
Abstract
In forensic casework, compromised samples often possess limited or degraded nuclear DNA, rendering mitochondrial DNA a more feasible option for forensic DNA analyses. The emergence of massively parallel sequencing (MPS) has enabled the recovery of extensive sequence information from very low quantities of DNA. We have developed a multiplex PCR method that amplifies the complete mitochondrial genome in a range of forensically relevant samples including single cells, cremated remains, bone, maggot and hairs isolated from dust bunnies. Following library preparation, MPS yields complete or nearly complete mitochondrial genome coverage for all samples. To confirm concordance between sample types and between sequencing platforms, we compared sequencing results from hair and buccal swabs from two references. Low initial DNA input into the multiplex PCR allows for conservation of precious DNA while MPS maximizes recovery of genetic information.
Lay abstract Compromised samples are routine in forensic casework, and evidence DNA is often limited and/or degraded. Mitochondrial DNA is often a suitable option for forensic analysis as it is available in multiple copies per cell. We have established a method that amplifies the complete mitochondrial genome in a range of compromised samples and utilizes new high-throughput sequencing technologies to recover maximal genetic information from a small amount of sample. Combination of the aforementioned amplification method and high-throughput sequencing allows the analyst to conserve DNA in forensic cases where DNA is often limited.
The enhancement of mitochondrial DNA (mtDNA) typing over the last 25 years has distinguished it as a viable application in forensic casework, particularly when dealing with compromised samples, which often contain degraded nuclear DNA [Citation1–3]. mtDNA is available in multiple copies per cell and confers an increased detection sensitivity compared with nuclear DNA [Citation4,Citation5]. Traditionally, studies have focused on the noncoding control region (CR) of the mitochondrial genome (mtGenome), which spans over 1100 base pairs and includes two hypervariable regions. There are cases, however, when the CR does not provide adequate discriminatory power for identification [Citation6]. In cases where identity cannot be resolved through CR sequencing, analysis of the complete mtGenome may provide greater power of discrimination [Citation7].
Currently Sanger-type sequencing (STS) is the method used in most casework laboratories. Although STS has been traditionally considered the gold standard, it is labor intensive, expensive and requires significant initial DNA input, which is not ideal when forensic sample DNA is frequently limited. Massively parallel sequencing (MPS) technologies offer an efficient way to generate high-throughput, in-depth, sequence information and have recently proven successful in recovering significant genomic DNA sequences from compromised sample types [Citation8–10].
Nearly 25 years after validation of mtDNA sequencing for forensic casework, laboratory methods for mtDNA analysis have changed very little. Here we present a novel multiplex PCR method for amplifying the full mtGenome from a variety of forensically relevant samples, including hairs from dust bunnies, bones, cremated remains, maggots and single whole cells. Subsequent MPS yields complete or near complete mtGenome coverage of at least 100×.
Dust bunny hairs were collected from various household locations between 2008 and 2015 (). A human femur of unknown age was obtained from a commercial vendor (Skulls Unlimited, OK, USA) where the bone was cleaned of tissue and treated with a peroxide solution. Cremated remains, including highly calcined bone fragments, were obtained from a private donor. A maggot was collected from a donor at the Forensic Osteology Research Station (FOREST) at Western Carolina University, l NC, USA. DNA from hair shafts was extracted following a solid-phase protocol developed in-house [Citation11]. DNA from remaining samples was extracted following protocols specialized for each sample type and are described in the associated protocol. Single whole cells were collected by micromanipulation [Citation12] and amplified directly. Following DNA extraction, the mtGenome copy number was quantified using a quantitative PCR assay targeting a 105 base pair segment of the mtGenome [Citation13]. For the multiplex PCR assay, we combined 46 previously published primer pairs (mitoSEQr Resequencing System, Applied Biosystems, CA, USA) into three 10 μl reactions (). PCR conditions and thermal cycling parameters are provided in the associated protocol. Primer concentrations are provided in Supplementary Table 1.
Table 1. Summary of relevant information and data collected for each sample.
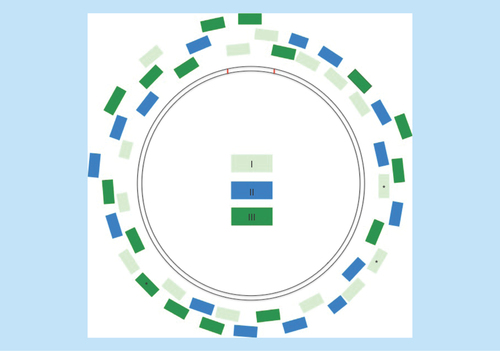
Figure 1. Orientation of 46 primers around the mitochondrial genome.
Primers were multiplexed into three reactions (I, II, III). 43 primers were modified from Applied Biosystems MitoSEQr Kit (Thermo Fischer, MA, USA); starred (*) primers were separately redesigned.
We evaluated amplification success of each multiplex by identification of appropriate peak size and pattern on an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA), and combined amplification products from each multiplex PCR were normalized to 0.2 ng/μl. All samples were subjected to library preparation following Nextera XT kit protocol (Illumina, CA, USA). MPS was performed on the Illumina MiSeq using paired-end 2 × 151 cycles. We performed secondary analysis using CLC Genomics Workbench version 8.5.1 (CLC bio, Aarhus, Denmark). Reads from each multiplex were simultaneously mapped to the revised Cambridge Reference Sequence (rCRS) and depth of coverage mapped across the mtGenome. We extracted a consensus sequence and uploaded FASTA files to a web-based mtDNA tool, mthap (https://dna.jameslick.com/mthap/), which compares sequences to the rCRS and evaluates sequence quality based on assignment to a mitochondrial haplogroup.
The number of mtGenome copies from challenging samples input into each multiplex PCR varied from 197 to 1.54 × 106 copies (). The most variability in recovered mtDNA among sample types occurred among cremated remains, which originated from a single donor. High DNA recovery from cremated remains was not expected and could be due to variables such as short incineration time resulting in less degraded DNA or handling of remains by persons with improper protection resulting in contamination. We treated the outside of each burned bone fragment with 5% bleach solution and did not find evidence of contamination in our MPS results. Additionally, all cremated samples matched a reference sequence from a maternal line relative.
Average coverage ranged from 455× in the single male cell to 16887× in cremated remains (); however, samples were sequenced across multiple MiSeq runs with varying numbers of samples per run, which likely impacted coverage per sample. We attempted to evaluate coverage in a more informative way by assessing percentage of genome covered by a minimum number of reads. We set minimum coverage at 100× and found all but two samples (MAG and MC-1) had >80% genome coverage ≥100× ().
In order to assess concordance of MPS with traditional STS, we compared buccal and hair samples from two donors sequenced on both platforms. Sequences generated using MPS were concordant with STS with the exception of a 43/57% CT point heteroplasmy detected in MPS samples of Donor 1 but not detected in STS. Additionally, we compared sequences from mid-shaft hair samples to more robust root-end hair fragments and buccal samples. Sequence concordance was found in cases with coverage ≥100× for all samples. These results are consistent with a recently conducted study examining concordance and reproducibility of MPS of mtGenome using an Illumina MiSeq [Citation14].
We used the mtDNA tool mthap to help provide an overview of sequence quality by evaluating sample fit into assigned mitochondrial haplogroups. Robust reference samples and some challenging samples were perfect or near perfect matches to their assigned haplogroups (). Some challenging samples were imperfect matches to their assigned haplogroups, containing missing or mismatched base calls. Such mismatched bases may be due to stochastic sampling effects exacerbated by PCR in low-copy number samples [Citation15,Citation16]. The patterns and magnitude of these incongruences warrant further investigation. The protocol for this method can be found in the Supplementary Material.
Conclusion
We have established a method which amplifies the entire mtGenome in a range of challenging sample types ranging from dust bunny hairs to cremated remains. The method requires minimal DNA input, thereby conserving precious DNA and provides full mtGenome coverage of at least 100× in most samples. Multiplex PCR combined with MPS offers a promising new tool for maximizing genetic information.
Future perspective
Massively parallel DNA sequencing is a powerful technique that has the potential to revolutionize forensic DNA typing. It offers the ability to generate expanded datasets in two complementary dimensions, one that easily extends the number of genetic targets amenable for typing, and a complementary depth dimension that provides replicate information at each sequenced base pair. Such data richness raises technical, philosophical, and interpretational issues that must be addressed before MPS is a routine technique in forensic science.
The wealth of data generated introduces a fundamental question as to how the data are to be treated. Some would argue that simplifying the data by using data divisors such as thresholds is the proper approach. The opposite perspective seeks to model the entire dataset. Variation within the data must also be addressed. For instance, sequencing reveals variants that are not observed using size-separation techniques, such as capillary electrophoresis. A question that may arise is whether or not these variants are a result of the amplification and sequencing process, and, if so, what are the implications of this for validly interpreting the MPS signal? With respect to human mitochondrial DNA analysis using MPS, the breadth dimension means that the entire mtDNA genome can be closed, and hence the power of this dimension is easily exploited. However, with this marker there are two sources of variation that must be further understood: biological variation present in the forensic sample that emanates from the sample donor and artifactual variation that results from the process of amplifying and sequencing the template molecules. These two sources must be more fully studied so that proper interpretational approaches can be developed. Once this is achieved, MPS can realize its huge promise in the field of forensic DNA typing.
In forensic cases where DNA is limited or degraded, analysis of the mitochondrial genome can provide higher quality data than nuclear DNA.
Analysis of the complete mitochondrial genome can provide greater power of discrimination than analysis of the control region alone.
Massively parallel sequencing technologies have the ability to yield in-depth sequence information from compromised samples.
We use a multiplex PCR method to amplify the complete mitochondrial genome in a variety of forensically relevant compromised samples and massively parallel sequencing to achieve complete or nearly complete coverage of the mitochondrial genome.
Authors’ contributions
MP Hickman refined and finalized multiplex assay, performed sequencing and data analysis, and authored manuscript. KS Grisedale helped supervise the experiments and edit the manuscript. BJ Bintz provided valuable laboratory support and performed the assay for maggot sample. ES Burnside developed initial protocol for the assay and provided valuable comments for the manuscript. EK Hanson performed micromanipulation for single whole cells. J Ballantyne helped develop initial idea for dust bunny and single whole cell testing. MR Wilson conceived initial idea for mtGenome multiplex assay, provided guidance throughout project and was principal investigator for grant that funded assay development.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
suppl_table.docx
Download MS Word (136.5 KB)Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.4155/fsoa-2018-0059
Acknowledgements
The authors would like to thank N Petraco for his contribution of dust bunny hairs, and J Foster for his assistance in designing the graphical layout for the multiplexes.
Financial & competing interests disclosure
This work was supported in part by the National Institute of Justice grant award #2013-DN-BX-K014. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Sullivan KM, Hopgood R, Gill P. Identification of human remains by amplification and automated sequencing of mitochondrial DNA. Int. J. Legal Med. 105(2), 83–86 (1992).
- Wilson MR, Polanskey D, Butler J, DiZinno JA, Replogle J, Budowle B. Extraction, PCR amplification and sequencing of mitochondrial DNA from human hair shafts. Biotechniques 18(4), 662–669 (1995).
- Holland M, Fisher D, Mitchell L et al. Mitochondrial DNA sequence analysis of human skeletal remains. J. Forensic Sci. 38(3), 542–553 (1993).
- Robin ED, Wong R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J. Cell. Physiol. 136(3), 507–513 (1988).
- Wilson MR, Stoneking M, Holland M, DiZinno J, Budowle B. Guidelines for the use of mitochondrial DNA sequencing in forensic science. Crime Lab. Dig. 20(4), (1993). Available from: www.ncjrs.gov/pdffiles1/Digitization/146991NCJRS.pdf.
- Parsons TJ, Coble MD. Increasing the forensic discrimination of mitochondrial DNA testing through analysis of the entire mitochondrial DNA genome. Croat. Med. J. 42(3), 304–309 (2001).
- Coble MD, Just RS, O’Callaghan J et al. Single nucleotide polymorphisms over the entire mtDNA genome that increase the power of forensic testing in Caucasians. Int. J. Legal Med. 118(3), 137–146 (2004).
- Loreille O, Koshinsky H, Fofanov VY, Irwin JA. Application of next generation sequencing technologies to the identification of highly degraded unknown soldiers’ remains. Forensic Sci. Int. Genet. Suppl. Ser. 3(1), e540–e541 (2011).
- Parson W, Huber G, Moreno L et al. Massively parallel sequencing of complete mitochondrial genomes from hair shaft samples. Forensic Sci. Int. Genet. 15, 8–15 (2015).
- Strobl C, Eduardoff M, Bus MM, Allen M, Parson W. Evaluation of the precision ID whole MtDNA genome panel for forensic analyses. Forensic Sci. Int. Genet. 35, 21–25 (2018). www.sciencedirect.com/science/article/pii/S1872497318300280.
- Burnside ES, Bintz BJ, Wilson MR. Improved extraction efficiency of human mitochondrial DNA from hair shafts and its implications for sequencing of the entire mtGenome from a single hair fragment. In: Proceedings of the American Academy of Forensic Sciences 65th Annual Meeting. WA, USA (February 2013).
- Farash K, Hanson EK, Ballantyne J. Enhanced genetic analysis of single human bioparticles recovered by simplified micromanipulation from forensic evidence. J. Vis. Exp. (97), 1–14 (2015). www.jove.com/video/52612/enhanced-genetic-analysis-single-human-bioparticles-recovered.
- Kavlick MF, Lawrence HS, Merritt RT et al. Quantification of human mitochondrial DNA using synthesized DNA standards. J. Forensic Sci. 56(6), 1457–1463 (2011).
- Peck MA, Brandhagen MD, Marshall C, Diegoli TM, Irwin JA, Sturk-Andreaggi K. Concordance and reproducibility of a next generation mtGenome sequencing method for high-quality samples using the Illumina MiSeq. Forensic Sci. Int. Genet. 24, 103–111 (2016).
- Caddy B, Taylor G, Linacre A. A review of the science of low template DNA analysis. Off. Forensic Regul. 1–35 (2008). www.gov.uk/government/publications/review-of-the-science-of-low-template-dna-analysis.
- Budowle B, Eisenberg J, van Daal A. Validity of low copy number typing and applications to forensic science. Croat. Med. J. 50(3), 207–217 (2009).