
During the drug-development process, it is essential to evaluate the potential for any drug–drug interaction (DDI) during early clinical Phase I stage based on the in vitro absorption distribution metabolism excretion (ADME) data gathered during the discovery-development transition phase leading to the IND filing of the new chemical entity [Citation1,Citation2]. Such an evaluation becomes critical for anticancer therapy due to a higher risk of the occurrence of clinical DDI if the novel chemical entity is a victim or a perpetrator drug of cytochrome P450 enzyme (CYP) and/or uptake (liver/renal) transporters because of the existence of polypharmacy in the treatment of cancer patients [Citation3,Citation4]. In this context, a recent article describes the dilemma observed in the PK data interpretation when rifampicin was used as a tool drug to study the induction of CYP3A4 enzyme in cancer patients [Citation5]. Because rifampicin contributes for DDI via additional mechanisms such as P-glycoprotein (Pgp)/UDP glucuronosyl transferase (UGT) induction and uptake transporter inhibition, the interpretation of the PK data poses a challenge [Citation5].
Because of difficulty in translatability of the observed findings obtained in healthy subjects from DDI studies for an unbiased dosing recommendation in cancer patients, it may be best to perform DDI studies in the relevant cancer patients. However, given the caveat that it may not be possible to conduct all DDI studies in cancer patients, one may have to resort to a population PK–PD analysis of several patient studies (e.g., Phase I/II) with sparse PK sampling where the relevant PK parameters, obtained via a modeling approach, is correlated with specific covariates that may include concomitant therapy, patient (genetic/clinical) factors, age, sex and so on [Citation6–8]. Based on the population PK/PK–PD versus covariate analyses, as deemed fit, appropriate conclusions may be drawn for dosing recommendation in clinical development and/or inclusion into the product label.
Scope
It is common that cancer patients use various acid reducing agents most notably proton pump inhibitors (PPIs), with prevalence usage rate between 20 and 55%, for palliation treatment of dyspepsia, gastritis or gastroesophageal reflux disease attributable to either tumor malignancy or the anticancer regimen [Citation9]. Moreover, the drugs in the oral targeted anticancer therapy have been shown to exhibit poor and variable oral bioavailability due to DDI with acid-reducing agents [Citation10].
The intent of this commentary is to provide perspectives on the proposed topic using several case studies of the clinical DDI PK data reported in healthy subjects and cancer patients, largely using omeprazole as the perpetrator drug.
Case studies
Ceritinib
To understand the potential DDI between ceritinib and esomeprazole (a PPI), Lau et al. conducted a PK study in both healthy and anaplastic lymphoma kinase (ALK) positive non-small-cell lung cancer patients with satisfactory sample size to ensure reliable interpretation of the data [Citation11]. The results suggested that esomeprazole decreased the systemic exposure area under the concentration versus time curve (AUC) of ceritinib by 76% in healthy subjects; whereas the decrease in the systemic availability by PPIs was by only 30% in cancer patients. However, the examination of steady state Cmin levels suggested that the impact of PPIs was only marginal and it was concluded that coadministration of PPIs was less likely to impact the efficacy of ceritinib therapy in cancer patients [Citation11].
Momelotinib
In a clinical Phase 1 study, the single dose relative bioavailability of the new tablet dosage form (300 mg) versus the earlier capsule formulation (300 mg) was investigated along with the assessments of the impact of concomitant omeprazole dosing and food effect on the PK of momelotinib [Citation12]. The PK data suggested that coingestion of food (high-fat meal) increased the Cmax and AUCinf values of momelotinib by approximately 28%, while coadministration of omeprazole reduced the Cmax and AUCinf values of momelotinib by 33–36%. Based on these findings, it was concluded that either food or coadministration of PPIs (i.e., omeprazole) would not alter safety and efficacy profile of momelotinib in a clinically meaningful manner [Citation12].
Erlotinib
The impact of PPI (i.e., omeprazole) and H2 receptor blockers (H2RB) on the plasma levels of erlotinib when coadministered in cancer patients was evaluated in a clinical study involving cancer patients [Citation13]. Both PPI and H2RB reduced the plasma levels of erlotinib; however, PPI produced an overall decrease of 61% as compared with 52% observed for H2RB. The results suggested that concomitant PPI dosing in the relevant patient population would lower the systemic exposure of erlotinib, which may have a consequence on the observed efficacy in the patients [Citation13]. Furthermore, it was noted that during concomitant dosing with PPI, consideration for erlotinib dosage adjustment guided by therapeutic drug monitoring may become necessary for ensuring clinical efficacy is not compromised [Citation13].
Kletzl et al. carried out a crossover study to determine the extent of DDI between erlotinib versus erlotinib + omeprazole in healthy subjects. There was a pronounced reduction in the mean Cmax and AUC of approximately 42 and 45%, respectively, for erlotinib when coadministered with PPI [Citation14]. Similar reduction in exposure was also observed with the key metabolite OSI-420 with the concomitant PPI intake. Using the DDI data, it was recommended that the concomitant intake of PPI with erlotinib be avoided. However, the staggering dosing strategy employed with ranitidine, permitted to minimize the exposure loss of erlotinib so that the product label was revised to permit the dosing of erlotinib 10 h after ranitidine and 2 h prior to the next ranitidine dosing [Citation14].
Nilotonib
Retrospective analyses were performed in patients with newly diagnosed Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia in chronic phase (CML-CP; n = 492) and in patients with imatinib-resistant or imatinib-intolerant Ph+ CML-CP (n = 256) treated with nilotinib [Citation15]. Regardless of the two patient populations treated with nilotinib, there appeared to be identical response rates of patients who were assigned to gastric acid suppressant versus nongastric acid suppressant groups. Also, the response rate appeared to be comparable between PPI versus H2RB patient groups. The steady-state nilotinib trough concentration measured in the study for cancer patients appeared to be similar with or without the coadministration of PPIs/H2RB. Therefore, it was concluded that coadministration of either PPI or H2RB did not affect the PK and efficacy of nilotinib in cancer patients [Citation15].
In a two-way crossover Phase I study in healthy subjects, the concomitant intake of esomeprazole reduced both Cmax and AUC values by 27 and 34%, respectively [Citation16]. The moderate reduction in the exposure observed with PPI was judged to have no clinical relevance in the oral cancer therapy of nilotinib in patients [Citation16].
Dasatinib
In a two-way crossover study in healthy subjects, concomitant treatment of omeprazole reduced the Cmax and AUC values of dasatinib by approximately 42 and 43%, respectively [Citation17]. The product label has suggested the use of antacids rather than PPIs or H2RB in the dasatinib therapy in cancer patients [Citation17].
Ibrutinib
A clinical Phase I two-way crossover study was conducted in healthy subjects to delineate the PK interaction between ibrutinib and omeprazole [Citation18]. The Cmax of ibrutinib in presence of omeprazole showed a reduction of approximately 62.5% while the extent of exposure showed a modest decrease and the terminal half-life values remained unaltered between the two treatments [Citation18]. The exposure of primary metabolite was reduced by approximately 20% with omeprazole coadministration. However, modeling data suggested that reduced Cmax did not influence target engagement. Based on the data it was concluded that dose adjustments of ibrutinib was not necessary when omeprazole or other PPI was added to the clinical therapy [Citation18].
Future perspective
Omeprazole remains on top of the list of widely prescribed medications and within the class of PPIs, it has the distinction of being most frequently associated with DDI. In vitro studies have shown that omeprazole can also alter the function of metabolic Phase I/II enzymes and transporters [Citation19,Citation20]. Therefore, the assessment of the impact of PPI (i.e., omeprazole) on oral absorption and bioavailability should also consider the possible role the PPI may have on metabolic and elimination pathway of the oral drugs in question.
Oral targeted small-molecule drugs such as ceritinib, momelotinib, nilotinib, erlotinib, dasatinib, ibrutinib and so on, owing to the drug structures (i.e., pKa values) exhibit varying degrees of pH dependency on individual drug solubility. Ceritinib displayed higher solubility in acidic pH (11.9 mg/ml at pH 1), which was drastically reduced at neutral pH values (0.01 mg/ml at pH 6.8). Momelotinib showed solubility of 1.3 μg/ml at pH 5, which increased substantially at <pH 3. Ibrutinib was practically insoluble at pH ≥3. Hence, it may have consequences on drug dissolution from dosage forms affecting drug absorption and oral bioavailability of these drugs in view of the altered pH that is likely to occur in the human gastrointestinal tract during the concomitant PPI administration.
It may be argued that since the physiology of gastrointestinal tract remains fairly consistent between healthy subjects and cancer patients, the use of DDI data with coadministered PPI from the healthy subjects may represent reasonable objectivity for making a dosing recommendation in cancer patients during clinical development and/or for label claims.
Since the intent of the commentary is to provide a snapshot of the comparative data between healthy subjects versus cancer patients to make judgment and sound conjectures on data extrapolation, a flow chart was created for an easy comparison of the data along with the product label instructions for the dosing (). Accordingly, the following deductions can be drawn based on the reviewed cases studies:
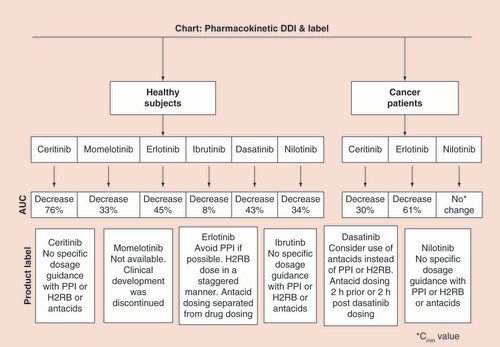
The cited data provide credence to the observed DDI (i.e., reduced exposure of the victim drug in the presence of PPI) remained consistent between healthy subjects versus cancer patients for ceritinib, erlotinib, etc., where PK data were available. However, in the case of nilotinib, there appeared to be a disagreement with what was reported in healthy subjects versus cancer patients [Citation15,Citation16]. It may be conceivable that Cmin value may not be a sensitive enough parameter (unlike AUC value) to show a difference between nilotinib versus nilotinib + PPI treatment groups in cancer patients [Citation15].
While the decrease of 76% in AUC in healthy subjects appeared to be very drastic as in the case of ceritinib for making the decision of avoidance of PPI, the decrease in exposure data (i.e., 30%) in cancer patients suggested that coadministration of PPI was unlikely to effect the efficacy of the drug [Citation11].
In case of ibrutinib, although the AUC decrease with concomitant PPI was found marginal (i.e., 8%), there was approximately a 62% decrease in the Cmax value in presence of PPI [Citation18]. Hence, extensive modeling exercise was carried out to show that the target engagement was less likely to be impacted despite the impact on the Cmax value of ibrutinib [Citation18].
The decrease of 43% in the AUC of dasatinib in presence of PPI in healthy subjects was considered adequate enough for a label claim to avoid PPI during the therapy in cancer patients [Citation17].
In the case of momelotinib, despite a 33% decrease in the AUC due to concomitant PPI, it was considered not clinically relevant because a wider 90% confidence interval, ranging between 70 and 143%, was used in the assessment of bioequivalence representing a 30% difference in the drug exposure, since the required target related response was achieved by a reduced tablet dose (200 mg) as compared with the capsule formulation (300 mg) [Citation12].
Conclusion
Based on the case studies and the above conjectures, if exposure reduction of oral targeted cancer drugs is <40% with concomitant PPI (i.e., omeprazole) dosing in healthy subjects, labeling restrictions on the use of PPI does not appear to be relevant in cancer patients. However, it is too risky for making a definitive call pertaining to the DDI of PPI with oral targeted cancer therapies, that have pH dependent solubility, if there is a exposure reduction >40% in healthy subjects. It appears prudent that if the exposure of the orally targeted cancer drugs is decreased by >40% with the concomitant PPI in healthy subjects, the need for the therapeutic monitoring of the oral cancer drug be put in place for cancer patient trials or the PK of the oral cancer drug is characterized in a subset of the patients (with and without PPI) prior to embarking on a longer duration clinical trial.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Wang X , ZhangZY , AroraSet al. Effects of rolapitant administered intravenously on the pharmacokinetics of a modified Cooperstown cocktail (midazolam, omeprazole, warfarin, caffeine, and dextromethorphan) in healthy subjects . J. Clin. Pharmacol. ( 2018 ). doi:10.1002/jcph.1114 ( Epub ahead of print ).
- Srinivas NR , MullangiR . Bioanalysis in oncology drug discovery . Biomark. Med.9 ( 9 ), 877 – 886 ( 2015 ).
- Wisinski KB , CantuCA , EickhoffJet al. Potential cytochrome P-450 drug–drug interactions in adults with metastatic solid tumors and effect on eligibility for Phase I clinical trials . Am. J. Health Syst. Pharm.72 ( 11 ), 958 – 965 ( 2015 ).
- Thomas-Schoemann A , BlanchetB , BardinCet al. Drug interactions with solid tumour-targeted therapies . Crit. Rev. Oncol. Hematol.89 ( 1 ), 179 – 196 ( 2014 ).
- Srinivas NR . Pharmacokinetic interaction of rifampicin with oral versus intravenous anticancer drugs: challenges, dilemmas and paradoxical effects due to multiple mechanisms . Drugs R D16 ( 2 ), 141 – 148 ( 2016 ).
- Hong Y , PassosVQ , HuangPH , LauYY . Population pharmacokinetics of ceritinib in adult patients with tumors characterized by genetic abnormalities in anaplastic lymphoma kinase . J. Clin. Pharmacol.57 ( 5 ), 652 – 662 ( 2017 ).
- Lane S , Al-ZubiediS , HatchEet al. The population pharmacokinetics of R- and S-warfarin: effect of genetic and clinical factors . Br. J. Clin. Pharmacol.73 ( 1 ), 66 – 76 ( 2012 ).
- Tate SC , SykesAK , KulanthaivelPet al. A population pharmacokinetic and pharmacodynamic analysis of abemaciclib in a Phase I clinical trial in cancer patients . Clin. Pharmacokinet.57 ( 3 ), 335 – 344 ( 2018 ).
- Smelick GS , HeffronTP , ChuLet al. Prevalence of acid-reducing agents (ARA) in cancer populations and ARA drug–drug interaction potential for molecular targeted agents in clinical development . Mol. Pharm.10 , 4055 – 4062 ( 2013 ).
- Budha NR , FrymoyerA , SmelickGSet al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the Achilles heel of targeted therapy? Clin. Pharmacol. Ther. 92 , 203 – 213 ( 2012 ).
- Lau YY , GuW , LinTet al. Assessment of drug-drug interaction potential between ceritinib and proton pump inhibitors in healthy subjects and in patients with ALK-positive non-small cell lung cancer . Cancer Chemother. Pharmacol.79 ( 6 ), 1119 – 1128 ( 2017 ).
- Xin Y , ShaoL , MaltzmanJet al. The relative bioavailability, food effect, and drug interaction with omeprazole of momelotinib tablet formulation in healthy subjects . Clin. Pharmacol. Drug Dev.7 ( 3 ), 277 – 286 ( 2018 ).
- Ohgami M , KaburagiT , KurosawaAet al. Effects of proton pump inhibitor co-administration on the plasma concentration of erlotinib in patients with non-small cell lung cancer . 40 ( 6 ), 699 – 704 ( 2018 ).
- Kletzl H , GiraudonM , DucrayPSet al. Effect of gastric pH on erlotinib pharmacokinetics in healthy individuals: omeprazole and ranitidine . Anticancer Drugs26 ( 5 ), 565 – 572 ( 2015 ).
- Yin OQ , GilesFJ , BaccaraniMet al. Concurrent use of proton pump inhibitors or H2 blockers did not adversely affect nilotinib efficacy in patients with chronic myeloid leukemia . Cancer Chemother. Pharmacol.70 ( 2 ), 345 – 350 ( 2012 ).
- Yin OQ , GallagherN , FischerDet al. Effect of the proton pump inhibitor esomeprazole on the oral absorption and pharmacokinetics of nilotinib . J. Clin. Pharmacol.50 ( 8 ), 960 – 967 ( 2010 ).
- SPRYCEL® (dasatinib) tablet for oral use (package insert) . https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021986s7s8lbl.pdf
- de Jong J , Haddish-BerhaneN , HellemansPet al. The pH-altering agent omeprazole affects rate but not the extent of ibrutinib exposure . Cancer Chemother. Pharmacol. doi:10.1007/s00280-018-3613-9 ( Epub ahead of print ) ( 2018 ).
- Furuta S , KamadaE , SuzukiTet al. Inhibition of drug metabolism in human liver microsomes by nizatidine, cimetidine and omeprazole . Xenobiotica31 ( 1 ), 1 – 10 ( 2001 ).
- Chioukh R , Noel-HudsonMS , RibesSet al. Proton pump inhibitors inhibit methotrexate transport by renal basolateral organic anion transporter hOAT3 . Drug Metab. Dispos.42 ( 12 ), 2041 – 2048 ( 2014 ).