
Abstract
Amyotrophic lateral sclerosis (ALS) is an incurable disease, caused by the loss of the upper and lower motor neurons. The lack of therapeutic progress is mainly due to the insufficient understanding of complexity and heterogeneity underlying the pathogenic mechanisms of ALS. Recently, we analyzed whole-genome expression profiles of motor cortex of sporadic ALS patients, classifying them into two subgroups characterized by differentially expressed genes and pathways. Some of the deregulated genes encode proteins, which are primary targets of drugs currently in preclinical or clinical studies for several clinical conditions, including neurodegenerative diseases. In this review, we discuss in-depth the potential role of these candidate targets in ALS pathogenesis, highlighting their possible relevance for personalized ALS treatments.
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, is a devastating, progressive and fatal neurodegenerative disease, characterized by the selective and gradual degeneration of both upper and lower motor neurons. Approximately 10% of cases has a known genetic cause that follow a familial, autosomal dominant inheritance (FALS) while the remaining cases (90%) are sporadic (SALS).
The figure shows as, by an unsupervised hierarchical clustering (similarity measure: Pearson centered; linkage rule: average), we were able to separate control (ten fresh-frozen motor cortex samples from individuals who had died from a non-neurological disease) from SALS patients (31 fresh-frozen motor cortex samples) on the basis of similarity in their expression patterns in motor cortex (considering the 9646 most informative genes, with an SD >1.5). In addition, this clustering allowed us to segregate sproadic amyotrophic lateral sclerosis (SALS) patients in two greatly divergent groups (SALS1 and SALS2). In the 2D presentation, each row represents a single gene and each column a motor cortex from control or SALS patients. Highly expressed genes are shown in red, downregulated genes in blue, no change in white. In the dendrograms shown (left and top), the length and the subdivision of the branches display the relatedness of the expression of the genes (left) and the motor cortex (top). The disease state is marked as follows: controls patients are indicated by blue rectangles and SALS patients by red rectangles. In the cluster panel, blue rectangle refers to control patients, yellow to SALS1 and orange to SALS2 patients. For further details, the reader is referred to [Citation7].
![Figure 1. Unsupervised hierarchical clustering of control and sporadic amyotrophic lateral sclerosis patients.The figure shows as, by an unsupervised hierarchical clustering (similarity measure: Pearson centered; linkage rule: average), we were able to separate control (ten fresh-frozen motor cortex samples from individuals who had died from a non-neurological disease) from SALS patients (31 fresh-frozen motor cortex samples) on the basis of similarity in their expression patterns in motor cortex (considering the 9646 most informative genes, with an SD >1.5). In addition, this clustering allowed us to segregate sproadic amyotrophic lateral sclerosis (SALS) patients in two greatly divergent groups (SALS1 and SALS2). In the 2D presentation, each row represents a single gene and each column a motor cortex from control or SALS patients. Highly expressed genes are shown in red, downregulated genes in blue, no change in white. In the dendrograms shown (left and top), the length and the subdivision of the branches display the relatedness of the expression of the genes (left) and the motor cortex (top). The disease state is marked as follows: controls patients are indicated by blue rectangles and SALS patients by red rectangles. In the cluster panel, blue rectangle refers to control patients, yellow to SALS1 and orange to SALS2 patients. For further details, the reader is referred to [Citation7].](/cms/asset/015996e5-60e0-42e2-ac3f-b05ebc1b1109/ifmc_a_12363292_f0001.jpg)
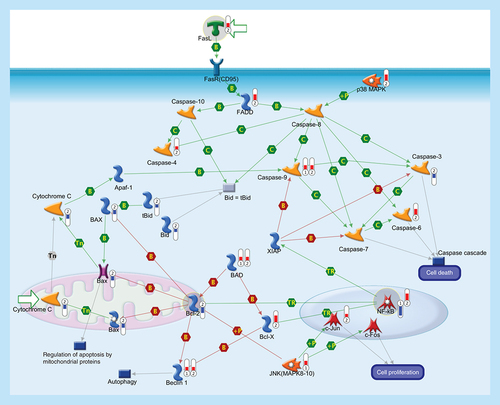
Apoptosis is a programmed cells death regulated through a variety of different pathways which interact and eventually lead to controlled cell death. The activation of the apoptotic response occurs through either the intrinsic or the extrinsic pathway, depending on the origin of the death stimuli. The extrinsic apoptosis is triggered by the interaction of an extracellular death ligand (e.g., FasL) to its cell-surface death receptor FasR (CD95), triggering movement of cytosolic adaptor proteins (e.g., FADD) to the intracellular portion of the receptor and, together with protein kinase p38 MAPK, transducing cell-death signals to downstream effector caspases that are responsible for the biochemical aspects of apoptosis. The intrinsic pathway is associated with mitochondrial membrane permeabilization and release of cytochrome c into the cytoplasm. The release of cytochrome c into the cytosol, in response to mitochondrial injury, triggers the recruitment of procaspase-9 to form a mitochondrial apoptosome and subsequent caspase activation. Sequential activation of caspases by the intrinsic or extrinsic signaling pathways plays a central role in the execution-phase of apoptosis. Bcl-2 family comprises a group of structurally related proteins (Bcl-2, Bcl-X, Bax, Bad, Bid) that play a fundamental role in the regulation of the intrinsic pathway by controlling mitochondrial membrane permeability and the release of the pro-apoptotic factor, cytochrome c. Caspase cascade is also involved in the autophagy, a catabolic process involved in cell death and in cell protective mechanism. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Activation of transcription factor NF-κB, a key mediator of genes involved in the control of the cellular proliferation and apoptosis, can promote neuronal survival by inducing the expression of genes encoding antiapoptotic proteins (e.g., XIAP). Activated JNK (MAPK8–10) induces cell apoptotic process (e.g., by phosphorylating mitochondrial proteins, such as the antiapoptotic proteins Bcl-XL and Bcl-2), or can mediate cell proliferation, regulation of apoptotic process and inflammatory response (mainly by phosphorylating transcription factors of the AP-1 family such as c-Jun, and c-Fos). Thermometers represent expression ratio in motor neuron among following conditions: (SALS1/control) and (SALS2/control). Upward thermometers have red color and indicate upregulated signals in SALS patients, downward (blue) ones indicate downregulated signals. Pathway objects and links are described separately in the Supplementary Figure 1.
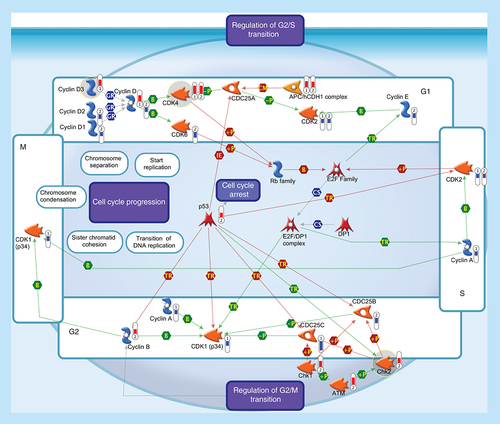
All somatic eukaryotic cells proceed through four phases in cell cycle: G1 (gap phase 1) in which the cell prepares for the upcoming events of S-phase; S (synthesis phase), in which DNA is replicated; G2 (gap phase 2) in which the cell prepares for the upcoming events of M-phase; and M (mitosis), in which chromosomes are separated over two new nuclei. Regulation of the cell cycle involves several protein kinases (such as the Checkpoint homologs Chk1, Chk2) that, activated by ATM, inhibit CDC25 phosphatase, preventing entry into mitosis and stabilize the tumor suppressor protein p53, leading to cell cycle arrest in G1. Progression through the cell-division cycle is driven by activation and inactivation of CDKs, which are controlled by periodic synthesis and degradation of cyclins and require association with a cyclin subunit for their activation. Cyclin/CDK complexes trigger the transition to next stage of the cell cycle: Cyclin D/CDK4 (or CDK6) for G1 progression, Cyclin E/CDK2 for the G1-S transition, Cyclin A/CDK2 for S-phase progression and Cyclin A/CDK1 and Cyclin B/CDK1 for entry into M-phase. Cyclin D, the first cyclin produced in the cell cycle in response to extracellular signals (e.g., growth factors), binds to existing CDK4, forming the active cyclin D-CDK4 complex that, in turn, phosphorylates the retinoblastoma family of tumor suppressor proteins (Rb family) liberating and activating the E2F transcription factors. These factors are associated with DP1 and their activation results in transcription of various genes like Cyclin E, Cyclin A, CDK1 and products that are necessary for the replication of DNA and beginning of the S phase. The activity of a Cyclin/CDK complex can be inhibited by phosphorylation at a pair of amino acids in the roof of the active site, while dephosphorylation of these sites by a phosphatase known as CDC25 (CDC25A, CDC25B and CDC25C) increases CDK activity. Events controlling cell division are governed by the degradation of different regulatory proteins by the ubiquitin-dependent pathway. Phosphorylated CDC25A may be exposed to ubiquitination by APC, one of ubiquitin ligases, which is known to be essential in maintaining normal cell cycle. Thermometers represent expression ratio in motor neuron among following conditions: 1 (SALS1/control) and 2 (SALS2/control). Upward thermometrs have red color and indicate expression ratios less than 1 while downward thermometers have blue color and indicate expression ratio less than 1. Pathway objects and links are described separately in the Supplementary Figure 1.
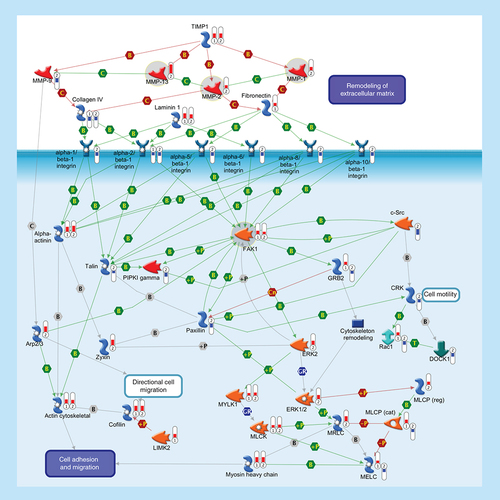
ECM remodeling is involved in normal physiological processes, such as cell motility and adhesion. MMPs are a family of proteolytic enzymes that degrade various components of the ECM in these processes. Endogenous TIMPs, such as TIMP1, reduce excessive proteolytic ECM degradation by MMPs. Integrins are heterodimeric cell surface adhesion receptors formed by two noncovalently associated subunits, alpha and beta. Most integrins recognize several ECM proteins, such as Laminin 1, Fibronectin and Collagen IV, whereas alpha-5/beta-1 integrin recognizes only Fibronectin. The ECM, integrins and the cell cytoskeleton interact at sites called focal contacts. The integrin-binding proteins Paxillin and Talin recruit FAK1 to focal contacts. Activated Talin binds to PI(4,5)P2 producing enzyme PIPKI gamma and activates it. PIPKI gamma also can be stimulated by tyrosine-protein kinase c-Src and FAK1 phosphorylation. Integrin clustering promotes FAK1 autophosphorylation, thereby creating a binding site for c-Src. Phosphorylation of FAK1 by c-Src maximizes catalytic activity of FAK1 and creates a binding site for GRB2, thereby leading to the activation of ERK1/2. ERK2 phosphorylates FAK1 and decreases Paxillin binding to FAK1. Within focal contacts, FAK1-c-Src-mediated phosphorylation of Paxillin promotes ERK2 binding. ERK2-mediated phosphorylation of Paxillin can facilitate FAK1 binding to Paxillin and enhance FAK1 activation. ERK2-mediated phosphorylation and activation of MYLK1 contributes to cell-matrix adhesion dynamics. Alpha-actinin is a cytoskeletal protein that crosslinks Actin in actomyosin stress fibers and tethers them to focal contacts. Phosphorylation of Alpha-actinin by FAK1 reduces the crosslinking of stress fibers and prevents maturation of the focal contacts. The Arp2/3 nucleates new Actin filaments from the sides of pre-existing filaments. Activation of LIMK2 leads to phosphorylation and inactivation of actin depolymerizing factor Cofilin, resulting in net increase in the cellular filamentous Actin cytoskeletal. Zyxin is an Alpha-actinin and stress-fiber-binding protein found in mature contacts. Inactivation of the MLCP attenuates phosphorylation of the MELC, MLCK and MRLC, inhibiting the formation of actomyosin stress fibers. CRK facilitates activation of Rac1 by DOCK1. Rac1 leads to activation of MRLC and the Arp2/3 complex. Thermometers represent expression ratio in motor neuron among following conditions: (SALS1/control) and (SALS2/control). Upward thermometrs have red color and indicate expression ratios less than 1 while downward thermometers have blue color and indicate expression ratio less than 1. Pathway objects and links are described separately in the Supplementary Figure 1.
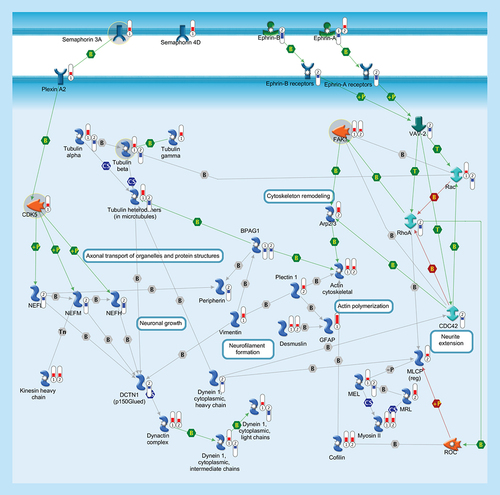
Cytoskeleton consists of three distinct, yet interconnected filament systems: microfilaments (actin filaments), intermediate filaments and microtubules. Microfilaments are the thinnest filaments of the cytoskeleton and are composed of linear polymers of actin subunits. They act as tracks for the movement of myosin molecules and are controlled by the Rho family of small GTP-binding proteins such as Rho, Rac and Cdc42. The activation of RhoA downstream effector Rho-associated kinase ROCK phosphorylates and inactivates the actin-associated protein cofilin, leading to reorganization of the actin cytoskeleton. Intermediate filaments (vimentin, desmusin, gfap, peripherin, NEFL, NEFM, NEFH) organize the internal tridimensional structure of the cell, anchoring organelles and serving as structural components of the nuclear lamina and sarcomeres. Intermediate filament binding protein Plectin 1 interacts with the Actin cytoskeleton to promote the formation of actin-rich structures that are important for cell motility. Neurofilaments are important protein cargoes for actin-associated motors, such as myosin, and microtubule-associated motor, such as kinesin in a complex with Dynactin. Microtubules are polymers of alpha and beta tubulin and, in association to other proteins such as Dynein and Dynactin, are important for axonal transport. Semaphorins act as axonal guidance factors. Semaphorin 3A binds to Plexin A2 and induces repulsive responses. Ephrins and their receptor protein-tyrosine kinases activate several factors (e.g., VAV-2) and regulate a variety of biological processes including the guidance of axon growth. Thermometers represent expression ratio in motor neuron among following conditions: (SALS1/control) and (SALS2/control). Upward thermometrs have red color and indicate expression ratios less than 1 while downward thermometers have blue color and indicate expression ratio less than 1. Pathway objects and links are described separately in the Supplementary Figure 1.
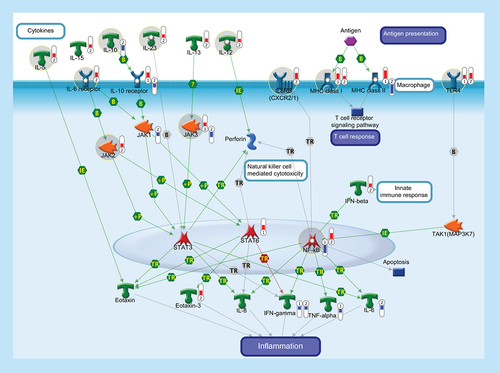
The function of the immune system depends on a large part on ILs, a group of cytokines involved in either immunologic or inflammatory response. ILs are mainly secreted by mononuclear cells that induce the growth and differentiation of lymphocytes and pluripotential stem cells. IL-5 is produced by Th2 cells and mast cells after activation by mitogens or antigens and is capable to induce the survival, growth and differentiation of eosinophils and B-cells. IL-10 is a pleiotropic cytokine with important immunoregulatory functions and potent anti-inflammatory properties. Binding of IL-10 to the extracellular domain of IL-10 receptor, together with the activation of IL6 receptor, activates phosphorylation of the receptor-associated JAK and STAT. Activation of JAK/STAT cascades is involved in the transcriptional regulation of several genes that regulate chemotaxis, inflammation, cell proliferation and migration (such as IL-8, Eotaxin, IFN-γ, TNF-α, Perforin and IL-6). IL-12 is a key immunoregulatory cytokine that coordinates innate and adaptive immune response. IL-13 plays a critical role in the regulation of immune responses and is also implicated in pathological conditions, such as asthma and allergy. IL-15 is a pleiotropic cytokine involved in several cellular process, such as proliferation, differentiation, immune response and cell survival. IL-23 plays an important role in differentiation and maintaining the Th17 cell population, a novel T-cell subset involved in antimicrobial immune response and establishment of many autoimmune diseases. IL-8 or CXCL8 is a chemokine produced by macrophages and other cell types that, through its binding to IL8 receptors (also known as CXCR1 and CXCR2), mediates several cellular functions, including neutrophil migration to sites of inflammation. IL-8 promotes NF-κB binding to DNA and transcriptionally regulates the expression of several factors involved in immune and inflammatory response. MHC class I molecules are specialized for presentation of endogenously synthesized proteins, bind ER chaperone calreticulin and then antigenic peptides. MHC class II molecules are found on antigen-presenting cells and present antigen derived from extracellular proteins (not cytosolic as in class I). TLR4, FCRL1 and FCRL2 are monocyte/macrophage-specific genes. Thermometers represent expression ratio in motor neuron among following conditions: (SALS1/control) and (SALS2/control). Upward thermometrs have red color and indicate expression ratios less than 1 while downward thermometers have blue color and indicate expression ratio less than 1. Pathway objects and links are described separately in the Supplementary Figure 1.
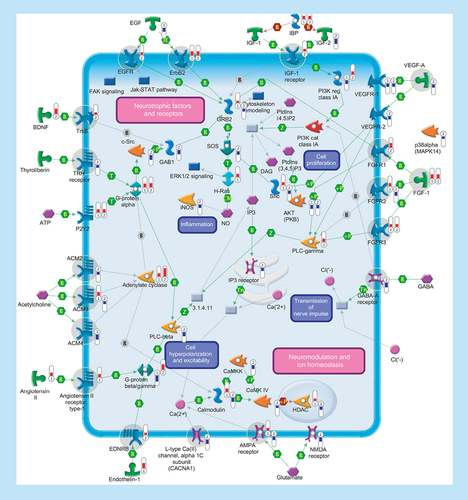
Several genes encoding ligands and receptors involved in signal transduction were differentially expressed in SALS patients. Signaling pathways are separated into two subsections (‘Neurotrophic factors and receptors’ and ‘Neuromodulation and ion homeostasis’), including primary (such as aAdenylate cyclase, phospholipase C or tyrosine kinase receptors) and secondary (such as Ca2+ release, PKC or protein kinase cascades) effectors that are regulated at multiple levels with different mechanisms (such as binding or phosphorylation). Thermometers represent expression ratio in motor neuron among following conditions: (SALS1/control) and (SALS2/control). Upward thermometers have red color and indicate expression ratios less than 1 while downward thermometers have blue color and indicate expression ratio less than 1. Pathway objects and links are described separately in the Supplementary Figure 1.
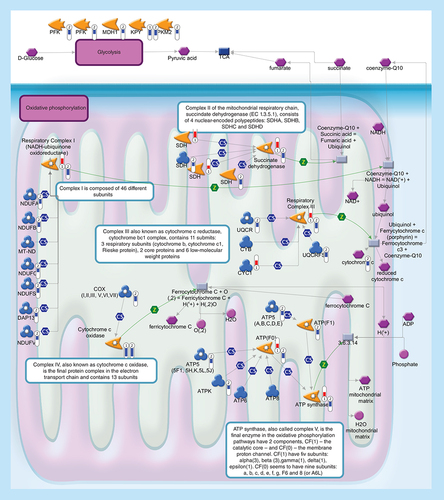
Glycolysis (upper level) converts glucose into pyruvate. During oxidative phosphorylation (lower level) electrons are transferred from electron donors to electron acceptors such as oxygen, in redox reactions. These redox reactions, carried out by five main protein complexes, release energy that is used to form ATP. The energy released by electrons flowing through this electron transport chain is used to transport protons across the inner mitochondrial membrane, in a process called chemiosmosis. This generates potential energy in the form of a pH gradient and an electrical potential across this membrane. Protons flow back across the membrane and down this gradient through ATP synthase, an enzyme that generates ATP from ADP, in a phosphorylation reaction. Thermometers represent expression ratio in motor neuron among following conditions: (SALS1/control) and (SALS2/control). Upward thermometrs have red color and indicate expression ratios less than 1 while downward thermometers have blue color and indicate expression ratio less than 1. Pathway objects and links are described separately in the Supplementary Figure 1.
In order to highlight a possible common pathogenic mechanism between familiar and sporadic ALS, we built up a pathway with 41 genes, some of which (37/41), besides being previously linked to FALS, were significantly deregulated in the cortex of SALS patients. Thermometers represent expression ratio in motor neuron among following conditions: (SALS1/control) and (SALS2/control). Upward thermometrs have red color and indicate expression ratios less than 1 while downward thermometers have blue color and indicate expression ratio less than 1. Pathway objects and links are described separately in the Supplementary Figure 1.
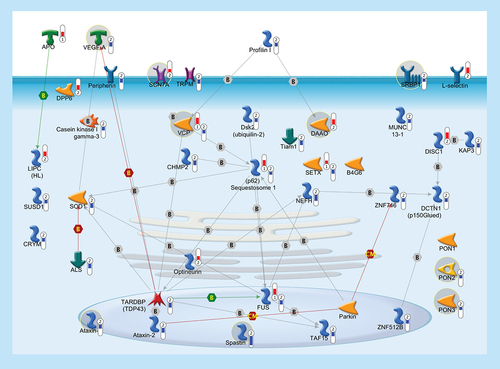
Important clues to mechanisms underlying ALS have been obtained through the identification of genes associated with FALS, such as copper/zinc SOD1, SETX, VAMPB, ALS2, SPG11, ANG, OPTN, VCP, C9ORF72, DCTN1 and TARDBP [Citation1]. However, despite intensive efforts, the pathogenic processes and precise genetic causes of ALS are not fully determined.
The lack of adequate knowledge about molecular and cellular players involved in the neurodegenerative progression of ALS made the development of effective treatments particularly difficult. In fact, to date, Riluzole is the only US FDA-approved drug for ALS that has been found to improve survival in patients but only to a modest extent [Citation2]. The failure of this and other putative therapies necessitates the development of new strategies for drug discovery targeting ALS pathogenesis as a system rather than on the level of the single protein molecule.
In the last few years, the advent of high-throughput technologies in sequencing of human genome and DNA microarray, have brought a deeper understanding of pathophysiological aspects of complex diseases, such as ALS, making possible to extend analysis to a great number of genes and pathways underlying disease pathogenesis [Citation3]. This ‘system biology’ approach is enabling us to overcome limitations arising from single-molecule analysis, offering noteworthy advantages for the discovery of several molecular mechanisms that, interacting with each other, may contribute to ALS pathogenesis [Citation4]. In addition, the advent of the systems biology and advances in genome research are opening the door to a more precise and accurate classification of disease, at the molecular level, that could revolutionize disease diagnosis, allowing for better clinical trial design and ultimately more individualized treatments for patients. The concept of personalized medicine is now undergoing a revolutionary change. While oncology offers multiple examples of how genomic medicine has changed disease understanding and therapy [Citation5], progresses in genomics of neurological diseases has been slow, mostly because of the poor availability and complex architecture of nervous tissue [Citation6].
During the last 10 years, our research group has made the most extensive database of whole genome expression profiles in the motor cortex of sporadic ALS patients. Although the use of postmortem brain tissues does not allow us to clarify whether investigated signaling cascades are a cause or a consequence of the disease process, their use provides essential features that cannot be obtained by using other approaches on a living patient.
In our previous work, the limited number of sample size has not enabled to differentiate patients in subgroups [Citation3]. Therefore, recently, we performed an unsupervised hierarchical clustering on a much larger sample size that allowed us to obtain a good separation of controls and SALS patients, segregating these latter into two greatly divergent groups () [Citation7]. This new molecular subclassification of SALS patients revealed etio-pathogenic mechanisms that were not put in evidence by considering SALS pathology as a single entity, providing a powerful means for defining molecular signatures for this disease. Moreover, the analysis of altered biological pathways in SALS provided a number of potential therapeutic targets for drugs currently in preclinical or clinical studies for several clinical conditions, including neurodegenerative diseases.
In the following sections, we rediscuss pathways emerged in our previous work, in light of the main targets within them, together with pharmacological compounds used for their regulation, highlighting their possible relevance to the development of more efficacious and individualized therapeutic interventions for ALS patients [Citation7]. In particular, proposed targets were selected taking into account encoded-proteins that are primary targets of drugs currently used in preclinical or clinical stages for treating several clinical diseases, with a particular focus on those that showed encouraging results for the treatment of neurodegenerative disorders.
Apoptosis & cell survival
A shift in the delicate balance between apoptosis- and survival-inducing genes plays a role in most neurodegenerative diseases, including ALS. Among genes associated with apoptosis and survival that were deregulated in cortex of SALS patients (), some encode for potential drug targets (Supplementary Table 1). Below, we describe in more details some of these targets (BCL-2; FasL; NF-κB) together with their possible pharmacological modulators.
BCL-2
BCL-2 is an antiapoptotic protein whose altered expression has been associated with neurodegenerative processes [Citation8]. In this regard, we observed downregulated expression of BCL-2 in SALS2 patients (), suggesting that the loss of this neuroprotective agent may contribute to trigger motor neurons degeneration.
Among drugs able to regulate BCL-2 activities, sodium phenylbutyrate has already been tested in Phase II clinical trials for ALS, showing promising results [Citation9]. This compound is a programmed cell death inhibitor and exerts its neuroprotective effects by pharmacological induction of BCL-2 with subsequent block both of cytochrome c release and caspase activation, contributing to slowing the motor neuron death in ALS mice and patients [Citation10].
Although the role of BCL-2 in ALS pathogenesis should be further investigated, the promising results obtained from its pharmacological modulation make this target a potential candidate for ALS therapy (Supplementary Table 1).
FasL
FasL or CD95L is a mediator of neuroinflammation and extrinsic apoptosis whose deregulation contributes to motor neuron degeneration in ALS [Citation11]. In accordance with these observations, we found increased expression of FasL in SALS2 patients (). Taken together, these studies support the role of FasL as a possible target for treating ALS (Supplementary Table 1).
Lenalidomide is a potent immunomodulatory agent that, by reducing the FasL expression, inactivates downstream effector caspases, extending survival in transgenic mouse models of ALS [Citation12]. These data encourage further clinical evaluation of lenalidomide as a therapeutic strategy to block or slow disease progression in human ALS patients.
NF-κB
NF-κB is a protein complex involved in various cellular mechanisms, including immune response and transcription regulation, cytokine production, cellular responses to oxidative stress as well as processes of synaptic plasticity and memory. The role of NF-κB on neuron survival is still controversial: while low levels of NF-κB have been related to loss of neuroprotection, high levels could be the result of its microglial induction occurring during neuroinflammatory processes [Citation13]. This apparent discrepancy may explain the opposite NF-κB expression found in SALS patients (decreased in SALS1 and increased in SALS2) ().
Despite the failure of some preclinical studies, NF-κB pharmacological inhibitors, like DL-3-n-butylphthalide (DL-NBP), seem to exert a neuroprotective role in ALS mainly by inhibiting programmed cell death and inflammation, reducing oxidative damage and improving mitochondrial function [Citation14]. Further studies aimed at defining the role of NF-κB in ALS pathogenesis are needed to elucidate its potentiality as a pharmacological target for personalized treatments of ALS patients (Supplementary Table 1).
Cell cycle
Alterations in gene expression and cellular distribution of cell-cycle regulating proteins have been characterized in several human neurodegenerative diseases, including ALS [Citation15], suggesting that the pharmacological modulation of this pathophysiological pathway could be beneficial in delaying the onset of ALS neurodegeneration (Supplementary Table 2).
In our recent work [Citation7], we observed the upregulation of some genes encoding proteins involved in G1 and G2 phases of cell cycle () and, among these, we identified some potential drug targets (CDK4; CHK2; cyclin D3) that will be discussed below in more details.
CDK4
SALS patients showed increased expression of CDK4 (), according to previous literature data [Citation16].
It is of interest to note that the pharmacological treatment with the CDK4 inhibitor minocycline attenuates microgliosis and slows down the motoneuronal degeneration in SOD1(G37R) mice [Citation17]. Additionally, other CDK4 inhibitor drugs (AT7519M, Alvocidib, R547, P276–00, Palbociclib), currently studied for cancer treatment, increase neuronal survival in a series of CNS disease models [Citation18]. Given this, CDK4 proves to be an interesting candidate therapeutic target for ALS (Supplementary Table 2) and future clinical trial should be undertaken truly to measure the efficacy of its inhibitors in ALS patients.
CHK2
Upregulation of CHK2 is found in SALS2 patients (). Although the role of CHK2 in ALS pathology remains to be elucidated, our results may support the hypothesis that increased CHK2 activity occurs after DNA damage induced by a variety of stress, leading to altered neuronal apoptosis [Citation19]. If so, pharmacological inhibition of CHK2 (e.g., by Rabusertib) may represent a potential therapeutic strategy to slow the progressive degeneration of motor neurons in ALS (Supplementary Table 2).
Cyclin D3
G1/S-specific cyclin-D3 is a protein that forms a complex with and functions as a regulatory subunit of CDK4. In SALS1 patients, we observed the upregulation of CCND3 (), supporting the theory that dysregulation of the cyclin system may contribute to ALS neurodegeneration [Citation20].
Drugs inhibiting cyclin D3 signaling, including the natural flavonoid silibinin, indirectly also decrease CDK4 activity, with a consequent reduction of oxidative stress and inflammation [Citation21]. Based on this, cyclin D3 inhibitors may thus be a new promising avenue for ALS therapy (Supplementary Table 2).
Cell adhesion
Alterations of the cell-adhesion system result in incorrect multicellular structure and cell–cell communications that, in some cases, may lead to the development of neurodegenerative processes, such as ALS [Citation22]. In support of this hypothesis, our analysis revealed differential expression of numerous genes involved in cell adhesion, mainly in SALS2 patients (). Among these, we discuss some potential candidates (MMPs; FAK-1) that may be attractive therapeutic intervention targets for the treatment of ALS (Supplementary Table 3).
MMP-1/MMP-2/MMP-13
Extracellular matrix metalloproteinases exert important functions in several neuroinflammatory processes as well as in synaptic remodeling and neuronal regeneration.
Upregulated expression of MMP-1, MMP-2 and MMP-13 has been observed in SALS2 patients (), in agreement with recent data [Citation23], supporting MMPs signaling cascade as a prominent therapeutic target for ALS (Supplementary Table 3).
Compounds inhibiting MMPs expression, synthesis and activity, exhibit beneficial effects in various neurodegenerative conditions [Citation24]. Particularly, thalidomide, a potent anti-inflammatory drug able to reduce MMP-2 levels, increases the lifespan of ALS transgenic mice [Citation25]. However, despite promising results obtained in ALS patients, the clinical use of this drug is limited by its side effects [Citation26]. Nonetheless, other MMPI inhibitors (marimastat, tanomastat, batimastat, ONO4817, doxycycline, rebimastat, pravastatin, S3304, halofuginone, melphalan), currently tested in preclinical or clinical trials as anticancer agents, have also gained interest for their therapeutic effects in neurodegenerative diseases, like Alzheimer's [Citation27,Citation28]. These encouraging results make MMP inhibitors attractive candidates for ALS therapy (Supplementary Table 3).
FAK-1
Upregulation of FAK-1 was found in SALS patients () and in other neurological diseases, such as Alzheimer's [Citation29], supporting its potential use as pharmacological target for neurological conditions, including ALS (Supplementary Table 3). Consistent with this hypothesis, oral administration of sulindac, a nonselective COX-inhibitor that also inhibits FAK-1 signaling cascade [Citation30], extends the survival of G93A SOD1 mice [Citation31]. Further studies are sorely needed to evaluate the potential use of sulindac and other FAK-1 inhibitors (such as PF562271) in ALS therapy (Supplementary Table 3).
Cytoskeleton remodeling & axonal transport
Our analysis uncovered differential expression of major components of the cytoskeleton in SALS patients (), in agreement with previous preclinical data [Citation32].
Below we discuss some potential drug targets (SEMA3A; CDK5; TUBBs) whose pharmacological modulation may be helpful in the recovery of the correct cytoskeleton remodeling and axonal transport might be of value in ALS.
SEMA3A
In SALS1 patients, we observed the upregulation of SEMA3A, a gene encoding a protein involved in the regulation of axon and dendrite growth guidance and neural system development (). Our findings support the hypothesis that increased expression of SEMA3A in ALS suppresses nerve terminal plasticity at specific neuromuscular synapses, contributing to early and selective motor neuronal loss [Citation33].
Although the role of SEMA3A in ALS pathology remains to be clarified, vinaxanthone and xanthofulvin, two selective SEMA3A inhibitors, have shown to promote neuronal regeneration [Citation34], suggesting a potential role of SEMA3A modulatory drugs as protective agents against neurodegenerative conditions, including ALS (Supplementary Table 4).
CDK5
Upregulated expression of CDK5 has been found in SALS1 patients (), according to previous data [Citation35].
Recent preclinical studies demonstrated that the pharmacological treatment with CDK5 inhibitors, such as olomoucine and roscovitine, attenuates neuronal cell death, reverses defective axonal transport and decreases microglial-dependent neurotoxicity, contributing to reduced ALS pathogenesis [Citation36,Citation37]. The robust effect of these and other CDK5 inhibitors (such as alvocidib) on the neuron survival spurs on future studies to evaluate the potentiality of this drug class in ALS therapy (Supplementary Table 4).
Tubulin proteins (TUBB1/TUBB3)
TUBB1 and TUBB3 belong to a protein family that, together with alpha-tubulin, makes up microtubules. TUBBs are expressed in neurons which are involved in neurogenesis as well as in axon guidance and maintenance.
In accordance with previous data [Citation3], the differential expression of genes encoding TUBB1/TUBB3 was observed in SALS patients (), suggesting that the tubulin system recovery might contribute to restore the altered nerve signal in ALS (Supplementary Table 4). In this regard, it is known that taxanes (paclitaxel, docetaxel) and other microtubule-targeting drugs (batabulin, vinorelbine, ABT751), widely used in cancer treatment, restore lost nerve signals in neurodegenerative diseases like Alzheimer's [Citation38]. Although the clinical use of this drug class is limited by their potential neurotoxicity, several novel compounds have been developed to prevent this side effect. Among these, olesoxime, a potent trophic factor stimulating neuronal survival and axonal sprouting, has demonstrated remarkable neuroprotective properties for motor neurons both in cell culture and in rodents [Citation39].
Immune response
Similar to what observed in both animal models and patients with ALS [Citation40], we found differential expression of an extensive number of immune-related genes in the cortex of SALS patients (). While further explorations are needed to clarify whether this system contributes to the disease pathogenesis or represents a response to damage, its potential as drug target is being explored [Citation41].
In the following subparagraphs, we describe some potential targets (TLR4; IL8R; MHC class I; ILs; JAK-2/JAK-3) whose pharmacological restoration may be useful for combating neuroinflammation in ALS patients (Supplementary Table 5).
TLR4
Similar to what is observed in reactive glial cells of ALS spinal cords [Citation42], SALS patients showed increased expression of TLR4 that encodes a protein involved in innate immunity response and microglial activation. (). In accordance with the hypothesis that the inhibition of TLR4 signaling could be helpful in reducing neuroinflammatory processes observed in ALS, several studies have demonstrated that the administration of TLR4 inhibitors (resatorvid, reparixin, E5531) improves the neurological functionality recovery after brain injury and neuroinflammation [Citation43–45] (Supplementary Table 5).
IL8R
SALS2 patients showed increased expression of IL8R (also known as CXCR2) encoding a chemokine receptor involved in neuroinflammatory cascade (). Our data are corroborated by previous findings showing a significant contribution to IL8R activation to motor neuron degeneration [Citation46]. Given this, drugs that inhibit IL-8R (reparixin, navarixin, elubrixin, SB332235) and suppress its pathogenic cascade, may reduce inflammation and provide neuroprotection in ALS (Supplementary Table 5).
MHC class I
Increased expression of genes encoding MHC class I molecules were found in SALS patients () and a similar expression was observed in the affected tissues in ALS [Citation47]. These data suggest that MHC antigens could promote susceptibility to ALS and, thus, represent potential drug targets for ALS treatment (Supplementary Table 5).
Among drugs that decrease MHC I expression, glatiramer acetate (GA) is an immunomodulator drug clinically used to treat multiple sclerosis that exerts a neuroprotective role in several neurological conditions [Citation48]. Phase II clinical trials of GA in patients with ALS are currently in progress (Supplementary Table 5).
IL-10
Downregulation of the gene encoding the potent anti-inflammatory cytokine IL-10 was observed in SALS2 patients (), sustaining the neuroprotective role of this factor against motor neuron injury in ALS [Citation49]. These studies suggest that the modulation of IL-10 activity may provide an avenue for new therapeutic approaches in ALS (Supplementary Table 5).
It is of interest to note that glatiramer acetate, in addition to its activity as MHC regulator (discussed above), also increases IL-10 levels [Citation50]. The multi-target action of this drug makes it a promising candidate for the treatment of ALS patients (Supplementary Table 5).
IL-5/IL-6R/IL-12/IL-23
Although the pathogenic role of cytokines in ALS is still unknown, recent studies have associated their abnormal expression to the clinical status [Citation51–53]. In line with these observations, we found upregulated expression of genes encoding some inflammatory mediators (IL-5, IL-6R, IL-12, IL-23) in SALS2 patients (), supporting their role as potential targets for the pharmacological treatment of ALS patients (Supplementary Table 5).
Among different IL inhibitors, tocilizumab, an anti-IL6R monoclonal antibody, showed an excellent activity in vitro against inflammatory activation in SALS patients [Citation54]. In addition, ustekinumab, an anti-IL-12/IL-23 monoclonal antibody, has reversed cognitive decline related to Alzheimer's disease [Citation55]. Further studies are needed to elucidate the potential clinical benefits of these and other inflammatory cytokine inhibitors (such as JTP-27536) in the clinical treatment of ALS (Supplementary Table 5).
JAK-2/JAK-3
SALS patients showed differential expression of genes encoding Janus kinase (JAK-1, KAK-2, JAK-3 and Tyk-2), a family of tyrosine kinases mainly involved in the regulation of gene expression (). These findings are in agreement with previous studies showing that the dysregulation of the JAK-STAT pathway occurs in inflammation and neurodegenerative diseases, including ALS [Citation56].
It is of interest to note that the pharmacological blockade of JAK-STAT pathway with JAK inhibitors has neuroprotective effects [Citation57]. Among members of this drug class, WHI-P131 has slowed disease progression and increased survival in an animal model of ALS [Citation58]. Further studies may clarify the therapeutic potential of this and other JAK inhibitors (AG490, AZD1480) in ALS treatment (Supplementary Table 5).
Signal transduction
Neurosignaling involves the largest class of proteins and receptors that transmit chemical signals from the cell surface to their intracellular targets. This section, for the multitude of drug targets identified, will be divided into two subsections (see the ‘Neurotrophic factors and receptors’ and ‘Neuromodulation and ion homeostasis’ sections), each of which will list their targets and relative pharmacological compounds.
Neurotrophic factors & receptors
Neurotrophin family includes proteins that influence the proliferation, differentiation, survival and death of neuronal and non-neuronal cells. One pathologic characteristic of neurodegenerative disorders, including ALS, is the loss of neurotrophins and their receptors, putting at risk the neuron health and stability [Citation59]. This notion is supported by our findings showing differential expression of several trophic factors and their receptors in SALS cortex ().
Despite the promising preclinical data, no neurotrophic factors succeeded yet in a clinical Phase III trial for ALS [Citation60]. This failure may be attributed to an incomplete knowledge of neurotrophin signaling cascades rather than to their limited value as potential targets for ALS therapy. In an effort to provide additional information about these signaling cascades and their potential for ALS therapy development, below we describe in details some neurotrophic factors (EGFR; TRHR; IGFR-1; FGFRs; TrkB; VEGF-A) together with their pharmacological modulators (Supplementary Table 6).
EGFR/ERBB2
SALS patients showed increased expression of genes encoding EGF and its receptors EGFR/ERBB2 (), consistently with the view that activation of this signaling pathway may trigger quiescent astrocytes into reactive astrocytes and, consequently, activate the neurodegenerative process [Citation61]. These results present EGF signaling cascade as a potential target to slow progression of ALS (Supplementary Table 6).
EGFR/ErbB2 inhibitors, clinically used in cancer treatment, have recently gained interest for their neuroprotective activities [Citation62]. Particularly, some of these drugs (erlotinib, genistein, masoprocol) significantly delay disease onset in ALS animal models [Citation63,Citation64]. Moreover, suramin, another important exponent of this drug class, is currently undergoing preclinical testing for neurological conditions, like Alzheimer's and Parkinson's [Citation65,Citation66]. In addition to its activity as EGF signaling inhibitor, suramin also inhibits the activity of HDAC, NOS and P2Y2 [Citation67,Citation68], three proteins which have been already extensively implicated in ALS [Citation69,Citation70].
Further developments and clinical validations of the above mentioned and other pharmacological agents belonging to EGFR/ErbB2 inhibitors class (PKI166; BMS690514; Canertinib; Gefitinib; PD153035) are needed to corroborate preclinical data, in the hope of translating them into clinical practice (Supplementary Table 6).
TRH receptor
Decreased expression of TRHR in SALS2 patients () is supported by similar findings in the spinal cord of ALS patients [Citation71]. These observations provide the rationale for future evaluations of TRHR signaling pathway as a potential target for ALS treatment (Supplementary Table 6). In this regard, masoprocol, besides its EGFR inhibitor activity, is also a TRHR agonist that exerts its neuroprotective effects through its anti-inflammatory and antioxidant properties as well as by reducing the glutamate neurotoxicity [Citation72]. Another TRH analogue, taltirelin, has shown neuroprotective effects in neurodegenerative disorders [Citation73]. Taken together, these observations contribute to make TRH agonists attractive candidates for restore damaged or maladaptive neural systems in ALS patients (Supplementary Table 6).
IGF1 receptor
Increased expression of IGFR-1 was found in SALS2 patients (), supporting the hypothesis that dysregulation of this signaling cascade may be critical for ALS pathogenesis [Citation74].
It has been observed that inhibition of the IGF signaling pathway increases lifespan and protects against neurodegeneration in model organisms [Citation75]. In this regard, simvastatin, an IGF receptor inhibitor, has been reported to reduce the excitotoxicity and neuronal apoptosis occurring in neurodegenerative diseases, like Alzheimer's and Parkinson's [Citation76,Citation77]. These data suggest that treatment with this or other IGF receptor inhibitors (such as masoprocol, BMS-754807 and linsitinib) may represent a potential strategy for the treatment of ALS (Supplementary Table 6).
FGF1/FGFR1/FGFR2/FGFR3
In SALS cortex samples, we observed the altered expression of genes encoding FGF1 and some of its receptors (FGFR1, FGFR2, FGFR3) (). According to our data, FGF-1 was believed to be released from motor neurons in response to oxidative stress, inducing FGFR1 accumulation and activating astrocytes that, in turn, could initiate motor neuron apoptosis in ALS [Citation78]. In support of this hypothesis, the FGFR inhibitor PD166866 has shown to prevent motor neuron apoptosis in ALS mouse model [Citation78]. Further studies are required to investigate the role of this and other inhibitors of FGF signal transduction (orantinib, brivanib, dovitinib, suramin, pentosan polysulfate) in ALS therapy (Supplementary Table 6).
TrkB
Increased expression of genes encoding BDNF and its receptor TrkB was found in SALS patients, in accordance with the observation that a prolonged TrkB activation may render motor neurons vulnerable to excitotoxic insult, contributing to developing ALS [Citation79]. These results suggest that inhibition of TrkB signaling could be considered a valid therapeutic strategy for treating ALS (Supplementary Table 6).
Although, due to the central role of BDNF in cognitive functions, the development of therapeutic compounds that interact with TrkB receptor has been difficult, some selective TrkB inhibitors (cyclotraxin-B and Ana-12) decrease neurotoxicity without affecting neuronal survival [Citation80,Citation81].
VEGF-A
Decreased expression of VEGF-A in SALS2 patients () is coherent with the view that reduced VEGF signaling may play a role in the pathogenesis of ALS [Citation82]. In support of this thesis, VEGF administration, in different models of ALS, has shown to strongly protect motor neurons from excitotoxicity [Citation83,Citation84]. In addition, some drugs activating VEGF (SB-509, celecoxib) have been tested in Phase II clinical trials for ALS, showing encouraging results [Citation85,Citation86]. All together, these observations propose VEGF-A signaling pathway as a promising new therapeutic target in ALS (Supplementary Table 6).
Neuromodulation & ion homeostasis
Deregulated expression of genes encoding proteins and/or their receptors involved in neuromodulation was observed in the motor cortex of SALS patients (). Among these, we discuss some potential candidates (GABRs; CACNA1C; GRIAs; ACMs; AGTR1; EDNR-B) that may be taken into account as targets in ALS therapy (Supplementary Table 7).
GABA-A receptor
SALS2 patients showed downregulated expression of gene encoding GABA-A (), coherently with several studies that highlight the presence of reduced GABA levels in the motor cortex of ALS patients [Citation87]. Drugs enhancing GABA-A signaling may thus represent potential strategies to treat or prevent the progressive motorneuron degeneration in ALS patients (Supplementary Table 7). In this regard, some GABA-A agonists (ganaxolone, clomethiazole, GT 1061, clorazepate, zolpidem, eszopiclone, pentobarbital, GT 1061) have shown to prevent excitotoxicity and neuronal loss, exerting neuroprotective effects in several neurological disorders, such as Alzheimer's and Parkinson's [Citation88–91].
CACNA1C & GRIA-1/GRIA-2
Decreased expression of CACNA1C and differential expression of genes encoding AMPA glutamate receptors subunits (GRIA-1, GRIA-2) were found in SALS patients (). Our findings support the hypothesis that toxic glutamate challenges, together with perturbation of neuronal Ca2+ homeostasis, may induce pathological changes in motor neuron circuitry, contributing to ALS pathogenesis [Citation92]. If so, appropriate pharmacological strategies that minimize glutamatergic neurotoxicity and restore Ca2+ homeostasis could be helpful in ALS treatment (Supplementary Table 7). In this regard, FPL 64176 and Bay K 8644, two CACNA1C agonists, have shown neuroprotective proprieties in preclinical studies for ALS [Citation93].
An alternative approach to re-establish Ca2+ homeostasis and reduce glutamate excitotoxicity is represented by the administration of AMPA receptor antagonists. Some of them (ZK 187638, NBQX, GYKI-52,466) have already been tested in ALS animal models, showing to protect motor neurons, improve motor function and prolong the survival [Citation94–96]. Another AMPA antagonist, talampanel, has shown promising preclinical data for ALS [Citation97], but only a limited effectiveness has been observed in clinical trial [Citation98]. The differential expression of GRIA1 and GRIA2 observed in SALS patients (GRIA1 upregulated in SALS1 and GRIA2 reduced in SALS2) may contribute to explain this clinical failure. In the light of this, further studies are needed to re-evaluate the potential therapeutic benefits of talampanel for the personalized treatment of ALS patients.
ACM2/ACM3/ACM4
SALS patients showed a differential expression of genes encoding muscarinic acetylcholine receptors (ACM2, ACM3, ACM4) (), according to previous studies [Citation99]. Therefore, pharmacological approaches aimed to repair the defective muscarinic signaling may contribute to reduce motor neuron death and ALS progression (Supplementary Table 7). In support of this hypothesis, several preclinical and clinical studies have shown that muscarinic agonists (such as cevimeline, xanomeline, bethanechol and clozapine) trigger the restoration of specific neuronal populations lost to a variety of CNS disorders, including ALS [Citation100,Citation101].
AGTR1
Consistent with previous studies that link dysregulation of angiotensin II system to ALS pathology [Citation102], we found increased expression of genes encoding angiotensin II and its receptor AGTR1 in SALS2 patients ().
Among AGTR1 blockers (ARBs), olmesartan has manifested neurotrophic properties on spinal motor neurons in vitro and in vivo [Citation103]. In addition, also other AGTR1 blockers (ARBs; telmisartan, olmesartan, candesartan), clinically used as antihypertensive agents, show neuroprotective effects in Alzheimer's and other neuronal diseases [Citation104–106]. These drugs exert their neuroprotective actions also by significantly enhancing GLT-1 expression [Citation107], whose loss of function has been implicated in the pathogenesis of ALS [Citation108].
All the above observations highlight a potential role of angiotensin signaling pathway as a new target in ALS therapy (Supplementary Table 7).
EDNR-B
Increased expression of genes encoding EDN-1 and its receptor EDNR-B was found in SALS2 patients (), coherently with previous studies that show a correlation between endothelin signal alterations and ALS pathology [Citation3]. If so, the pharmacological modulation of this signal pathway might represent a new therapeutic strategy for ALS (Supplementary Table 7). In favor of this hypothesis, several studies showed that EDNR-B antagonists (such as BQ-788, bosentan, IRL-2500) exert neuroprotective properties besides being able to reduce neuroinflammation [Citation109–111].
Mitochondrial functions & energy metabolism
Energy metabolism is an extremely complicated series of chemical reactions that break down organic matter to harvest energy. Mitochondrial dysfunctions, as well as disruption of energy metabolism, were reported to play an important role in ALS pathology [Citation112]. Dysfunctional mitochondria, in fact, exhibit a reduction in bioenergetics efficiency, putting neurons at risk of death when energy demands exceed cellular energy production. In accordance with these observations, we found a coordinated decrease of some genes encoding proteins involved in the mitochondrial oxidative phosphorylation pathway in SALS2 patients ().
Some drugs that are able to reverse mitochondrial dysfunction (acetyl-l-carnitine, creatine, minocycline, olesoxime, dexpramipexole, cyclosporine A) are currently in clinical trials for ALS, showing to be effective and well tolerated [Citation113–116]. In addition, other members of this drug class (uridine, pyruvate, dichloroacetate) have shown promising data in preclinical testing for ALS [Citation117–119]. The success of these studies supports the idea that mitochondria-targeted therapies may be helpful for the management of bioenergetics defects observed in SALS2 patients (Supplementary Table 8).
Genes altered in sporadic & familial ALS
SALS patients showed differential expression of a large number of genes, whose mutation has been previously associated to FALS (). This association provides a potential common pathogenic link between familiar and sporadic ALS that may affect the development of new strategies to treat both forms of ALS (Supplementary Table 9).
In the following subparagraphs will be discussed in details some candidate targets (VEGF, SRBP1, SCN7, ATXN1, SPAST, VCP, DAAO, PONs) with their respective pharmacological compounds.
VEGF
As already mentioned, VEGF exerts a protective role on motor neurons and decreased expression of VEGF was found in SALS2 patients ().
Low levels of VEGF have also been observed in individuals homozygous for a variety of mutations in the VEGF promotor region, suggesting that the lack of this neuroprotective factor may also contribute to FALS [Citation120]. Based on these assumptions, the pharmacological activation of the VEGF signaling pathway could be a promising therapeutic strategy to treat both forms of ALS (Supplementary Table 9).
SRBP1
Mutations in SRBP1 gene have been associated with juvenile ALS [Citation121]. In addition, our analysis has revealed a reduced expression of SRBP1 in SALS2 patients (), supporting the neuroprotective role of this protein in maintaining physiological motor neuron functions, including proliferation, survival and death.
SRBP1 pharmacological ligands (such as igmesine, dextromethorphan, PRE-084) exert neuroprotective effects in a variety of neurodegenerative diseases, including ALS [Citation122,Citation123], suggesting SRBP1 signal pathway as promising target for ALS therapy (Supplementary Table 9).
SCN7
In SALS2 patients, we found upregulated expression of SCN7 (). In addition, an abnormal increase in the persistent Na+ currents has been observed in a mouse model of a familial form of ALS [Citation124]. If so, drugs that restore physiological Na+ currents and reduce the consequent hyperexcitability may be helpful to treat both forms of ALS (Supplementary Table 9). In support of this hypothesis, it has been seen that riluzole, the only FDA-approved drug for ALS, is able to block sodium currents reducing the firing frequency and hyperexcitability observed in ALS [Citation125]. Moreover, other sodium channel blockers (topiramate, mexiletine, lamotrigine, valproic acid, zonisamide, riluzole, tetrodotoxin) have shown promising preclinical and clinical results against several neurodegenerative diseases [Citation126–128], including ALS [Citation129,Citation130] (Supplementary Table 9).
ATXN1
ATXN1 is mainly implicated in mRNA processing by forming an RNA-dependent complex with PQBP-1. ATXN1 normally contains a polyglutamine (polyQ) tract with 22–23 repeats and evidences suggest that an augmented number of polyQ repeats (23–34) may increase genetic risk for ALS [Citation131]. In addition to these observations, SALS2 patients showed decreased expression of ATXN-1 (), suggesting that alterations of ATXN-1 functions may be involved in the development of both sporadic and familial ALS.
Among agents able to prevent mutant ATXN-1 aggregation and the consequent neurotoxicity, bromocriptine, an FDA-approved drug for the treatment of Parkinson's disease, has also shown promising results in preclinical stages for ALS [Citation132].
All together, these results propose ATXN-1 and its pharmacological modulators as potential therapeutic strategies against ALS (Supplementary Table 9).
SPAST
Spastin is an ATPase microtubules binding necessary for normal neurite outgrowth. Recent evidences suggest that mutations in SPAST (also known as SPG4) are responsible for a loss of spastin function and may represent a genetic risk factor in ALS [Citation133]. In addition, we found decreased expression of SPAST in SALS2 patients (). These observations highlight how spastin may be a valid new therapeutic target in both forms of ALS (Supplementary Table 9). Consistent with this hypothesis, some microtubule-targeting-drugs (noscapine, vinblastine) have shown to increase motor neuron survival, delay symptoms and life extension in symptomatic SOD1G93A mice [Citation134]. Further clinical studies are required to verify if this drug class may represent a pharmacological strategy for ALS (Supplementary Table 9).
VCP
VCP is a member of an ATP-binding protein family, implicated in a number of cellular events, including vesicle transport and protein degradation.
VCP mutations were identified as the genetic cause of the frontotemporal dementia but, recently, a specific mutation (p.R191Q) was also observed in approximately 1–2% of familial ALS cases [Citation135]. In addition to its potential role as genetic risk factor in FALS patients, the differential expression of VCP was observed in SALS patients (), consistently with the view that altered VCP functionality is linked with the toxic gain of function of TDP-43 protein [Citation136].
TDP-43 is a DNA/RNA binding protein that exerts an important role for transcription, pre-mRNA splicing, mRNA stability and mRNA transport [Citation137]. Different insult damage, such as calcium dysregulation and oxidative stress, can cause migration of TDP-43 from the nucleus to the cytoplasm where it sequesters cytoplasmic RNAs stress granule, producing loss and gain of splicing functions. These aberrant nuclear or cytoplasmic TDP-43 inclusions have been also observed in nervous tissue of ALS patients [Citation138]. Despite several studies have investigated the role of TDP-43 in ALS pathogenesis, its complex involvement deserves additional investigations. Meanwhile, the pharmacological modulation of VCP may reduce motorneuron-toxicity TDP-43-mediate and thus represents a therapeutic strategy for ALS (Supplementary Table 9). In this regard, some VCP inhibitors (Vesnarinone, Xanthohumol) [Citation139,Citation140] have demonstrated to prevent neuroinflammation and apoptosis associated with several neurological diseases [Citation141,Citation142].
DAAO
DAAO is a flavoenzyme localized in motor neurons that metabolize D-serine.
A mutation in DAAO (R199W) has been associated with classical adult-onset FALS [Citation143]. In addition to this, we found the upregulated expression of DAAO in SALS2 patients (), in agreement with the observation that altered DAAO activity may lead to increased accumulation of D-serine observed in ALS mouse models and patients [Citation144]. It is, therefore, plausible that drugs reducing D-serine or controlling DAAO activity may be helpful in treating both forms of ALS (Supplementary Table 9). In this respect, recent studies demonstrate that DAO inhibitors (AS057278, sodium benzoate, chlorpromazine) are helpful in treatment of several neuronal conditions, like Alzheimer's [Citation145–148].
PON2/PON3
Mutations in PON2 and PON3 two genes encoding enzymes involved in the prevention of oxidative damage [Citation149], have been associated with familial and sporadic ALS [Citation150]. Moreover, decreased expression of PON2 and PON3 was observed in SALS2 patients (), supporting the neuroprotective role exerted by these factors. Among drugs that are able to improve PON-2/PON-3 activity, genistein (previously mentioned for its activity as EGFR/ErbB2 inhibitor) has been tested in preclinical studies for ALS, showing promising results [Citation64]. Other drugs PONs-activating (montelukast, berberine) manifest neuroprotective effects in animal models of various neuronal diseases [Citation151,Citation152]. Taken together, these findings suggest that PON-activating drugs might to be evaluated as potential therapeutic strategies to prevent or reduce neurodegeneration in ALS (Supplementary Table 9).
Conclusion
ALS is a fatal motor neuron disease where different mechanisms interact to promote the disease progression. Despite intensive research and dozens of preclinical and clinical strategies, ALS remains incurable and the only available treatment, Riluzole, has showed only modest benefit to patients. This lack of progress may be mainly because the current classification of ALS diseases does not take into account the complexity and heterogeneity of molecular pathogenetic mechanisms in different patients.
In our previous work, we used unsupervised hierarchical clustering to identify two SALS patient subgroups, characterized by their gene expression pattern, and revealed new clues to pathogenesis and potential therapeutic targets.
As it is already happening in the cancer genomics field, the emerging molecular taxonomy of SALS patients may open new avenues toward genotype-specific therapeutic interventions, providing opportunities for developing more effective and individualized diagnosis and treatments for SALS patients, with consequent reduced toxicity and increased treatment efficacy. Moreover, some drugs that were abandoned at clinical stages because of their low efficacy and/or toxicity, may be re-evaluated for their potential therapeutic role (an approach known as ‘drug repositioning’), with the consequent possibility to reduce both the time and costs associated with drug discovery and development. At last, the identification of a possible common pathogenic mechanism between familiar and sporadic ALS might open a new vista for a therapy helpful for both forms of this disease.
The purpose of this review is to provide a useful guide for the rational selection of the most promising drug targets whose pharmacological modulation may be helpful in the development of efficacious therapeutic interventions based on molecular subclassification of ALS disease.
Future perspective
Future preclinical or clinical validation of the most promising targets investigated in this paper will be crucial to provide useful information for the development of an effective and personalized therapeutic plan. Unfortunately, a disconnection exists between preclinical target validation and their translation into clinical practice. This is mainly due to the use of monogenic animal models (e.g., SOD1G93A, TDP-43, TAUP301L and Wobbler mice) that do not fully reproduce the complex, multifactorial nature of SALS. However, their use in the preclinical stage of pharmacological research is inevitable. Therefore, it becomes evident the need to use several strategies, such as meta-analysis on gene expression data from human and ALS animal models, in order to identify common genomic changes and, consequently, permit the prioritization and strategy selection of targets in view of their subsequent preclinical and clinical validation.
Amyotrophic lateral sclerosis: Devastating, progressive neurodegenerative disease, caused by the loss of motor neurons in the brain and spinal cord. ALS is divided into familial ALS (FALS – where there is a family history of others with the disease) and sporadic ALS (SALS – where there is no family history).
Systems biology: Biology-based interdisciplinary field of study that focuses on complex interactions within biological systems, using a holistic approach (holism instead of the more traditional reductionism) to biological and biomedical research.
Personalized medicine: The use of marker-assisted diagnosis and targeted therapies derived from an individual's molecular profile. The aim of patient-oriented research is to identify the best treatment strategy guided by disease heterogeneity diagnostics.
Expression profiles: Determination of the pattern of thousands of genes expressed, at the level of transcription, under specific circumstances or in a specific cell to give a global picture of cellular function.
Drug target: A key molecule involved in a particular metabolic or signaling pathway that is specific to a disease condition whose activity is modified by a drug resulting in a desirable therapeutic effect.
Signaling pathways: Represent the way in which the combined effect of gene activity elicits cell-level responses by activating/deactivating specific functionalities in response to a particular stimulus through a chain of intermediate molecules. Different software tools, such as Metacore, have been developed to help visualizing, editing and superimposing gene expression values over the pathway, in order to transform expression data, of often difficult interpretation into meaningful functional information.
Molecular taxonomy: Classification of diseases based on underlying molecular and environmental causes, rather than on physical signs and symptoms.
Amyotrophic lateral sclerosis (ALS) is a devastating, progressive neurodegenerative disease, caused by the loss of motor neurons in the brain and spinal cord. Although the identification of disease-linked mutations has played an important role in understanding ALS pathology, the development of an effective therapy remains an elusive goal.
With the advent of high-throughput technologies, such as genome sequencing and DNA microarray, the transcriptional cascades and networks regulating ALS neurodegeneration are being elucidated, providing a genome portrait of the disease.
In view of the better understanding of molecular and cellular players in ALS, it became necessary to reformulate strategies for drug development, moving pharmacological research toward treatments targeting the ALS pathogenesis as a system rather than at the level of the single protein molecules.
Our research group has, recently, analyzed whole-genome expression profiles of motor cortex samples from SALS patients and identified two SALS subtypes, each associated with deregulation of different genes and pathways. The analysis of altered biological pathways in SALS also provided a number of potential therapeutic targets for drugs currently in preclinical or clinical studies for several clinical conditions, including neurodegenerative diseases.
As it is already happening in the cancer genomics field, the molecular taxonomy of SALS patients, emerged in our work, may open the avenue toward genotype-specific therapeutic interventions, providing opportunities for determining treatment selection and directing patients to clinical trials, with consequent reduced toxicity and increased treatment efficacy.
In this review, we rediscuss pathways identified in our previous work, in light of the main targets contained in them, together with pharmacological compounds used for their regulation.
This ‘system biology/pharmacology’ approach may reveal new therapeutic indications for existing drugs that, alone or in a ‘cocktail’ therapy, may be re-evaluated for their potential therapeutic role in ALS, opening the way toward developing of genotype-specific and more efficient therapeutic interventions.
Future preclinical or clinical validation of the most promising targets investigated in this paper, will be crucial to provide useful information for the development of an effective and personalized therapeutic plan, offering a new hope of a more optimistic future for ALS patients.
Open acess
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Supplementary Figure 1
Download MS Word (493.6 KB)Supplementary Information
Download MS Excel (2.6 MB)Acknowledgements
The authors gratefully acknowledge Cristina Calì, Alfia Corsino, Maria Patrizia D'Angelo and Francesco Marino for their administrative and technical assistance.
Financial & competing interests disclosure
SC acknowledges support from Italian Ministry of Education, University and Research (MIUR) grant number CTN01_00177_817708. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Pasinelli P , BrownRH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci.7 (9), 710–723 (2006).
- Turner MR , PartonMJ, LeighPN. Clinical trials in ALS: an overview. Semin. Neurol.21 (2), 167–175 (2001).
- Lederer CW , TorrisiA, PantelidouM, SantamaN, CavallaroS. Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC Genomics8 (1), 26 (2007).
- Ferraiuolo L , KirbyJ, GriersonAJ, SendtnerM, ShawPJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol.7 (11), 616–630 (2011).
- Gasparini G , LongoR. The paradigm of personalized therapy in oncology. Expert Opin. Ther. Targets16 (Suppl. 1), S7–S16 (2012).
- Sharp FR , XuH, LitLet al. The future of genomic profiling of neurological diseases using blood. Arch. Neurol.63 (11), 1529–1536 (2006).
- Aronica E , BaasF, IyerA, Ten AsbroekAL, MorelloG, CavallaroS. Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiol. Dis.74C, 359–376 (2014).
- Glantz LA , GilmoreJH, LiebermanJA, JarskogLF. Apoptotic mechanisms and the synaptic pathology of schizophrenia. Schizophr. Res.81 (1), 47–63 (2006).
- Cudkowicz ME , AndresPL, MacdonaldSAet al. Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph. Lateral. Scler.10 (2), 99–106 (2009).
- Ryu H , SmithK, CameloSIet al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem.93 (5), 1087–1098 (2005).
- Raoul C , BuhlerE, SadeghiCet al. Chronic activation in presymptomatic amyotrophic lateral sclerosis (ALS) mice of a feedback loop involving Fas, Daxx, and FasL. Proc. Natl Acad. Sci. USA103 (15), 6007–6012 (2006).
- Neymotin A , PetriS, CalingasanNYet al. Lenalidomide (revlimid) administration at symptom onset is neuroprotective in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol.220 (1), 191–197 (2009).
- Camandola S , MattsonMP. Nf-kappa b as a therapeutic target in neurodegenerative diseases. Expert Opin. Ther. Targets11 (2), 123–132 (2007).
- Feng XH , YuanW, PengY, LiuMS, CuiLY. Therapeutic effects of dl-3-n-butylphthalide in a transgenic mouse model of amyotrophic lateral sclerosis. Chin. Med. J. (Engl.)125 (10), 1760–1766 (2012).
- Vincent I , PaeCI, HallowsJL. The cell cycle and human neurodegenerative disease. Prog. Cell Cycle Res.5, 31–41 (2003).
- Wang W , BuB, XieM, ZhangM, YuZ, TaoD. Neural cell cycle dysregulation and central nervous system diseases. Prog. Neurobiol.89 (1), 1–17 (2009).
- Nguyen MD , BoudreauM, KrizJ, Couillard-DespresS, KaplanDR, JulienJP. Cell cycle regulators in the neuronal death pathway of amyotrophic lateral sclerosis caused by mutant superoxide dismutase 1. J. Neurosci.23 (6), 2131–2140 (2003).
- Liu DZ , AnderBP. Cell cycle inhibition without disruption of neurogenesis is a strategy for treatment of aberrant cell cycle diseases: an update. TheScientificWorldJournal2012, 491737 (2012).
- Zhang Y , QuD, MorrisEJet al. The Chk1/Cdc25A pathway as activators of the cell cycle in neuronal death induced by camptothecin. J. Neurosci.26 (34), 8819–8828 (2006).
- Becker EB , BonniA. Cell cycle regulation of neuronal apoptosis in development and disease. Prog. Neurobiol.72 (1), 1–25 (2004).
- Tota S , KamatPK, ShuklaR, NathC. Improvement of brain energy metabolism and cholinergic functions contributes to the beneficial effects of silibinin against streptozotocin induced memory impairment. Behav. Brain Res.221 (1), 207–215 (2011).
- Boutahar N , WierinckxA, CamdessancheJPet al. Differential effect of oxidative or excitotoxic stress on the transcriptional profile of amyotrophic lateral sclerosis-linked mutant sod1 cultured neurons. J. Neurosci. Res.89 (9), 1439–1450 (2011).
- Niebroj-Dobosz I , JanikP, SokolowskaB, KwiecinskiH. Matrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Eur. J. Neurol.17 (2), 226–231 (2010).
- Jarvelainen H , SainioA, KouluM, WightTN, PenttinenR. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol. Rev.61 (2), 198–223 (2009).
- Kiaei M , PetriS, KipianiKet al. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci.26 (9), 2467–2473 (2006).
- Stommel EW , CohenJA, FadulCEet al. Efficacy of thalidomide for the treatment of amyotrophic lateral sclerosis: a phase II open label clinical trial. Amyotroph. Lateral Scler.10 (5–6), 393–404 (2009).
- Costa R , SperettaE, CrowtherDC, CardosoI. Testing the therapeutic potential of doxycycline in a drosophila melanogaster model of Alzheimer disease. J. Biol. Chem.286 (48), 41647–41655 (2011).
- Dong W , VuleticS, AlbersJJ. Differential effects of simvastatin and pravastatin on expression of Alzheimer's disease-related genes in human astrocytes and neuronal cells. J. Lipid Res.50 (10), 2095–2102 (2009).
- Grace EA , BusciglioJ. Aberrant activation of focal adhesion proteins mediates fibrillar amyloid beta-induced neuronal dystrophy. J. Neurosci.23 (2), 493–502 (2003).
- Weyant MJ , CarothersAM, BertagnolliME, BertagnolliMM. Colon cancer chemopreventive drugs modulate integrin-mediated signaling pathways. Clin. Cancer Res.6 (3), 949–956 (2000).
- Kiaei M , KipianiK, PetriSet al. Integrative role of cPLA with COX-2 and the effect of non-steriodal anti-inflammatory drugs in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurochem.93 (2), 403–411 (2005).
- De Vos KJ , GriersonAJ, AckerleyS, MillerCC. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci.31, 151–173 (2008).
- De Winter F , VoT, StamFJet al. The expression of the chemorepellent semaphorin 3a is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol. Cell. Neurosci.32 (1–2), 102–117 (2006).
- Axelrod A , EliasenAM, ChinMR, ZlotkowskiK, SiegelD. Syntheses of xanthofulvin and vinaxanthone, natural products enabling spinal cord regeneration. Angew. Chem. Int. Ed. Engl.52 (12), 3421–3424 (2013).
- Bajaj NP . Cyclin-dependent kinase-5 (CDK5) and amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord.1 (5), 319–327 (2000).
- Appert-Collin A , HugelB, LevyRet al. Cyclin dependent kinase inhibitors prevent apoptosis of postmitotic mouse motoneurons. Life Sci.79 (5), 484–490 (2006).
- Mapelli M , MassimilianoL, CrovaceCet al. Mechanism of cdk5/p25 binding by cdk inhibitors. J. Med. Chem.48 (3), 671–679 (2005).
- Shemesh OA , SpiraME. Rescue of neurons from undergoing hallmark tau-induced Alzheimer's disease cell pathologies by the antimitotic drug paclitaxel. Neurobiol. Dis.43 (1), 163–175 (2011).
- Sunyach C , MichaudM, ArnouxTet al. Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology62 (7), 2346–2352 (2012).
- Mccombe PA , HendersonRD. The role of immune and inflammatory mechanisms in ALS. Curr. Mol. Med.11 (3), 246–254 (2011).
- Calvo A , MogliaC, BalmaM, ChioA. Involvement of immune response in the pathogenesis of amyotrophic lateral sclerosis: a therapeutic opportunity?CNS Neurol. Disord. Drug Targets9 (3), 325–330 (2010).
- Casula M , IyerAM, SplietWGet al. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience179, 233–243 (2011).
- Villa P , TriulziS, CavalieriBet al. The interleukin-8 (IL-8/CXCL8) receptor inhibitor reparixin improves neurological deficits and reduces long-term inflammation in permanent and transient cerebral ischemia in rats. Mol. Med.13 (3–4), 125–133 (2007).
- Suzuki Y , HattoriK, HamanakaJet al. Pharmacological inhibition of TLR4-NOX4 signal protects against neuronal death in transient focal ischemia. Sci. Rep.2, 896 (2012).
- Akashi S , SaitohS, WakabayashiYet al. Lipopolysaccharide interaction with cell surface Toll-like receptor 4-MD-2: higher affinity than that with MD-2 or CD14. J. Exp. Med.198 (7), 1035–1042 (2003).
- De Paola M , BuanneP, BiordiL, BertiniR, GhezziP, MenniniT. Chemokine MIP-2/CXCL2, acting on CXCR2, induces motor neuron death in primary cultures. Neuroimmunomodulation14 (6), 310–316 (2007).
- Lampson LA , KushnerPD, SobelRA. Major histocompatibility complex antigen expression in the affected tissues in amyotrophic lateral sclerosis. Ann. Neurol.28 (3), 365–372 (1990).
- Scorisa JM , FreriaCM, VictorioSC, BarbizanR, ZanonRG, OliveiraAL. Glatiramer acetate treatment increases stability of spinal synapses and down regulates MHC I during the course of eae. Int. J. Biol. Sci.7 (8), 1188–1202 (2011).
- Xin J , WainwrightDA, MesnardNA, SerpeCJ, SandersVM, JonesKJ. Il-10 within the CNS is necessary for CD4+ t cells to mediate neuroprotection. Brain. Behav. Immun.25 (5), 820–829 (2011).
- Arnon R , AharoniR. Mechanism of action of glatiramer acetate in multiple sclerosis and its potential for the development of new applications. Proc. Natl Acad. Sci. USA101 (Suppl. 2), 14593–14598 (2004).
- Moreau C , DevosD, Brunaud-DanelVet al. Elevated IL-6 and TNF-alpha levels in patients with ALS: inflammation or hypoxia? Neurology 65 (12), 1958–1960 (2005).
- Shi N , KawanoY, TateishiTet al. Increased IL-13-producing t cells in ALS: positive correlations with disease severity and progression rate. J. Neuroimmunol.182 (1–2), 232–235 (2007).
- Rentzos M , RombosA, NikolaouCet al. Interleukin-17 and interleukin-23 are elevated in serum and cerebrospinal fluid of patients with ALS: a reflection of th17 cells activation? Acta Neurol. Scand. 122 (6), 425–429 (2010).
- Mizwicki MT , FialaM, MagpantayLet al. Tocilizumab attenuates inflammation in ALS patients through inhibition of IL6 receptor signaling. Am. J. Neurodegener. Dis.1 (3), 305–315 (2012).
- Vom Berg J , ProkopS, MillerKRet al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat. Med.18 (12), 1812–1819 (2012).
- Nicolas CS , AmiciM, BortolottoZAet al. The role of JAK-STAT signaling within the CNS. JAKSTAT2 (1), e22925 (2013).
- Ignarro RS , VieiraAS, SartoriCR, LangoneF, RogerioF, ParadaCA. JAK2 inhibition is neuroprotective and reduces astrogliosis after quinolinic acid striatal lesion in adult mice. J. Chem. Neuroanat.48–49, 14–22 (2013).
- Trieu VN , LiuR, LiuXP, UckunFM. A specific inhibitor of janus kinase-3 increases survival in a transgenic mouse model of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun.267 (1), 22–25 (2000).
- Chao MV , RajagopalR, LeeFS. Neurotrophin signalling in health and disease. Clin. Sci. (London)110 (2), 167–173 (2006).
- Henriques A , PitzerC, SchneiderA. Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: where do we stand?Front. Neurosci.4, 32 (2010).
- Liu B , ChenH, JohnsTG, NeufeldAH. Epidermal growth factor receptor activation: an upstream signal for transition of quiescent astrocytes into reactive astrocytes after neural injury. J. Neurosci.26 (28), 7532–7540 (2006).
- Mizuno M , SotoyamaH, NambaHet al. Erbb inhibitors ameliorate behavioral impairments of an animal model for schizophrenia: implication of their dopamine-modulatory actions. Transl. Psychiat.3, e252 (2013).
- Boston-Howes W , WilliamsEO, BogushA, ScolereM, PasinelliP, TrottiD. Nordihydroguaiaretic acid increases glutamate uptake in vitro and in vivo: therapeutic implications for amyotrophic lateral sclerosis. Exp. Neurol.213 (1), 229–237 (2008).
- Trieu VN , UckunFM. Genistein is neuroprotective in murine models of familial amyotrophic lateral sclerosis and stroke. Biochem. Biophys. Res. Commun.258 (3), 685–688 (1999).
- Chan CF , Lin-ShiauSY. Suramin prevents cerebellar granule cell-death induced by dequalinium. Neurochem. Int.38 (2), 135–143 (2001).
- Heneberg P . Use of protein tyrosine phosphatase inhibitors as promising targeted therapeutic drugs. Curr. Med. Chem.16 (6), 706–733 (2009).
- Xu K , DaiXL, HuangHC, JiangZF. Targeting HDACs: a promising therapy for Alzheimer's disease. Oxid. Med. Cell. Longev.2011, 143269 (2011).
- Goureau O , LepoivreM, BecquetF, CourtoisY. Differential regulation of inducible nitric oxide synthase by fibroblast growth factors and transforming growth factor beta in bovine retinal pigmented epithelial cells: inverse correlation with cellular proliferation. Proc. Natl Acad. Sci. USA90 (9), 4276–4280 (1993).
- Janssen C , SchmalbachS, BoeseltS, SarletteA, DenglerR, PetriS. Differential histone deacetylase mRNA expression patterns in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol.69 (6), 573–581 (2010).
- Carlesi C , PasqualiL, PiazzaSet al. Strategies for clinical approach to neurodegeneration in amyotrophic lateral sclerosis. Arch. Ital. Biol.149 (1), 151–167 (2011).
- Manaker S , CaineSB, WinokurA. Alterations in receptors for thyrotropin-releasing hormone, serotonin, and acetylcholine in amyotrophic lateral sclerosis. Neurology38 (9), 1464–1474 (1988).
- Lu JM , NurkoJ, WeakleySMet al. Molecular mechanisms and clinical applications of nordihydroguaiaretic acid (NDGA) and its derivatives: an update. Med. Sci. Monit.16 (5), RA93–100K (2010).
- Khomane KS , MeenaCL, JainR, BansalAK. Novel thyrotropin-releasing hormone analogs: a patent review. Expert Opin. Ther. Pat.21 (11), 1673–1691 (2011).
- Chung YH , JooKM, ShinCMet al. Brain Res. 994 (2), 253–259 (2003).
- Renna M , BentoCF, FlemingAet al. IGF-1 receptor antagonism inhibits autophagy. Hum. Mol. Genet.22 (22), 4528–4544 (2013).
- Yan J , XuY, ZhuCet al. Simvastatin prevents dopaminergic neurodegeneration in experimental parkinsonian models: the association with anti-inflammatory responses. PLoS ONE6 (6), e20945 (2011).
- Ramirez C , TerceroI, PinedaA, BurgosJS. Simvastatin is the statin that most efficiently protects against kainate-induced excitotoxicity and memory impairment. J. Alzheimers Dis.24 (1), 161–174 (2011).
- Cassina P , PeharM, VargasMRet al. Astrocyte activation by fibroblast growth factor-1 and motor neuron apoptosis: Implications for amyotrophic lateral sclerosis. J. Neurochem.93 (1), 38–46 (2005).
- Mojsilovic-Petrovic J , JeongGB, CrockerAet al. Protecting motor neurons from toxic insult by antagonism of adenosine A2a and Trk receptors. J. Neurosci.26 (36), 9250–9263 (2006).
- Cazorla M , PremontJ, MannA, GirardN, KellendonkC, RognanD. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J. Clin. Invest.121 (5), 1846–1857 (2011).
- Cazorla M , JouvenceauA, RoseCet al. Cyclotraxin-B, the first highly potent and selective TrkB inhibitor, has anxiolytic properties in mice. PLoS ONE5 (3), e9777 (2010).
- Brockington A , WhartonSB, FernandoMet al. Expression of vascular endothelial growth factor and its receptors in the central nervous system in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol.65 (1), 26–36 (2006).
- Tovar-Y-Romo LB , TapiaR. Delayed administration of VEGF rescues spinal motor neurons from death with a short effective time frame in excitotoxic experimental models in vivo. ASN Neuro4 (2), e00081 (2012).
- Zheng C , NennesmoI, FadeelB, HenterJI. Vascular endothelial growth factor prolongs survival in a transgenic mouse model of ALS. Ann. Neurol.56 (4), 564–567 (2004).
- Nefussy B , DroryVE. Moving toward a predictive and personalized clinical approach in amyotrophic lateral sclerosis: novel developments and future directions in diagnosis, genetics, pathogenesis and therapies. EPMA J.1 (2), 329–341 (2010).
- Gordon PH , CheungYK, LevinBet al. A novel, efficient, randomized selection trial comparing combinations of drug therapy for ALS. Amyotroph. Lateral. Scler.9 (4), 212–222 (2008).
- Foerster BR , CallaghanBC, PetrouM, EddenRA, ChenevertTL, FeldmanEL. Decreased motor cortex gamma-aminobutyric acid in amyotrophic lateral sclerosis. Neurology78 (20), 1596–1600 (2012).
- Adeosun SO , HouX, JiaoYet al. Allopregnanolone reinstates tyrosine hydroxylase immunoreactive neurons and motor performance in an MPTP-lesioned mouse model of parkinson's disease. PLoS ONE7 (11), e50040 (2012).
- Kumar A , GoyalR. Possible gabaergic modulation in the protective effect of zolpidem in acute hypoxic stress-induced behavior alterations and oxidative damage. Neurochem. Res.33 (3), 370–377 (2008).
- Fung SJ , XiMC, ZhangJHet al. Eszopiclone prevents excitotoxicity and neurodegeneration in the hippocampus induced by experimental apnea. Sleep32 (12), 1593–1601 (2009).
- Ishimaru H , TakahashiA, IkarashiY, MaruyamaY. Effects of MK-801 and pentobarbital on cholinergic terminal damage and delayed neuronal death in the ischemic gerbil hippocampus. Brain Res. Bull.43 (1), 81–85 (1997).
- Shaw PJ , IncePG. Glutamate, excitotoxicity and amyotrophic lateral sclerosis. J. Neurol.244 (Suppl. 2), S3–S14 (1997).
- Armstrong GA , DrapeauP. Calcium channel agonists protect against neuromuscular dysfunction in a genetic model of TDP-43 mutation in ALS. J. Neurosci.33 (4), 1741–1752 (2013).
- Tortarolo M , GrignaschiG, CalvaresiNet al. Glutamate ampa receptors change in motor neurons of SOD1G93Atransgenic mice and their inhibition by a noncompetitive antagonist ameliorates the progression of amytrophic lateral sclerosis-like disease. J. Neurosci. Res.83 (1), 134–146 (2006).
- Van Damme P , LeyssenM, CallewaertG, RobberechtW, Van Den BoschL. The AMPA receptor antagonist NBQX prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci. Lett.343 (2), 81–84 (2003).
- Albo F , PieriM, ZonaC. Modulation of AMPAreceptors in spinal motor neurons by the neuroprotective agent riluzole. J. Neurosci. Res.78 (2), 200–207 (2004).
- Paizs M , TortaroloM, BendottiC, EngelhardtJI, SiklosL. Talampanel reduces the level of motoneuronal calcium in transgenic mutant SOD1 mice only if applied presymptomatically. Amyotroph. Lateral. Scler.12 (5), 340–344 (2011).
- Pascuzzi RM , ShefnerJ, ChappellASet al. A Phase II trial of talampanel in subjects with amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler.11 (3), 266–271 (2010).
- Whitehouse PJ , WamsleyJK, ZarbinMA, PriceDL, TourtellotteWW, KuharMJ. Amyotrophic lateral sclerosis: alterations in neurotransmitter receptors. Ann. Neurol.14 (1), 8–16 (1983).
- Van Kampen JM , EckmanCB. Agonist-induced restoration of hippocampal neurogenesis and cognitive improvement in a model of cholinergic denervation. Neuropharmacology58 (6), 921–929 (2010).
- Turner BJ , RembachA, SparkR, LopesEC, CheemaSS. Opposing effects of low and high-dose clozapine on survival of transgenic amyotrophic lateral sclerosis mice. J. Neurosci. Res.74 (4), 605–613 (2003).
- Kawajiri M , MogiM, HigakiNet al. Reduced angiotensin II levels in the cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neurol. Scand.119 (5), 341–344 (2009).
- Iwasaki Y , IchikawaY, IgarashiO, KinoshitaM, IkedaK. Trophic effect of olmesartan, a novel AT1R antagonist, on spinal motor neurons in vitro and in vivo. Neurol. Res.24 (5), 468–472 (2002).
- Saavedra JM . Angiotensin II AT(1) receptor blockers as treatments for inflammatory brain disorders. Clin. Sci. (London)123 (10), 567–590 (2012).
- Dominguez-Meijide A , Villar-ChedaB, Garrido-GilP, Sierrra-ParedesG, GuerraMJ, Labandeira-GarciaJL. Effect of chronic treatment with angiotensin type 1 receptor antagonists on striatal dopamine levels in normal rats and in a rat model of parkinson's disease treated with L-DOPA. Neuropharmacology76 (Pt A), 156–168 (2014).
- Tsukuda K , MogiM, IwanamiJet al. Cognitive deficit in amyloid-beta-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-gamma activation. Hypertension54 (4), 782–787 (2009).
- Wu X , KiharaT, HongoH, AkaikeA, NiidomeT, SugimotoH. Angiotensin receptor type 1 antagonists protect against neuronal injury induced by oxygen-glucose depletion. Br. J. Pharmacol.161 (1), 33–50 (2010).
- Rothstein JD . Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol.65 (Suppl. 1), S3–S9 (2009).
- Tonari M , KurimotoT, HorieT, SugiyamaT, IkedaT, OkuH. Blocking endothelin-b receptors rescues retinal ganglion cells from optic nerve injury through suppression of neuroinflammation. Invest. Ophthalmol. Vis. Sci.53 (7), 3490–3500 (2012).
- Filipovich T , Fleisher-BerkovichS. Regulation of glial inflammatory mediators synthesis: possible role of endothelins. Peptides29 (12), 2250–2256 (2008).
- Koyama Y , KotaniM, SawamuraT, KuribayashiM, KonishiR, MichinagaS. Different actions of endothelin-1 on chemokine production in rat cultured astrocytes: reduction of CX3CL1/fractalkine and an increase in CCL2/MCP-1 and CXCL1/CINC-1. J. Neuroinflamm.10, 51 (2013).
- Cozzolino M , CarriMT. Mitochondrial dysfunction in ALS. Prog. Neurobiol.97 (2), 54–66 (2012).
- Kira Y , NishikawaM, OchiA, SatoE, InoueM. L-carnitine suppresses the onset of neuromuscular degeneration and increases the life span of mice with familial amyotrophic lateral sclerosis. Brain Res.1070 (1), 206–214 (2006).
- Tarnopolsky MA , BealMF. Potential for creatine and other therapies targeting cellular energy dysfunction in neurological disorders. Ann. Neurol.49 (5), 561–574 (2001).
- Martin LJ . Olesoxime, a cholesterol-like neuroprotectant for the potential treatment of amyotrophic lateral sclerosis. IDrugs13 (8), 568–580 (2010).
- Cudkowicz ME , Van Den BergLH, ShefnerJMet al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (empower): a randomised, double-blind, Phase 3 trial. Lancet Neurol.12 (11), 1059–1067 (2013).
- Amante DJ , KimJ, CarreiroSTet al. Uridine ameliorates the pathological phenotype in transgenic G93A-ALS mice. Amyotroph. Lateral. Scler.11 (6), 520–530 (2010).
- Park JH , HongYH, KimHJet al. Neurosci. Lett. 413 (3), 265–269 (2007).
- Miquel E , CassinaA, Martinez-PalmaLet al. Modulation of astrocytic mitochondrial function by dichloroacetate improves survival and motor performance in inherited amyotrophic lateral sclerosis. PLoS ONE7 (4), e34776 (2012).
- Kunst CB . Complex genetics of amyotrophic lateral sclerosis. Am. J. Hum. Genet.75 (6), 933–947 (2004).
- Al-Saif A , Al-MohannaF, BohlegaS. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol.70 (6), 913–919 (2011).
- Niitsu T , IyoM, HashimotoK. Sigma-1 receptor agonists as therapeutic drugs for cognitive impairment in neuropsychiatric diseases. Curr. Pharm. Des.18 (7), 875–883 (2012).
- Mancuso R , OlivanS, RandoA, CasasC, OstaR, NavarroX. Sigma-1r agonist improves motor function and motoneuron survival in ALS mice. Neurotherapeutics9 (4), 814–826 (2012).
- Nutini M , SpalloniA, FlorenzanoFet al. Increased expression of the beta3 subunit of voltage-gated Na+ channels in the spinal cord of the SOD1G93A mouse. Mol. Cell. Neurosci.47 (2), 108–118 (2011).
- Bellingham MC . Pre- and postsynaptic mechanisms underlying inhibition of hypoglossal motor neuron excitability by riluzole. J. Neurophysiol.110 (5), 1047–1061 (2013).
- Edmonds HL Jr. , JiangYD, ZhangPY, ShankR. Topiramate as a neuroprotectant in a rat model of global ischemia-induced neurodegeneration. Life Sci.69 (19), 2265–2277 (2001).
- Bermejo PE , AncionesB. A review of the use of zonisamide in Parkinson's disease. Ther. Adv. Neurol. Disord.2 (5), 313–317 (2009).
- Teng YD , WrathallJR. Local blockade of sodium channels by tetrodotoxin ameliorates tissue loss and long-term functional deficits resulting from experimental spinal cord injury. J. Neurosci.17 (11), 4359–4366 (1997).
- Feng HL , LengY, MaCH, ZhangJ, RenM, ChuangDM. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience155 (3), 567–572 (2008).
- Ryberg H , AskmarkH, PerssonLI. A double-blind randomized clinical trial in amyotrophic lateral sclerosis using lamotrigine: effects on csf glutamate, aspartate, branched-chain amino acid levels and clinical parameters. Acta Neurol. Scand.108 (1), 1–8 (2003).
- Conforti FL , SpataroR, SprovieroWet al. Ataxin-1 and ataxin-2 intermediate-length polyq expansions in amyotrophic lateral sclerosis. Neurology79 (24), 2315–2320 (2012).
- Tanaka K , KannoT, YanagisawaYet al. Bromocriptine methylate suppresses glial inflammation and moderates disease progression in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol.232 (1), 41–52 (2011).
- Meyer T , SchwanA, DullingerJSet al. Early-onset ALS with long-term survival associated with spastin gene mutation. Neurology65 (1), 141–143 (2005).
- Fanara P , BanerjeeJ, HueckRVet al. Stabilization of hyperdynamic microtubules is neuroprotective in amyotrophic lateral sclerosis. J. Biol. Chem.282 (32), 23465–23472 (2007).
- Gonzalez-Perez P , CirulliET, DroryVEet al. Novel mutation in VCP gene causes atypical amyotrophic lateral sclerosis. Neurology79 (22), 2201–2208 (2012).
- Rodriguez-Ortiz CJ , HoshinoH, ChengDet al. Neuronal-specific overexpression of a mutant valosin-containing protein associated with IBMPFD promotes aberrant ubiquitin and TDP-43 accumulation and cognitive dysfunction in transgenic mice. Am. J. Pathol.183 (2), 504–515 (2013).
- Budini M , BaralleFE, BurattiE. Regulation of gene expression by TDP-43 and FUS/TLS in frontotemporal lobar degeneration. Curr. Alzheimer Res.8 (3), 237–245 (2011).
- Lagier-Tourenne C , ClevelandDW. Rethinking ALS: The fus about tdp-43. Cell136 (6), 1001–1004 (2009).
- Hotta K , NashimotoA, YasumuraEet al. Vesnarinone suppresses TNFalpha mRNA expression by inhibiting valosin-containing protein. Mol. Pharmacol.83 (5), 930–938 (2013).
- Sasazawa Y , KanagakiS, TashiroEet al. Xanthohumol impairs autophagosome maturation through direct inhibition of valosin-containing protein. ACS Chem. Biol.7 (5), 892–900 (2012).
- Suzumura A , SawadaM. Effects of vesnarinone on cytokine production and activation of murine microglia. Life Sci.64 (14), 1197–1203 (1999).
- Yen TL , HsuCK, LuWJet al. Neuroprotective effects of xanthohumol, a prenylated flavonoid from hops (humulus lupulus), in ischemic stroke of rats. J. Agric. Food Chem.60 (8), 1937–1944 (2012).
- Mitchell J , PaulP, ChenHJet al. Familial amyotrophic lateral sclerosis is associated with a mutation in d-amino acid oxidase. Proc. Natl Acad. Sci. USA107 (16), 7556–7561 (2010).
- Sasabe J , MiyoshiY, SuzukiMet al. D-amino acid oxidase controls motoneuron degeneration through d-serine. Proc. Natl Acad. Sci. USA109 (2), 627–632 (2012).
- Lin CH , ChenPK, ChangYCet al. Benzoate, a d-amino acid oxidase inhibitor, for the treatment of early-phase Alzheimer disease: a randomized, double-blind, placebo-controlled trial. Biol. Psychiatry75 (9), 678–685 (2013).
- Adage T , TrillatAC, QuattropaniAet al. In vitro and in vivo pharmacological profile of AS057278, a selective d-amino acid oxidase inhibitor with potential anti-psychotic properties. Eur. Neuropsychopharmacol.18 (3), 200–214 (2008).
- Smith SM , UslanerJM, HutsonPH. The therapeutic potential of d-amino acid oxidase (DAAO) inhibitors. Open Med. Chem. J.4, 3–9 (2010).
- Wu J , SongR, SongWet al. Chlorpromazine protects against apoptosis induced by exogenous stimuli in the developing rat brain. PLoS ONE6 (7), e21966 (2011).
- Giordano G , ColeTB, FurlongCE, CostaLG. Paraoxonase 2 (PON2) in the mouse central nervous system: a neuroprotective role?Toxicol. Appl. Pharmacol.256 (3), 369–378 (2011).
- Saeed M , SiddiqueN, HungWYet al. Paraoxonase cluster polymorphisms are associated with sporadic ALS. Neurology67 (5), 771–776 (2006).
- Kalonia H , KumarP, KumarA, NehruB. Protective effect of montelukast against quinolinic acid/malonic acid induced neurotoxicity: possible behavioral, biochemical, mitochondrial and tumor necrosis factor-alpha level alterations in rats. Neuroscience171 (1), 284–299 (2010).
- Bae J , LeeD, KimYK, GilM, LeeJY, LeeKJ. Berberine protects 6-hydroxydopamine-induced human dopaminergic neuronal cell death through the induction of heme oxygenase-1. Mol. Cells35 (2), 151–157 (2013).