unicef. www.polioeradication.org/content/general/Final_English.GPEIProgrammeofWork2009.pdf
- World Health Organisation Country Office Tajikistan, WHO Regional Office for Europe, European Centre for Disease Prevention and Control. Outbreak of poliomyelitis in Tajikistan in 2010: risk for importation and impact on polio surveillance in Europe? Eurosurveillance, 15, issue 17, April 29, 2010 www.eurosurveillance.org/Public/Articles/Archives.aspx
- International Consortium om Antivirals www.icav-citav.org