
Abstract
Glioblastoma (GBM) is a deadly malignancy with a poor prognosis. An important factor contributing to GBM recurrence is high resistance of GBM cancer stem cells (GSCs). While temozolomide (TMZ), has been shown to consistently extend survival, GSCs grow resistant to TMZ through upregulation of DNA damage repair mechanisms and avoidance of apoptosis. Since a single-drug approach has failed to significantly alter prognosis in the past 15 years, unique approaches such as multidrug combination therapy together with distinctive targeted drug-delivery approaches against cancer stem cells are needed. In this review, a rationale for multidrug therapy using a targeted nanotechnology approach that preferentially target GSCs is proposed with discussion and examples of drugs, nanomedicine delivery systems, and targeting moieties.
Patients with glioblastoma (GBM) have a dismal prognosis and limited treatment options. Reasons for GBM resistance to therapy include its highly infiltrative nature, genetic heterogeneity, propensity for genetic mutations, and presence of an intrinsic self-renewing population of cancer stem cells (CSCs) [Citation1]. Lack of progress in treating GBM for the past 15 years has necessitated new therapy strategies [Citation2]. Among these new approaches, combination therapy, CSC targeting and nanosystems development are all under intense investigation. In this review, an overview of GBM is provided, including the role of GBM CSCs in resistance development. Limitations of current chemotherapy options are described in brief followed by select examples of drugs that may be limited in isolation but overcome certain properties of CSC drug resistance in combination. Combination therapy may be facilitated using nanosystems that are enhanced with tumor surface-targeted delivery agents.
Glioblastoma overview
Epidemiology
Glioblastoma (GBM) is the most aggressive malignant primary intracranial tumor, affecting nearly 15,000 people per year according to the American Brain Tumor Association. According to the Ivy Brain Tumor Center, GBM or grade IV glioma is the most common form of brain cancer. While incidence rates for malignant tumors are declining, only 36% of patients survive more than 5 years after diagnosis [Citation3]. Current treatment for GBM patients often includes maximal surgical resection followed by temozolomide (TMZ) chemotherapy and radiation therapy (RT) (Stupp Regimen). The direct cost for each patient with GBM is projected to be around $8,500 a month [Citation4]. However, the total cost of GBM across the globe was approximately $465 million in 2016 and is expected to reach $1 billion in 2025 [Citation4]. Given the impact on patient quality of life and life expectancy, as well as high overall society costs, there is an urgent need to find treatments that have even a small chance of extending patient survival and improving quality of life.
Initial GBM patient diagnosis
Initial workup of a suspected brain tumor includes contrast-enhanced magnetic resonance imaging (MRI) of the brain, which provides information about tumor location, size, effect on normal structures and extent of associated edema. The average size of a GBM tumor at diagnosis is approximately 4 cm and often is an indication of advanced, incurable disease once that large [Citation5]. GBM rarely occurs outside the central nervous system and is extremely rare in the spinal cord. More common brain regions include the frontal lobe (25%), temporal lobe (20%), parietal lobe (13%) and occipital lobe (3%) [Citation6]. In addition to MRI, computed tomography (CT) may be used as a diagnostic tool to raise suspicion of a possible brain tumor [Citation6]. However, CT lacks the high tissue contrast needed for complete characterization.
Surgical resection of GBM
Following imaging to define essential characteristics of the tumor, biopsy and/or surgical resection ensue. Complete resection is difficult to attain because the highly infiltrative margins of the tumor are often incompletely visible on conventional MR imaging and intra-operatively [Citation6]. The decision of how much to resect can also be based on assessment of morbidity verses mortality when operating near eloquent areas, which are necessary for basic functioning.
Pathologic analysis of GBM characteristics
On histopathologic diagnosis from biopsied or surgically resected tissue, specific markers correlate with the various cell types in a single glioma. Normal brain cells include neurons, glial cells, oligodendrocytes and immune cells. GBM is believed to arise from precursors to neurons and glia. Indeed, GBM tumors are comprised of a heterogeneous number of cell types based on the biopsy analysis of a variety of markers. In normal astrocytes, the most specific marker is the glial fibrillary acidic protein (GFAP) [Citation7]. Loss of GFAP is a marker of increased malignancy of the tumor, which aids in grading the tumor. The WHO grades tumors from I through IV based on various histologic parameters. GBM is a grade IV tumor and has the highest level of GFAP loss. GBM is classified by the presence of characteristic mutations that can be categorized into three tumor-intrinsic transcriptional subtypes, regardless of the level of intertumoral heterogeneity [Citation8]. These subtypes are proneural, classical and mesenchymal [Citation8]. The proneural subtype has been associated with a more favorable outcome, while the mesenchymal subtype is more debilitating and often associated with recurrence [Citation9]. Cell irregularity is due to polymorphism (genetic variation within a population), anaplasia (poor cellular differentiation), and anisokaryosis (larger than normal variation). Histopathologic analysis also provides additional morphologic information such as calcification, necrosis and microcystic change [Citation10,Citation11].
GBM biomarkers for personalized medicine
Important markers involved in assessment of GBM for personalized medicine include identification of genetic biomarkers, such as the IDH1 or2 mutation, degree of methylation of MGMT and 1p/19q codeletion [Citation12]. The presence of a mutated IDH or methylated MGMT both independently correlate with longer survival time [Citation13] and help predict responsiveness of the tumor to TMZ. However, recent studies indicate that MGMT hypermethylated glioma cells expressed significant activity of MGMT via promoter independent pathways which in turn produces resistance to TMZ [Citation14]. Despite all that has already been studied about glioma histology, a clear consensus is still lacking among pathologists regarding what should ultimately define a glioma [Citation15].
Chemoradiation therapy
After surgery, radiation and chemotherapy will begin anywhere from 1 to 4 weeks later [Citation6]. Radiation is often administered stereotactically in smaller fractions for a maximum dose of 60 Gray. TMZ is given orally according to the Stupp regimen at a dose of 75 mg/m2 daily for 6 weeks concurrently with radiation [Citation16]. The patient will take a 1-month rest and then again start on six cycles of adjuvant TMZ at a dose of 150–200 mg/m2 for 5 days every 28 days [Citation16]. TMZ was approved for treatment of adult GBM patients in 2005 and remains the only first-line chemotherapy to date. A newer noninvasive therapy option, tested by Stupp et al., called tumor treating fields (worn as a cap with weak electrical fields pulsing through the skin of the scalp) appeared to extend overall survival time from 16 to 20.9 months when used in combination with radiotherapy and chemotherapy [Citation17]. Therefore, tumor treating fields are increasingly used as part of the standard of care.
Second-line therapy examples
The tumor initially responds positively to therapy, but often recurs. While, the standard of care is well defined after the initial diagnosis, the regimen of therapy options is less defined after recurrence [Citation18]. A number of immunotherapy modulators, such as PD-1/PD-L1 have proven to be effective in a number of cancers. However, they have been reported to be associated with ‘immune-related adverse events’ (irAE) [Citation19]. Bevacizumab is a VEGF inhibitor, which may be integrated into treatment but has had mixed results regarding overall survival. Bevacizumab appears to allow an improved quality of life before the tumor becomes resistant to the drug [Citation18]. On the other hand, addition of bevacizumab to radiotherapy with concurrent TMZ did not improve survival in patients [Citation20]. Serial MRI is performed for continuous monitoring of the treatment course as shown in . This figure shows a typical progression through treatment and diagnosis for a GBM patient. Multidisciplinary care of the patient often includes a neuro-oncologist, neurosurgeon, radiation oncologist, neuropathologist, and a neuroradiologist who will often meet to discuss the next course of action to best benefit the patient.
The above figure demonstrates a representative timeline of events that a patient diagnosed with glioblastoma may undergo during their course of treatment.
TMZ: Temozolomide.

Glioblastoma cancer stem cells
Numerous studies have identified a key subpopulation of cells in solid tumors that have properties similar to the body's normal stem cells, which are termed CSCs. The first CSCs were detected in acute myeloid leukemia in 1994 [Citation21] and then subsequently in GBM in 2004 [Citation1]. Intratumoral heterogeneity and therapy resistance are believed to be promoted by the self-renewal and differentiation of GBM stem cells [Citation22]. GBM stem cells can survive in unfavorable and complex hypoxic microenvironments that are a characteristic feature of advanced tumors. For instance, CSCs promote increased invasiveness through epithelial mesenchymal transformation and facilitate neovascularization when cellular growth outstrips the vascular supply [Citation23–27]. An interesting theory is the migration of GBM CSCs as precursors to GBM beginning with underlying driver mutations at the subventricular zone along ventricles. These special mutated CSCs eventually migrate away from the subventricular zones into the deeper brain regions and mutate further to develop the glioma [Citation28]. Such a hypothesis could explain why despite aggressive treatments, the tumor can easily recur in nearby, non-treated brain regions over time in multi-focal, multi-lobar and even contralateral brain regions.
A biomarker is defined as, “A characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” [Citation29]. Although MGMT promoter methylation has been shown to be an independent prognostic factor and a predictor of success of TMZ therapy, there is a lack of standardization of the assay method [Citation30]. The critical factors for failure of cancer treatments are metastasis, recurrence, heterogeneity, resistance to chemo and radiotherapy and immunological approaches [Citation31] for which CSCs are the principal agents. The search for a curative approach against CSCs has been hindered by absence of a specific defining molecular biomarker unique only to CSCs that can be targeted [Citation32]. The ideal targetable biomarker would be critical for CSCs to maintain basic function and remain viable. In addition, the targetable molecule on the CSC would be amenable to irreversible binding and inhibition, thereby leading to reduced cell proliferation and increased apoptosis. Along these lines, there are several candidate markers that are found more frequently than others in CSCs but, to date, there has been no one molecule that purely defines a CSC. A leading approach is to identify CSC specific glycans attached to proteins or lipids on the cell surface that exist in higher frequency than normal tissue [Citation33]. The most often cited and studied examples include CD133 [Citation1], CD44 [Citation34], CD15 [Citation35], CD24 [Citation36], CD90 [Citation37], CD49f [Citation38], CXCR4 [Citation39], L1CAM [Citation40] and ABC efflux transporters [Citation41]. Cytoplasmic proteins have been delineated as potential CSC markers which include Nestin [Citation42], Musashi-1 [Citation43] and aldehyde dehydrogenase (i.e., ALDH1) [Citation44]. Nuclear proteins are also promising as CSC markers, with Oc 4, Nanog, and Sox2 as examples [Citation45]. Certain signal cascades identified commonly in adult neural stem cells are also present in CSCs such as the Notch, Sonic or Wnt/B-catenin pathways. In addition, a number of microRNAs, which are small non-coding RNA molecules approximately 21–25 nucleotides in length, have been reported to be potential biomarkers for diagnosis and promising potential miRNA-targeted therapies in TMZ-resistant GBM [Citation46–48].
The central role of GBM CSCs (GSCs) in a theoretical hierarchy that contributes to chemoradiotherapy resistance and regrowth of tumor, has led to heightened interest in finding unique targets that specifically eliminate the CSC population, and thus destroy the regenerative potential of the tumor. Biomarkers present at more concentrated levels within CSCs and rarely found at much lower levels within normal cells would permit selective curative therapeutic benefit without incurring typical side effects that befall many other promising chemotherapies.
Unless novel markers exist that are yet to be identified, it is unlikely that a single marker or same set of markers can be used to identify any given CSC. It is more likely that a combination of biomarkers will increase the specificity for a given CSC type depending on the type of cancer. It is also likely that a given niche even within the same tumor microenvironment could cause a shift in expression or dominance of various markers. An example would be the higher invasive profile of CD44 positive GSCs as part of the epithelial–mesenchymal transition (EMT) niche. Although a number of past studies indicated that CD133 positive are responsible for recurrence of GBM tumor while CD133 negative cells are not [Citation1,Citation49], evidence suggest that CD133 negative cells could regenerate heterogeneous tumors, showing different genetic expression [Citation50]. Due to this controversy, two additional CSC markers are discussed (Nestin and CD44) as dual-expression markers that may better define a GSC in a specific environment or situation.
CD133
CD133 also known as AC133, and prominin-1 was the first well-characterized CSC surface marker [Citation1]. CD133 is a 97 kDa transmembrane glycoprotein with multiple proposed functions [Citation51]. It is an important biomarker used to identify the CSC population within various tumor types and may have a role in cell growth, proliferation and pathophysiology of growing tumors [Citation52]. Cells that express CD133 have a capacity to generate neurospheres in vitro and induce tumor formation in in vivo models, suggesting its central role in GBM CSC function and success [Citation53]. Due to its usual location in plasma membrane protrusions and microvilli, CD133 is likely involved in membrane organization but new evidence suggests a critical modulator of important signal transduction cascades necessary for stem cell function [Citation51]. A key example would be the demonstration of CD133 modulation of the PI3K-Akt-mTOR, Wnt/B-catenin and TERT pathways, which are all heavily involved in cancer stem cell biology [Citation54]. In a recent study, recurrent pilocytic astrocytic tumors with reduced CD133 expression was associated with a decrease in the multiple drug resistant 1 (MDR1) receptor perhaps from downstream inhibition of the PI3K-Akt-NF-κB cascade [Citation55]. The findings are in line with another prior report where CD133 knockdown potentially inhibited PI3K/Akt pathway activity and reduced self-renewal and tumorigenicity of GSCs [Citation56]. In essence, signal transduction cascades involving CD133, may hold a key to finding to novel therapies that specifically target at least the GSCs that express CD133.
As mentioned earlier, not all tumor cells express the CD133 cell surface ligand so targeting of CD133 will need to be accompanied with a more sophisticated multi-targeting approach discussed below. The percentage of CSCs with the CD133 marker is not consistent, either. For instance, when three different primary GBM tumor cell lines were analyzed, the percentages of CD133+ cells by analysis of flow cytometry among the cultures were 10.2, 69.7 and 27.5% [Citation57]. In another study, as few as 100 CD133+ CSCs were sufficient to form tumors [Citation1]. Even small subsets of CD133+ CSCs would show neutrosphere-like growth and heightened tumor progression in the first few weeks. This phenomenon is shown in , comparing the number of spheres formed by CD133+ and CD133- CSCs [Citation58]. Not surprisingly, significantly greater number of tumor spheres correlates to the poor prognosis associated with tumors containing CD133+ CSCs [Citation58].
The number of spheres formed by CD133+ and CD133- cells after 14 days is given (clonogenic index: spheres per 100 cells plated).
Reproduced with permission from [Citation58].
![Figure 2. Cancer stem cell lines derived from tumors with poor prognosis were sorted for CD133 expression and plated with 10 cells/well.The number of spheres formed by CD133+ and CD133- cells after 14 days is given (clonogenic index: spheres per 100 cells plated).Reproduced with permission from [Citation58].](/cms/asset/a73130c1-39de-4138-a2a2-f59a0b0b4e30/itde_a_12365486_f0003.jpg)
Nestin
Nestin has many features that provide a strong complement to CD133 for identification of GSCs, especially during more aggressive GBM periods of stem cell proliferation. Nestin is an intermediate filament protein expressed primarily in neural progenitor/stem cells with higher expression early in development of the central nervous system. Nestin is also expressed in many human malignancies, including GBM, and has been proposed as another CSC marker [Citation59,Citation60]. Enhanced Nestin expression correlates with dedifferentiation, invasiveness and tumor grade [Citation61]. Mechanistically, Nestin likely facilitates aggressive growth and self-renewal capacity of CSCs through cytoskeletal organization, cell signaling, and cell metabolism [Citation62]. Nestin represents a promising marker that could facilitate identification of CSCs through dual marker identification along with CD133. For instance, prognostic accuracy significantly improved with co-identification of Nestin with CD133 [Citation63]. Typically, separation of CSCs using cell sorting rely on cell surface expression of a marker for identification. Despite its classical role in the cytoplasm, Nestin was expressed on the cell surface of 14 different glioma CSCs, but not on non-CSC cells, indicating potential specificity for identifying and sorting CSCs with Nestin cell surface expression [Citation64]. While a promising concept, it remains to be determined whether other GBM or cancer cell types may also express cell surface Nestin.
CD44
CD44 is a transmembrane glycoprotein and hyaluronic acid receptor, and is another example of a promising CSC marker particularly identifying the invasive subtype of CSCs [Citation65]. For instance, patient derived neurospheres enriched with CSCs with high CD44 expression displayed a substantially greater self-renewal capability and produced secondary tumors of the same heterogenous histopathological traits as tumor derived from patients [Citation66]. CD44 is implicated in epithelial-mesenchymal transition (EMT) and tumor invasion [Citation24] and predicts resistance to chemotherapy in mesenchymal-like glioma [Citation25,Citation32]. Enhanced expression of two mesenchymal GBM subgroup gene regulators, CEBP-beta and signal transducer and STAT3, were associated with increased expression of CD44 [Citation67]. Microarray analysis of human GBM tumors correlated with decreased survival rate in CD44 positive cases [Citation68]. Several studies have identified crosstalk between CD44 and EGFR, which is upregulated in 60% of GBM. For instance, activation of both EGFR and CD44 signal transduction pathways led to co-stimulation and GBM tumor promotion [Citation69]. Overexpression of EGF in high-grade glioma increased CD44 mRNA expression [Citation70], while inhibition of MAPK/ERK pathway with the compound, U0126, led to a reduction of CD44 expression [Citation70]. In addition, expression of CD44 in GSCs and its involvement in cancer stem cells niche signaling is associated with poor prognosis of GBM [Citation66]. CD44+ CSCs are found mainly in the edge area of tumor. Lower numbers are found in the intermediate zone and the lowest number are within the core of tumor, which is indicative of core-to-edge transition of the cells within a tumor. The cells in the edge of tumor are more resistant to therapy [Citation71]. On the other hand, longer median survival time in GBM patients with higher CD44 expression was reported [Citation72]. It was postulated that stem cell traits in low expression of CD44 in GBM cells were induced by Oct4 and Nestin [Citation73]. It was recently reported that knockdown of CD44 expression in GSCs inhibited migration/invasion as well as reduced the expressions of stem cell markers such as Nestin and Sox2 of these cells both in vitro and in vivo mouse xenograft model [Citation74]. Therefore, CD44 is a potential target for effective elimination of GSCs.
CSCs can survive in various conditions within the tumor microenvironment (TME) by residing in distinct hypoxic and perivascular niches in several types of solid tumors [Citation75]. A hypoxic TME induces quiescence in CSCs, as opposed to the perivascular niche, where environmental conditions are more favorable for more metabolically demanding invasive and proliferative activities. The issue of identifying GSCs in a given tumor microenvironmental niche is of high interest when considering novel treatment approaches against GBM. For instance, an attractive dual-drug treatment approach to treat GSCs may facilitate the study of drugs that can kill not only proliferating GSCs, but also CSCs in the quiescent hypoxic state [Citation76]. In addition, different TME conditions also drive expression of candidate CSC markers which can be targeted. For instance, hypoxia induces a CD44+ to CD133+ shift and chemo-radiotherapy also induces a CD133+ to CD44+ shift [Citation77]. Studies have shown that CD133+/CD144- glioma CSCs differentiate into cancer cells and endothelial progenitor cells and finally form mature vascular endothelial cells (ECs) [Citation78]. Within the perivascular niche, GSCs are located around the blood vessels and can differentiate into cancer vascular progenitor cells. For instance, Calabrese et al., showed that a direct contact exists between ECs and CSCs in brain tumors linking them directly to angiogenesis or to form vasculogenic mimicry that promotes the microcirculation in tumors [Citation79].
Glioblastoma drug treatment
For most drugs, the blood–brain barrier (BBB) is a primary obstacle to treating central nervous system diseases. The BBB is comprised of endothelial cells that form tight junctions to separate the brain from the body's main circulatory system [Citation80,Citation81]. For the small number of drugs that can cross the BBB, other issues such as offsite absorption and binding as well as multiple drug resistance efflux pumps serve as additional factors that reduce availability of drug to the tumor. For instance, although 100% of the TMZ is absorbed into the blood following oral delivery, only 17% of the drug makes it to the target location into the brain interstitium [Citation82,Citation83]. While a comprehensive analysis of all chemotherapeutic drugs is beyond the scope of this review, a few drugs are highlighted and discussed that have been shown to be therapeutic against GBM, including TMZ, idasanutlin (RG7388), and paclitaxel (PTX). For further information on current treatments and strategies in targeting GBM and glioma stem cells refer to the following review articles by Wu et al., Xiong et al. and Abadi et al. [Citation84–86].
Temozolomide
TMZ is a deoxyribonucleic acid (DNA) alkylator that nonspecifically methylates DNA [Citation87]. TMZ methylates at N3-adenine, N7-guanine and O6-guanine sites, arresting the cell at the G2/M phase [Citation87]. As a DNA alkylator, TMZ not only arrests the cell cycle of tumor cells but also non-specifically alkylates normal hematopoietic stem cells causing an unwanted toxic side effect [Citation88]. In addition, TMZ has poor serum stability with a short half-life of 1.8 h, necessitating multiple doses for maximal efficacy [Citation89]. Tighter control of the treatment regimen is required to prevent systemic side effects since TMZ is known to cause lymphopenia, thrombocytopenia and myelodysplasia [Citation90].
TMZ in conjunction with radiation is usually successful initially, but the majority of GBM recurs in the first year [Citation6,Citation80]. Within the genome of a GBM cell, the IDH gene leads to treatment resistance and the presence of wild-type IDH is a predictor of a poor response to a high dose of TMZ [Citation91]. Patients with normal or wild-type IDH1 correlate to a shorter survival, compared with those with IDH1 mutations [Citation15]. IDH1 and IDH2 both work to block stem cell differentiation and increase both VEGF and hypoxia within the tumor environment [Citation92]. All are contributing factors to TMZ resistance.
The high rate of GBM recurrence after TMZ treatment can also be attributed to GSCs [Citation93]. CSCs have a highly developed DNA damage response (DDR) system which can repair DNA damage caused by TMZ and other chemotherapeutic drugs and avoid apoptosis. From CSCs, resistance is thought to be due to either increased expression of MGMT, which can reverse the TMZ-induced methylation, or from reduced expression of tumor suppressor p53 as a result of high inhibition [Citation87]. The MGMT gene encodes a DNA-repair protein to fix any DNA alkylation from TMZ [Citation94]. When the promoter for this gene is methylated, it leads to a more positive prognosis because MGMT is not over-produced [Citation13]. CSCs typically have an over-expression of MGMT leading to rapid DNA repair after TMZ alkylation. Given initial success rate of TMZ against GBM, there is great interest in developing promising chemotherapies that can prevent or overcome the eventual resistance of GBM to TMZ. One study demonstrated that through increased TMZ concentrations for a longer term, TMZ therapy was found to preferentially deplete CSCs [Citation95]. The need for discovering additional therapies to target the population of CSCs in a different manner exists to reduce the overall treatment time for patients with debilitating symptoms.
TMZ resistance is thought to be due to the upregulation of a complex interplay of DNA damage response (DDR) system. Various strategies have been developed in attempt to prevent GBM from developing resistance to TMZ by blocking the DDR. In addition to the overexpression of MGMT resulting in TMZ resistance, CSCs also over-express MDM2, which negatively regulates tumor suppressor protein p53. However, when MDM2 is overexpressed, p53's ability to reduce the tumor's oncogenic effects and send the cell down an apoptotic pathway is inhibited [Citation96]. This dysregulation of the p53 pathway is found in 85.3% of primary GBM with 15% of those resulting from overexpression of MDM homologs (1/2/4) [Citation97]. Not only is p53 involved in tumor suppression, it plays a role in regulation of many other cellular processes including cell-cycle progression, senescence, DNA repair and metabolism [Citation98]. Loss of p53 permits expansion of cells, while in normal cells p53 presence will permit the cell to exit mitosis and allow time for DNA repair [Citation99,Citation100]. Because p53 is inhibited by MDM2 by many pathways, a small molecule blocking MDM2 would prevent p53 inhibition and induce tumor suppression. An understanding of TMZ resistance brought about from CSCs and the microenvironment allows for treatment development to strategically diminish further GBM resistance [Citation101].
Idasanutlin
Idasanutlin (RG7388) is an example of a potent MDM2 inhibitor with >100-fold selectivity compared with its predecessor, RG7122 [Citation102]. Idasanutlin is a viable treatment option against GBM cell lines or tumors in vivo as long as P53 is not mutated [Citation96]. Idasanutlin has great systemic exposure, is metabolically stable in vivo, passes through BBB, and is non-genotoxic [Citation102–104]. Idasanutlin competes with p53 for binding to MDM2 inside the cell nucleus resulting in accumulation of p53 and induction of antitumor activity [Citation98]. While Idasanutlin has many advantages, it possesses challenges as well. Idasanutlin is potent against wild type but not mutant p53 tumors [Citation105]. Additionally, therapy solely with Idasanutlin may lead to a secondary resistance, since prolonged treatment with Idasanutlin has led to generation of p53-mutated resistant populations that occurred de novo and may inhibit elimination of cancer cells [Citation106]. Therefore, it may be essential to pair MDM2 inhibitors with other agents that target tumor cells with mutated p53 [Citation98]. Unfortunately, idasanutlin also has dose-limiting toxicity leading to thrombocytopenia and neutropenia [Citation98].
Short-term treatment with Idasanutlin (100 nmol/l) inhibited clonogenicity in p53 wild-type GBM cells lines and cultures. Another study demonstrated effective Idasanutlin therapy against p53 wild-type tumor cells lines in nanomolar concentrations [Citation107]. Idasanutlin has been studied preclinically in several cancers such as neuroblastoma, childhood sarcoma, and ovarian carcinoma. It had also progressed to a phase III clinical trial for patients with relapsed or refractory AML (NCT02545283). Currently one clinical trial includes Idasanutlin against GBM, which is actively recruiting patients as of Nov 2021 (NCT03158389). The most common adverse effects reported for Idasanutlin were diarrhea, nausea and vomiting, and myelosuppression causing febrile neutropenia and thrombocytopenia. Overall, Idasanutlin was well tolerated over multiple studies [Citation108].
Paclitaxel
PTX is a chemotherapy medication that is used for the treatment of several cancers. PTX is an anti-microtubule agent that stabilizes microtubules by binding to the β-tubulin subunit, resulting in disruption of microtubule dynamics and mitotic apparatus during cell division. A downstream result is inhibition of cell cycle transition from the G2 phase to the M phase in malignant cells [Citation109–111]. Based on its mechanism, PTX serves as an excellent anti-cancer agent due to the joint antiangiogenic and cytotoxic properties [Citation112]. A recent study also found that PTX can stimulate autophagy and induce apoptosis [Citation113]. PTX can promote apoptosis and suppress growth and proliferation in two GBM cells lines, U251 and U87MG, in both in vitro and in vivo models [Citation114]. It is known that MMP-9 pathway inhibition can prevent the growth and metastasis of tumors and PTX inhibits MMP-9 and p38/JNK [Citation115].
Although, PTX is an example of a very potent drug against GBM, it is rapidly removed from the brain through multiple drug resistance (MDR) receptors, greatly limiting its application in vivo. To overcome this limitation, PTX is often studied in conjunction with nanosystems, which will be discussed in the nanosystems section later in this review. The main challenge of treating PTX in its pure form either intravenously or orally, is that PTX is a strong p-glycoprotein substrate, and thus has limited distribution across the BBB [Citation116]. In addition, lack of ionizable groups limits PTX solubility in altered pH solutions [Citation112]. Current forms of PTX available are also challenged by these chemical constraints as the low solubility risks drug precipitation and side effects such as bronchospasms, hypotension, and hypersensitivity [Citation112]. Because of these challenges, PTX may benefit by an alternate drug delivery method to better target GBM and limit systemic travel. PTX can be challenged by tumor resistance, like TMZ and RG7388. PTX resistance can occur with overexpression of multi-drug resistance (MDR-1) gene, molecular changes in β-tubulin, and changes in apoptotic and mitosis checkpoint proteins [Citation117].
For systemic delivery of PTX, lipophilic agents such as Cremophor EL (CrEL) has been used as a vehicle [Citation118,Citation119]. Combination of PTX and its solubilizing agent CrEL produced a toxic effect due to induction of oxidative stress that is largely responsible for the in vivo oxidative damage induced by PTX [Citation120]. Additionally, for treatment of brain cancer and to achieve maximal accumulation of PTX in the brain tumor, either stereotaxic infusion or convection-enhanced delivery (CED) is required [Citation121]. These delivery approaches are less favorable clinically due to invasiveness. Therefore, there is a great demand for noninvasive approaches that could provide effective delivery of PTX to the brain for the treatment of GBM.
Combination therapy strategy
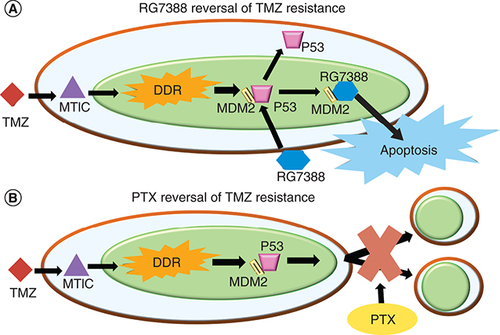
Because of the high rate of resistance of GBM against TMZ and other mono-chemotherapy approaches, combination therapies are needed to increase the likelihood of combating solid heterogenous tumors. Combination with therapies like RG7388 or PTX, could offer additional treatment options to overcome TMZ resistance and eliminate residual CSCs. Each drug works in a different delivery pathway and has shown not to exhibit drug interference [Citation122]. Preliminary data from Wang et al. has shown that TMZ and Nutlin 3A in combination produced a greater than expected, or a synergistic effect, in a primary GBM10 cell line [Citation96]. Therefore, TMZ and MDM2 inhibitors provide a promising option for a combination therapy. As shown in A, TMZ enters the cell and converts to the active methyldiazonium ion MTIC that alkylates the DNA in the nucleus inducing apoptosis. Because of the increased DDR in the tumor, the cells become TMZ resistant. PTX combined with TMZ also work synergistically against GBM. PTX and TMZ were co-loaded into mPEG-PLGA NPs and used to treat both U87 and C6 glioma cells [Citation123]. The two drugs co-loaded into the NPs were more cytotoxic than the drugs loaded individually. displays the two different pathways taken by TMZ (A) and PTX (B) when treating GBM. As with TMZ and RG7388, the two different pathways can reduce potential drug interference and increase synergistic behavior.
(A) The mechanism that TMZ and RG7388 undergo independently and in combination to send the cell population toward apoptosis. (B) The two separate pathways that TMZ and PTX undergo to send the cell population toward either apoptosis or cytokinesis failure.
DDR: DNA damage response; PTX: Paclitaxel; TMZ: Temozolomide.
Nanomedicines for stem cell resistant GBM treatment
A number of studies have reported that GBM stem cells are more chemo-resistant than differentiated cancer cells due to their intrinsic properties as well as varied resistant mechanisms [Citation124]. GBM cells are found to be resistant to a wide range of chemically dissimilar chemotherapeutic drugs due to multiple drug resistance caused by inhibition of apoptosis, activated detoxification systems, diminished drug entrance, increased drug efflux, altered therapeutic targets, and activated DNA repair systems [Citation125]. In addition, the BBB poses a unique challenge in allowing drugs to enter the GBM cells due to its highly selective structure with practically no paracellular diffusion [Citation126–130]. Transcellular BBB absorption could take place via both passive and active transports. It is believed that nanoparticles mostly follow vesicular transport route via specific (receptor mediated transcytosis) or non-specific (absorptive mediated transcytosis) pathways [Citation131–133]. Moreover, the BBB transport and the intrabrain distribution of a drug are not be considered same, while the intrabrain distribution is dependent on a complex distribution kinetics and disposition in the target site [Citation134,Citation135].
It is estimated that 98% of low-molecular-weight molecules and 100% of high-molecular weight macromolecules fail in entering the central nervous system due to physiological and physiochemical challenges [Citation136,Citation137]. In general, lipophilic, low molecular weight (<400 Da), non-ionized molecules cross BBB. On the other hand, highly lipophilic compounds were retained in the lipid layer of BBB, causing non-specific uptake leading to toxicity or removal by efflux transporters [Citation138]. In addition, ionization of drugs at the biological pH also alters the diffusivity of a number of drugs across BBB. Therefore, while many small molecules are unable to cross the BBB, nanosystems may overcome the brain penetration issues by eliminating potential interactions along the drug delivery route or contain therapeutic molecules in a carrier that provide a stealth component. The stealth property decreases recognition of the drug carrier system by macrophages, thereby allowing more prolonged circulation times. This can lead to better BBB penetration and help maintain stability in biological milieu [Citation139–141]. Other advantages of nanosystems include manipulation of therapeutic doses of a drug by selection of suitable biomaterials, improved endothelial permeability and extravasation, and decreased clearance from phagocytes [Citation142]. Nanosystems may also preserve the pharmacological action of the drug loaded into the system. During transport, nanosystems can protect a drug to potentially increase the drug's effective concentration. Nanosystems include nanoemulsions, nanoparticles, nanogels, and dendrimers [Citation137,Citation143]. While many options are available for different delivery and targeting methods, nanoparticles will be discussed in this review as they are the most popular carriers for delivery of anticancer drugs [Citation144–146].
Different nanoparticulate drug carriers
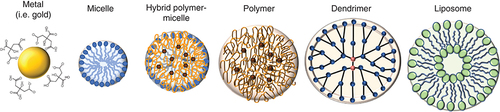
Nanoparticles (NPs) can be solid or vesicular with diameters ranging from 1 to 1000 nm and are actively being developed for both therapy and diagnostics of GBM [Citation147]. NPs with diameters smaller than 400 nm have enhanced permeability and retention (EPR) effect [Citation148], which could enable passive targeting of drug-loaded NPs into the GBM [Citation149]. More specifically, NPs less than 100 nm have many advantages including enhanced solubility, increased bioavailability of poorly soluble drugs, increased surface area, and a potential decrease in required dose [Citation150]. NPs may be composed of biodegradable and non-biodegradable constituents and are further categorized as polymeric, polymeric micelles and inorganic, among others as shown in [Citation142]. The size and surface characteristics of NPs can be easily manipulated for passive or active drug targeting [Citation151]. Other advantages include the capability for site-specific targeting, ability to utilize various delivery routes, and the option for a more controlled and longer sustained release of drugs at the target site [Citation151]. There are a few disadvantages, such that small size and large surface area can lead to particle-particle aggregation and drug-loading may be limited with a burst release [Citation151]. To construct NPs, the appropriate method must be chosen based on the materials and drugs used. An important consideration is the extent of biodegradability leading to optimal drug-release characteristics. Additional considerations include the desired size and size distribution, surface characteristics, drug solubility, and drug stability [Citation150].
Metal nanoparticle (NP) types include gold, gadolinium and silica and can achieve the smallest sizes including 1–10 nm. Micellar NPs are made of a lipid monolayer and can be on the order of 10 nm. Polymer NPs are versatile structures with a wide range of different polymer types. The most often used polymers are PLA and PLGA. Aside from PLA and PLGA, other polymers that can be used and can also be incorporated into micelles, such as PEO, PPO, PS and PCL. Dendrimers are highly branched, star-shaped macromolecules with nanometer-scale dimensions. Liposomes are lipid bilayers used often in cancer drug delivery.
There are many methods for fabricating NPs, including solvent evaporation, nanoprecipitation and microfluidics, to name a few [Citation143,Citation152,Citation153]. Solvent evaporation, adopting the principles of either single or double emulsions are the most common methods used in fabrication of nanoparticles. The single emulsion is chosen typically for encapsulation of hydrophobic drugs. This type would be characterized by an oil in water (o/w) emulsion and involves dissolving both drug and polymers into an organic solvent and emulsifying them, using ultrasonication, in an aqueous medium [Citation150]. The double emulsion can be used to encapsulate hydrophilic drugs or both hydrophilic and hydrophobic drugs together. Typically, a hydrophilic drug is dissolved in an aqueous medium followed by emulsification into a non-aqueous solvent containing the hydrophobic drug and carrier polymers. The resulting o/w emulsion is then emulsified into an aqueous medium, forming a water in oil in water (w/o/w) emulsion. With every emulsion it is essential that each solution is completely immiscible with each other to allow the solvents to disperse homogeneously forming a smooth two-phase system [Citation154]. A surfactant can be used to reduce the dispersed droplet size, reduce droplet aggregation, and impart hydrophilicity leading to increased circulation blood in vivo [Citation151,Citation155]. Selection of surfactant concentration is important because a low concentration of surfactant could lead to an increased polydispersity and particle aggregation, while a high surfactant concentration can decrease drug-loading [Citation156].
Nanoprecipitation, otherwise known as the solvent displacement method, is a popular method to fabricate NPs due to its simplicity [Citation157,Citation158]. Nanoprecipitation is based on the principle of miscibility of solvents and the polymers to fabricate the NPs. When the organic solvent is evaporated it will cause phase separation of the polymer [Citation157]. During this step of fabrication, the organic solvent typically containing the polymers is added to the aqueous solvent and the NPs form spontaneously. The drug is dissolved in either the organic or the aqueous solution, depending on its water solubility. Nanoprecipitation has been studied as a method encapsulating both hydrophobic compounds (paclitaxel, triptolide, dexamethasone and anti-inflammatory drugs) as well as hydrophilic compounds (DNA, protein, and vancomycin) [Citation157–160]. NPs have also been fabricated using the nanoprecipitation method for TMZ, which some regard as amphiphilic and others as hydrophilic [Citation87,Citation161]. Hydrophobic drugs have been found to have a greater encapsulation efficiency in comparison to hydrophilic compounds [Citation160]. The nanoprecipitation method includes limited variability between batches and limited scalability, making its transfer from lab to industry more feasible [Citation157]. A limitation of this procedure is the solubility of the drug to be encapsulated. If the drug is more soluble in the non-solvent, the aqueous solvent, the drug-loading percentage may be low [Citation158].
While previously discussed methods may be considered a more conventional approach, they may suffer from the generation of NPs with wide size distribution and large batch-to-batch variations [Citation149]. This is one of the major reasons for slow translation of nanoparticulated drugs to the clinic compared with non-carrier mediated delivery systems. Many studies may be successful for small-scale in experimental settings, but they may be significantly challenged at the industrial level. Microfluidics techniques have shown to automate the fabrication process by generating NPs with narrow size distribution; for example, the average size of polymeric NPs fabricated through microfluidics is 50 nm with a polydispersity index (PDI) of 0.1 [Citation149]. Microfluidics is based on the principle of manipulating very small amounts of fluids, through channels having dimensions of tens to hundreds of micrometers [Citation162]. Two different methods are used when synthesizing NPs by microfluidics and both are dependent on the flow type. Single-phase flow, otherwise known as continuous flow, is the most commonly used and is driven by laminar flow [Citation162]. Single-phase flow solves many challenges with nanoprecipitation as the method is more controllable and reproducible [Citation162]. The second method, multi-phase flow or droplet-based, involves at least two immiscible fluids. Droplets are manipulated and formed inside microchannels. Parameters such as flow rates, channel dimensions, and geometry allow control over the process. PLGA has been used to fabricate NPs by microfluidics using both methods. These NPs formed spheres as small as 20 nm [Citation149]. It is to be noted that microfluidic principles could also be applied to the previously discussed fabrication techniques.
Polymeric NPs
Polymeric NPs are a type of NPs made of organic materials that can become multifunctional because of further conjugation after the initial formulation of NPs [Citation163]. Polymeric NPs are also the simplest type often using single-chain polymers [Citation164]. Many polymer chains contain groups such as carboxylic acids or amino groups that allow molecules to be conjugated to them for various applications. In addition, polymers can be chosen depending on the time needed to release a drug from the system. Polymers with specific functional groups, such as methyl groups, will slowly hydrolyze in the body, hence slowing the release. Some polymer examples used include polylactic acid (PLA), polyethylene glycol (PEG), polycaprolactone (PCL) and chitosan. PLGA (poly lactide-co-glycolide) is a common polymer used in the fabrication of polymeric NPs and combines both PLA and PGA. PLGA is biodegradable as well as biocompatible and is approved by FDA. Varying the ratio of PLA to PGA allows for fine tuning of drug release, compared with each polymer alone, because as the ratio of PLA:PGA increases, the rate of hydrolysis decreases, slowing the release of drug. One group fabricated PLGA NPs using an emulsion-solvent evaporation technique and loaded them with TMZ for treatment of GBM [Citation90]. The NPs were then conjugated to a transferrin receptor, OX26 mAb. By encapsulating TMZ in the NPs, an in vitro study showed that the NPs slowed the release profile of TMZ. A slower release profile would allow for more of the drug to be delivered to the tumor and less of adverse systemic effects ().
Free TMZ was used as control. Results are represented as mean ± SD (n = 3).
mAb: Monoclonal antibody; NP: Nanoparticle; TMZ: Temozolomide.
Reprinted with permission from [Citation90].
![Figure 5. In vitro release of temozolomide from monoclonal antibody-modified and non-modified PLGA nanoparticles in phospate-buffered saline (pH 7.4, 0.01 M) at 37°C.Free TMZ was used as control. Results are represented as mean ± SD (n = 3).mAb: Monoclonal antibody; NP: Nanoparticle; TMZ: Temozolomide.Reprinted with permission from [Citation90].](/cms/asset/ca1ef04e-24b7-4d69-816b-530d39bacf32/itde_a_12365486_f0006.jpg)
NPs for the delivery of a TMZ-ester for increased drug delivery for GBM were also developed by Chu et al. [Citation165]. The PLGA NPs were constructed using an emulsion-solvent evaporation technique. PLGA and the TMZ-ester were dissolved in acetone and dichloromethane (DCM) and then added to a 1% polyvinyl alcohol (PVA) for sonication. These NPs were then conjugated to an anti-human ephrin type-A receptor 3 tyrosine kinase antibody to target GBM intranasally, which is a membrane-associated receptor over-expressed in the vasculature in GBM tumors. The conjugated NPs were approximately 146 nm and targeted the brain more effectively than unlabeled NPs as measured by fluorescence imaging in the in vivo rat models. While 20–50 nm particle size range has been found to be most favorable for crossing the BBB, carrier material type, particle shape, surface morphology with or without receptor binding all determine the optimal BBB crossing of the particulate systems [Citation166,Citation167].
Micellar nanoparticles
Micelles are amphiphilic surfactant molecules that spontaneously aggregate into spherical NPs [Citation168]. In an aqueous environment, the hydrophobic tail group of micelles collects toward the center of the spherical particle and the hydrophilic head group of the micelles collects toward the outer edges of the particle. Micelles are formed by self-assembly through the hydrophobic and hydrophilic groups. A recent review covered the details of fabrication and characterization of polymeric micelles for anticancer drug delivery [Citation169]. Micelles are beneficial because of their advantages in delivering hydrophobic drugs as well as their small size. Some examples of polymers that have been used to formulate micelles include poly(ethylene oxide) (PEO), poly(propylene oxide) (PPO), polystyrene (PS) and PCL [Citation154,Citation168]. Micelles are heralded for their small size and can also be integrated into polymers and provide a simpler assembly as opposed to liposomes. While their small size is beneficial when crossing the BBB, it may lead to rapid clearance and limit drug-loading capacity [Citation151,Citation170]. In addition, compared with other NPs such as polymer NPs, micelles lack strong intermolecular interactions with the encapsulated drug causing premature leakage [Citation170].
One example of micelles utilized for the treatment of GBM includes the use of PCL and methoxy-PEG copolymer [Citation171]. Methoxy-PEG-PCL encapsulated both doxorubicin and honokiol for co-delivery. PCL formed the hydrophobic core while PEG was effective at stabilizing the shells of the micelles in an aqueous environment. The micelles were 34 nm in size and showed strong anti-cancer effects such as tumor cell apoptosis, decreasing cell proliferation and tumor angiogenesis compared with single drug micelles [Citation171]. Another group fabricated PEG-PLA micelles and conjugated cyclic-arginine-glycine, Aspartic acid-D-tyrosine-lysine (c-RGDyK) to target integrin αvβ3 which is over-expressed on GBM neovasculature [Citation172]. The micelles were loaded with PTX and were administered to mice. displays the percent survival of treating the mice with the micelles and shows that the drug-loaded micelles allowed for a significantly improved median survival time [Citation172]. Mice were implanted with 5 × 106 U87MG cells and once the tumors reached 40–100 mm3, the mice were given 100 μl of one of the following: saline, free PTX (Taxol), PEG-PLA-PTX micelles, or c(RGDyK)-PEG-PLA-PTX micelles containing 7.5 mg of PTX/kg body weight.
Mice received four doses treatment of c(RGDyK)-PEG-PLA-PTX micelle survived significantly longer than mice that received i.v. administrations of PEG-PLA-PTX micelle (p < 0.05, log-rank analysis), Taxol® (p < 0.01) or saline (p < 0.01).
PTX: Paclitaxel.
Reprinted with permission from [Citation63].
![Figure 6. Kaplan–Meier survival curve of mice bearing intracranial U87MG glioblastoma.Mice received four doses treatment of c(RGDyK)-PEG-PLA-PTX micelle survived significantly longer than mice that received i.v. administrations of PEG-PLA-PTX micelle (p < 0.05, log-rank analysis), Taxol® (p < 0.01) or saline (p < 0.01).PTX: Paclitaxel.Reprinted with permission from [Citation63].](/cms/asset/f920cd25-6dfc-4e9f-9790-e63ebc95c943/itde_a_12365486_f0007.jpg)
Polymer-micellar nanoparticles
Polymer-micellar NPs could combine many of the advantages of polymer NPs and micelles while potentially avoiding some of their respective disadvantages. For instance, the polymer component provides structural stability while the micelle component allows decreased size [Citation168,Citation170]. In addition, compared with traditional micelles, the combination of both components escapes rapid excretion, seen with micelles, as polymeric NPs could sustain drug release over time. Micelles are known to possess good renal clearance because their hydrodynamic diameters are similar to globular proteins, and it has been shown that globular proteins of approximately 5–6 nm are associated with the ability to clear via renal filtration or urinary excretion [Citation173]. The larger size of polymer-micelles prevents rapid renal clearance. Nabar et al. produced polymer-micellar NPs containing both PLGA and poly (styrene-block-ethylene oxide) (PS-b-PEO) [Citation170]. PLGA/PS-b-PEO particles with a polymer: micelle ratio equal to five achieved a particle size of about 50 nm. These particles achieved a sustainable drug release characteristic across 25 days. However, the NPs lacked functional characteristics such as the ability to target a specific tissue or disease or to track via imaging.
Smiley et al. developed similar polymer-micellar NPs and loaded them with both TMZ and RG7388 [Citation161]. These NPs were also conjugated with a 15-nucleotide base-pair CD133 aptamer to target the CSC population for in vitro studies. shows that targeted TMZ + RG7388 NPs had a higher percent cytotoxicity to GBM CSCs compared with non-targeted TMZ + RG7388 NPs, TMZ containing NPs and empty NPs [Citation161].
Percent cytotoxicity is depicted following treatment with control nanoparticles (yellow), TMZ-containing nanoparticles (blue), non-targeted TMZ + RG7388 NPs (green) and targeted TMZ + RG7388 (orange). Lower concentrations of NPs with and without TMZ/RG7388 (5.58, 11.16, 22.31, 44.63 and 89.25 μg) had no effect and so are not represented on the graph; n = 3 ± SEM.
NP: Nanoparticle; TMZ: Temozolomide.
Reprinted with permission from [Citation161].
![Figure 7. Treatment of nanoparticles in cancer stem cells.Percent cytotoxicity is depicted following treatment with control nanoparticles (yellow), TMZ-containing nanoparticles (blue), non-targeted TMZ + RG7388 NPs (green) and targeted TMZ + RG7388 (orange). Lower concentrations of NPs with and without TMZ/RG7388 (5.58, 11.16, 22.31, 44.63 and 89.25 μg) had no effect and so are not represented on the graph; n = 3 ± SEM.NP: Nanoparticle; TMZ: Temozolomide.Reprinted with permission from [Citation161].](/cms/asset/90c5c43f-097a-4075-97d3-ee82dca622d7/itde_a_12365486_f0008.jpg)
Solid lipid nanoparticles
Solid lipid NPs are also among the options for encapsulating the drug combinations for GBM treatment. Typically, the nanosystems designed contain synthetic polymers that were previously listed including PLGA or PEO. While they have many advantages such as functionalization capability and versatility in their physicochemical properties, many present toxicity issues from acidic byproducts that may make them inappropriate for extended brain delivery [Citation174]. Lipid-based NPs have been at the forefront of latest research due to becoming a pivotal role in COVID-19 vaccine success [Citation175]. Solid lipid NPs have advantages such as incorporating biocompatible lipids in their synthesis, high drug-loading capabilities, and controlled drug release [Citation174,Citation176]. Another advantage is that they have been regarded as easy-to-produce formulations [Citation177].
While solid lipid NPs overcome many disadvantages associated with other nanosystems, they are challenged with the expulsion of the encapsulated drug, their tendency to gelate, and their low encapsulation efficiency due to the lack of empty spaces in the lipid core [Citation174]. Solid lipid nanoparticles can be produced by methods such as high-pressure homogenization, solvent emulsification/evaporation, supercritical fluid extraction of emulsions, ultrasonication, and spray drying [Citation174,Citation178]. The MDM2 inhibitor, Nutlin 3A and superparamagnetic NPs were both encapsulated in solid lipid NPs to not only treat GBM, but also to direct the NPs to the tumor site using magnetic fields [Citation176]. These NPs that were prepared through a solvent evaporation technique demonstrated the ability to cross an in vitro BBB model and pro-apoptotic activity in U87 GBM cells superior to free drug [Citation176]. In the study, increased expression of p53, p21 and MDM2 all correlated to a cytotoxic effect of drug treatment [Citation176]. shows that treatment with Nutlin 3A-loaded magnetic NPs significantly increased expression of p53 and p21 compared with control, empty NPs, and free drug in U87MG cells [Citation176].
(A) Expression of p53 and its downstream proteins (MDM2 and p21) was analyzed on U-87 MG cells after 72 h of treatment with 100 μg ml-1 of Mag-SLNs, 1.33 μM of nutlin-3a, or 100 μg ml-1 of Nut-Mag-SLNs (corresponding to 1.33 μM of drug) and compared with control cultures. (B) Quantitative evaluation of western blotting results.
SLN: Solid lipid nanoparticles.
Adapted from [Citation176] Creative Commons Open Access.
![Figure 8. Western blotting analysis of markers involved in apoptosis. (A) Expression of p53 and its downstream proteins (MDM2 and p21) was analyzed on U-87 MG cells after 72 h of treatment with 100 μg ml-1 of Mag-SLNs, 1.33 μM of nutlin-3a, or 100 μg ml-1 of Nut-Mag-SLNs (corresponding to 1.33 μM of drug) and compared with control cultures. (B) Quantitative evaluation of western blotting results.SLN: Solid lipid nanoparticles.Adapted from [Citation176] Creative Commons Open Access.](/cms/asset/535d7d89-b684-4397-987d-cf4888f9dd83/itde_a_12365486_f0009.jpg)
Liposomes
Liposomes are an important member of the nanoparticle community that have been investigated for treatment of GBM tumors. Doxil®, a PEGylated liposomal-based doxorubicin (100 nm with 10,000–15,000 doxorubicin per liposome), was the first nano-drug to be approved by the US FDA in 1995 for the treatment of AIDS-related Kaposi's sarcoma, in 1999 for the treatment of ovarian cancer, and in 2007 for the treatment of multiple myeloma. Liposomes are lipid-bilayer vesicles known for their ability to encapsulate a wide variety of drugs, their biocompatibility, non-immunogenicity and commercial availability [Citation93]. They are artificial drug carriers that can be made of cholesterol and natural phospholipids [Citation179]. Using lipids provides the opportunity to carry both hydrophobic and hydrophilic drugs because lipids are amphipathic. Liposomes form lipid bilayers resemble that of the cell membrane; they have polar ends and the components chosen to form the liposome govern the degree of charge and their size. One advantage to using liposomes is their stability once a drug is encapsulated. This stability is crucial when systemically delivering NPs as it keeps healthy tissues clear of potential toxic chemotherapy drugs. Other than stability, liposomes are also effective drug delivery vehicles for their site avoidance effect and their flexibility in design by conjugating site-specific ligands [Citation179]. Disadvantages of liposomes include low solubility, short half-life and high cost production [Citation179].
Liposomes are formed in a similar manner as micelles by self-assembly through the hydrophobic and hydrophilic groups. However, liposomes form a bilayer, like a cell membrane, and have the capability to contain both hydrophilic and hydrophobic drugs [Citation168]. An example of liposomes for the treatment of GBM comes from the fabrication of polyethyleneglycol-carbamyl distearoylphosphatidylethanolamine (DSPE-PEG[2000]-NHS) loaded with chemotherapeutic drugs doxorubicin and erlotinib, then further conjugated with transferrin for mediated transcytosis and penetratin for enhanced cell penetration [Citation180]. These liposomes were fabricated using a thin film hydration, but compared with micelle formation with solvent evaporation, this method involved additional steps. For instance, they used three different phospholipids with DSPE-PEG(2000)-penetratin, a pH gradient to encapsulate doxorubicin and a G100 sephadex column [Citation180]. This fabrication resulted in a particle size greater than 150 nm and a polydispersity index (PDI) of 0.193. While the construct was effective at decreasing tumor volume over time in mouse in vivo models, the synthesis of such a construct can take time and be quite costly with the materials needed. In addition, this construct lacked a targeting moiety to effectively target the GBM tumor direct and resulted in offsite binding in other organs such as the spleen and heart [Citation180]. Another group fabricated PTX-loaded liposomes composed of cholesterol, DSPE-PEG and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) [Citation181]. To enhance delivery across the BBB, the liposomes were extruded through a membrane to form microbubbles and during in vivo studies, the BBB was disrupted by focused ultrasound. compares different treatment groups given to mice bearing intracranial GBM tumors. Compared with the control, PTX-loaded liposomes that were encapsulated in microbubbles and aided across the BBB with focused ultrasound were the most effective at increasing the percent survival [Citation181].
Kaplan-Meier survival curves of mice bearing intracranial glioblastoma tumors treated with either control solution, MBs enhanced across the BBB with FUS, PTX-loaded LIPO, and a combination of PTX-loaded LIPO, encapsulated into MB and with FUS.
BBB: Blood–brain barrier; FUS: Focused ultrasound; LIPO: Liposome; MB: Microbubble; PTX: Paclitaxel.
Reprinted with permission from [Citation181].
![Figure 9. Percentage survival of mice treated with paclitaxel-loaded liposomes.Kaplan-Meier survival curves of mice bearing intracranial glioblastoma tumors treated with either control solution, MBs enhanced across the BBB with FUS, PTX-loaded LIPO, and a combination of PTX-loaded LIPO, encapsulated into MB and with FUS.BBB: Blood–brain barrier; FUS: Focused ultrasound; LIPO: Liposome; MB: Microbubble; PTX: Paclitaxel.Reprinted with permission from [Citation181].](/cms/asset/c61e0f5d-36ec-441c-bd3b-f06eea17502e/itde_a_12365486_f0010.jpg)
Nanoparticle for targeting of GBM CSCs
Nanoparticle functionalization
A major benefit to using NPs for drug delivery, is the capability to functionalize the NPs. They may be functionalized with a number of targeting or internalization moieties for a variety of purposes. Most common reasons include targeting the delivery system to a specific population of cells or to aid in diagnostics. One way to functionalize NPs with additional molecules is through covalent bonding. Covalent conjugation can occur through the following chemical groups outlined by Werengowska-Ciećwierz et al.: that use two thiol groups, two primary amines, a carboxylic acid and primary amine, a maleimide and a thiol, or a hydrazide and aldehyde [Citation182]. Covalent bonding may be more beneficial than noncovalent because while toxic reaction agents may be used, covalent bonding is a stronger bond and provides more control over the reaction. The amide formation reaction is a two-step procedure that utilize a carboxylic acid N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) reaction [Citation182,Citation183]. PLGA with a carboxylic acid group is activated with EDC to facilitate binding to NHS. This allows the covalent bonding of primary amines containing molecules such as a peptide or an aptamer to the NHS group. EDC reacts with the carboxylic acid and forms an O-acylisourea intermediate that is displaced by the primary amine. NHS is used to improve the efficiency of the binding by forming a more stable intermediate. The polymer containing the carboxylic acid could be combined with a polymer containing an NHS group [Citation161]. Because the amine reactive NHS ester is already within the NPs, this eliminates the need for the use of EDC which may provide toxicity to in vitro studies if not fully removed.
The versatility of NPs allows a vast array of options for developing imaging-based contrast agents. One example is using zirconium-89 (89Zr) for positron emission tomography (PET) imaging given its high 511 keV gamma emission [Citation142]. 89Zr was originally utilized for antibody labelling because its longer half-life of 78 h corresponds with the 3-day half-life of circulating therapeutic antibodies. This allows an optimal amount of time for labeling the NPs and the ability to monitor the biodistribution of NPs in vivo over time. To effectively chelate the 89Zr to the NPs, deferoxamine (DFO) is used as a chelating agent. DFO is a hexadentate siderophore that has the ability to chelate metals through three hydroxamate groups [Citation184]. DFO is covalently conjugated to an amino group on the NPs through amide formation. The 89Zr is then contained on the inside of the DFO in a ring-like structure to allow for imaging of the NPs. The goal is to maintain the 89Zr covalent bond to the NPs throughout their time travelling to the tumor and this can be studied using labelling efficiency studies. Using this process, Veronesi et al. was able to achieve a 60% binding efficiency when labelling polymer micellar NPs using DFO [Citation185].
Nanoparticle targeting molecules
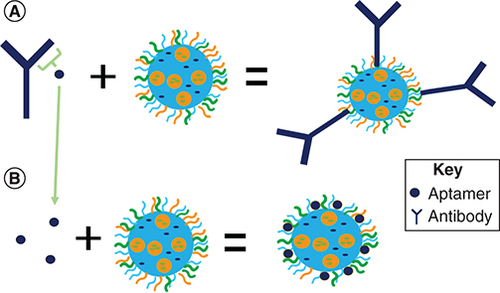
While GSCs appear to be a driver of GBM tumor recurrence and drug resistance, they may also hold the key to improved therapy, if not a cure, if they can be selectively targeted. Along with CD133, GSCs also express other biomarkers including CD44, CD95, Nestin and GFAP, which provide additional potential options for GSC targeting [Citation186]. Recent studies have shown the AC133 epitope on CD133 is a more specific marker for CSCs [Citation1]. During CSC differentiation, the AC133 epitope becomes sequestered, which is therefore only present during its undifferentiated state. This provides an important opportunity for targeting the undifferentiated CSCs in a selective manner. Given the difficulties associated with developing antibodies to the AC133 epitope, aptamers are instead being developed because of their ability to conform to any three-dimensional shape to reproduce the active binding site of the target ligand. Additional advantages of aptamers include reduced immunogenicity which prevents premature clearance, high reproducibility, stable conformation and much smaller size compared with antibodies, which may increase the likelihood of crossing cellular barriers. shows an approximate size increase by conjugation of an antibody versus an aptamer to nanoparticle surface. Conjugating the entire antibody could result in an increase in size while a fifteen base pair aptamer specific to the epitope that can be conjugated to the NPs and potentially maintain the original size.
(A) The antibody being conjugated to the nanoparticles, potentially increasing size. (B) The aptamer being conjugated to the nanoparticles, potentially maintaining size.
Aptamers have been previously conjugated and used as a targeting agent in drug delivery via nanosystems. Gui et al. used an aptamer for CD133 to conjugate lipid polymer NPs for drug delivery to osteosarcoma initiating cells [Citation187]. Addition of the aptamer increased the size of NPs by only 7 nm. In an in vivo model of BALB/c nude mice bearing an osteosarcoma xenograft of Saos-2 cells, the targeted particles reduced tumor size more avidly compared with nontargeted NPs [Citation187]. Shigdar et al. identified and produced a 15 nucleotide RNA aptamer (5′-CCCUCCUACAUAGGG-3′) conformation that binds to the CD133 epitope for greater penetration and more stable uptake into tumor spheres generated by either colorectal cancer cells or embryonic kidney cells [Citation188]. Therefore, using this aptamer in a drug delivery system can potentially enable a highly efficacious, site-specific therapy, thereby reducing non-specific unintended side effects.
Theranostics
The term ‘theranostic’ was coined in 2002 by Funkhouser and is defined as a material that combines the modalities of therapy and diagnostic imaging [Citation189]. This is an emerging field where both therapeutics and diagnostic imaging agents for pretreatment, planning and real-time monitoring or posttreatment assessment are co-delivered [Citation142]. There are many different compounds that can be used as the diagnostic imaging agent. Imaging agents such as iodine-123, yttrium-86/yttrium-90, zirconium-89, or terbium isotopes can be used for theranostic imaging [Citation161,Citation190]. Examples such as iodine-131 and lutetium-17 can be used as both the therapeutic and the diagnostic agent [Citation190]. Radioactive isotopes can be labelled directly to different therapies. For example, iodine-123 and iodine-131 was labelled to metaiodobenzylguanidine (MIBG) has been used for theranostics in neuroblastoma [Citation191]. Iodine-131 can provide therapy and imaging, while the labelling with iodine-123 enhances the imaging effectiveness [Citation192]. Typically, direct imaging of small-molecule therapies, such as a combination therapy of TMZ and PTX, poses a challenge for in vivo analysis. In general, the diagnostic approaches together with therapeutic agent provide information of trafficking pathways, kinetics of delivery and real-time assessment of therapeutics efficacy [Citation193]. Adding imaging tracers directly to the molecule increases the risk that the imaging agent interferes with an active site and may reduce the therapeutic efficacy [Citation142]. In addition, conjugating an imaging agent to the therapeutic molecule may negatively alter the biodistribution and pharmacodynamic/pharmacokinetic profiles [Citation142]. To reduce the potential for interfering with a drug's active site after conjugating an imaging agent, the use of nanosystems could be employed. Nanosystems can be engineered to tailor to the overall goal of the theranostic. By labelling a drug carrier, the overall fate of the therapy could still be studied while potentially avoiding a drug's effectiveness.
Conclusion
The poor prognosis and dismal few months of life for a GBM patient necessitates that we think in new and innovative ways with a sense of urgency. GSCs represent a key means of recurrence and resistance in need of new thinking about how we approach GBM treatment. TMZ stands as a lone beacon of light that quickly fades in the presence of GBM's insatiable capacity to overcome any single drug approach to date. Multidrug combination therapy is becoming a standard approach to many cancers and is most likely going to be necessary to overcome GBM's many survival tactics. In this review, we discussed one of many possible rationales for thinking about multidrug therapy and an integrated role for a targeted nanotechnological approach against GSCs.
Future perspective
With the advent of checkpoint inhibitors, the tide against many cancers has turned. It is now only a matter of time before new approaches are found for the cancers that remain resistant to therapeutic intervention, including GBM. Cancer resistance to promising therapies is a major issue of our time and new ways around this resistance need a laser-like focus. Development is needed on all fronts using all available resources. No stone must remain unturned in the quest for the answer. Smaller TME niches where certain key CSCs reside is a topic worthy of further investigation and understanding, and approaches that shut down the continuous self-renewal capacity of the most resistant malignancies represent a promising area of research. Nanotechnology is still in an infantile period of discovery and exploration. Nanoparticle technology is a key element of the success of the Covid-19 vaccines, which has revolutionized our fight against the SARS-CoV-2. As of 3 April 2022, 331.65 million doses of Pfizer/BioNTech and 210.69 million doses of Moderna Covid-19 vaccines have been administered in the USA, that are based on nanoparticle technology based lipid vesicles of mRNA [Citation194]. A nano-sized device that can offer countless possibilities should and must be explored toward a new frontier in discovery and innovation against GBM.
Glioblastoma (GBM) patients experience a poor prognosis from the tumor's resistance to therapy; therefore, the need for new treatment options is imperative.
Glioblastoma overview
GBM is an aggressive tumor with a very low survival rate that often taxes patients financially.
The tumor is diagnosed and assessed by imaging techniques such as magnetic resonance imaging (MRI), to determine the size and extent of edema.
GBM is characterized pathologically by the highest loss of glial fibrillary acidic protein (GFAP) and cell irregularity. Key biomarkers include isocitrate dehydrogenase (IDH1 or 2) mutations, methylated O6-methylguanine methyltransferase (MGMT) and 1p/19q codeletion.
Current treatment is limited to surgical resection, radiation, and chemotherapy; however, the tumor often inevitably recurs with undefined second-line therapies.
Glioblastoma cancer stem cells
GBM cancer stem cells (CSCs) are thought to be the driving force in a theoretical hierarchy for treatment resistance.
No single molecule defines CSCs as the specific tumor and surrounding tumor microenvironment are thought to govern the expression of specific markers. The identification of CSC biomarkers allows for tumor targeting.
CD133, thought to be involved in membrane formation, is a glycoprotein on CSCs. As few as 100 CD133+ CSCs have been found to induce tumor formation.
Nestin is another biomarker of CSCs that has been found on CSCs, but not non-CSC cells to allow for specificity.
High expression of CD44 is indicative of tumors with greater self-renewal capacity and increased production of secondary tumors.
Glioblastoma drug treatment
TMZ is the standard chemotherapy against GBM; however, side effects from nonspecific DNA methylation and poor serum stability warrant new treatment options.
The blood–brain barrier is an obstacle for TMZ to treat the tumor, but CSCs are thought to be the main cause of GBM recurrence due to their ability to repair the DNA damage caused by TMZ.
Idasanutlin (RG7388) is an MDM2 inhibitor and a treatment option for GBM tumors with normal p53 as it passes through the blood–brain barrier. Nanomolar concentrations have been found to provide therapy in tumor cell lines and the drug is currently in clinical trials.
Paclitaxel (PTX) is an anti-microtubule agent and has been used to treat several cancers. PTX is very potent against GBM but has limited solubility. Use of PTX in GBM tumor treatment will benefit from the incorporation of nanosystems in their delivery.
Previous studies have shown that TMZ in combination with RG7388 or PTX work synergistically against GBM cell lines; their co-treatment may overcome TMZ resistance and eliminate residual CSCs.
Nanomedicines for stem cell resistant GBM treatment
Chemotherapies for the treatment of GBM tumors may benefit from the use of nanosystems as delivery vehicles. Nanosystems help transport drugs across the blood–brain barrier that may have otherwise been limited by size or polarity.
Nanoparticles (NPs) are a type of nanosystem characterized by small size and enhanced permeability and retention (EPR) effect.
NPs may increase the bioavailability of poorly soluble drugs, have increased surface area, and may be composed of a variety of biodegradable and non-biodegradable materials.
The differing types of NPs (polymeric NPs, micellar NPs, polymer-micellar, solid lipid NPs, liposomes) and various fabrications methods (nanoprecipitation, emulsion, microfluidics) allow for customization to the drug and desired characteristics (drug release profile, size and biocompatibility).
Nanoparticle targeting of GBM CSCs
By using NPs as drug delivery systems, GBM CSCs can be targeted directly. NPs may be functionalized with several molecules for different purposes by methods such as covalent bonding or amide formation.
Aptamers are advantageous targeting molecules because of their ability to conform to any shape, small size compared with antibodies and stable conformation.
NP conjugation also opens the door of theranostics. By conjugating imaging-based contrast agents such as zirconium-89 (89Zr), treatment systems may be developed to not only eliminate the tumor, but for diagnostic imaging.
Conclusion
Current standard of care treatment is no longer a viable option for GBM patients due to tumor recurrence driven by GBM CSCs. There are several options available through the concept of combination therapy integrated with a targeted nanotechnological approach.
Future perspective & opinion
Cancer resistance is a major problem for our time and targeting the smallest niche of CSCs and tumor origin may be the key with further investigation and understanding. Nanotechnology has proven vital in the success of the Covid-19 vaccines and may provide an answer to limited advancement in GBM tumors.
Acknowledgments
Indiana University School of Medicine (Indiana University School of Medicine Biomedical Research Grant). The content is solely the responsibility of the authors and does not necessarily represent the official views of their employers.
A part of the manuscript was submitted towards partial fulfillment of the Master's thesis for Shelby Smiley.
Financial & competing interests disclosure
S Das is a member of the Therapeutic Delivery Editorial Board. They were not involved in any editorial decisions related to the publication of this article, and all author details were blinded to the article's peer reviewers as per the journal's double-blind peer review policy. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Singh SK , HawkinsC , ClarkeIDet al. Identification of human brain tumour initiating cells. Nature432(7015), 396–401 (2004).
- Vasilev A , SofiR , TongL , TeschemacherAG , KasparovS. In search of a breakthrough therapy for glioblastoma. Neuroglia1(2), 292–310 (2018).
- Miller KD , OstromQT , KruchkoCet al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin.71(5), 381–406 (2021).
- Alphandery E . Glioblastoma treatments: an account of recent industrial developments. Front. Pharmacol.9, 879 (2018).
- Urbańska K , SokołowskaJ , SzmidtM , SysaP. Glioblastoma multiforme - an overview. Contemp. Oncol. (Poznan, Poland)18(5), 307–312 (2014).
- Davis ME . Glioblastoma: overview of disease and treatment. Clin. J. Oncol. Nurs.20(Suppl. 5), S2–S8 (2016).
- The Huntington's Disease Collaborative Research Group . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell72(6), 971–983 (1993).
- Gimple RC , BhargavaS , DixitD , RichJN. Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev.33(11–12), 591–609 (2019).
- Wang Q , HuB , HuXet al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell32(1), 42–56.e46 (2017).
- Walid MS . Prognostic factors for long-term survival after glioblastoma. Permanente J.12(4), 45–48 (2008).
- Pourgholi F , HajivaliliM , FarhadJN , KafilHS , YousefiM. Nanoparticles: novel vehicles in treatment of Glioblastoma. Biomed. Pharmacother.77, 98–107 (2016).
- Van Den Bent MJ , WellerM , WenPY , KrosJM , AldapeK , ChangS. A clinical perspective on the 2016 WHO brain tumor classification and routine molecular diagnostics. Neuro-oncology19(5), 614–624 (2017).
- Songtao Q , LeiY , SiGet al. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci.103(2), 269–273 (2012).
- Chen X , ZhangM , GanHet al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun.9(1), 1–14 (2018).
- Hsu JB , ChangTH , LeeGA , LeeTY , ChenCY. Identification of potential biomarkers related to glioma survival by gene expression profile analysis. BMC Med. Genom.11(Suppl. 7), 34 (2019).
- Stupp R , MasonWP , VanDen Bent MJet al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N England J. Med.352(10), 987–996 (2005).
- Stupp R , TaillibertS , KannerAet al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA318(23), 2306–2316 (2017).
- Gramatzki D , RothP , RushingEJet al. Bevacizumab may improve quality of life, but not overall survival in glioblastoma: an epidemiological study. Ann. Oncol.29(6), 1431–1436 (2018).
- Roth P , WinklhoferS , MüllerAMet al. Neurological complications of cancer immunotherapy. Cancer Treat. Rev.97, 102189 (2021).
- Chinot OL , WickW , MasonWet al. Bevacizumab plus radiotherapy–temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med.370(8), 709–722 (2014).
- Lapidot T , SirardC , VormoorJet al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature367(6464), 645–648 (1994).
- Lathia JD , MackSC , Mulkearns-HubertEE , ValentimCL , RichJN. Cancer stem cells in glioblastoma. Genes Development29(12), 1203–1217 (2015).
- Yang L , ShiP , ZhaoGet al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target Ther.5(1), 8 (2020).
- Cheng L , WuQ , GuryanovaOAet al. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun.406(4), 643–648 (2011).
- Auffinger B , SpencerD , PytelP , AhmedAU , LesniakMS. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert. Rev. Neurother.15(7), 741–752 (2015).
- Prager BC , BhargavaS , MahadevV , HubertCG , RichJN. Glioblastoma stem cells: driving resilience through chaos. Trends Cancer6(3), 223–235 (2020).
- Carnero A , LleonartM. The hypoxic microenvironment: a determinant of cancer stem cell evolution. BioEssays38(Suppl. 1), S65–S74 (2016).
- Lee JH , LeeJE , KahngJYet al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature560(7717), 243–247 (2018).
- Group BDW , AtkinsonAJJr , ColburnWAet al. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin. Pharmacol. Therapeut.69(3), 89–95 (2001).
- Mason S , McdonaldK. MGMT testing for glioma in clinical laboratories: discordance with methylation analyses prevents the implementation of routine immunohistochemistry. J. Cancer Res. Clin. Oncol.138(11), 1789–1797 (2012).
- Batlle E , CleversH. Cancer stem cells revisited. Nat. Med.23(10), 1124–1134 (2017).
- Alison MR , LimSM , NicholsonLJ. Cancer stem cells: problems for therapy?J. Pathol.223(2), 147–161 (2011).
- Karsten U , GoletzS. What makes cancer stem cell markers different?Springerplus2(1), 301 (2013).
- Si D , YinF , PengJ , ZhangG. High expression of CD44 predicts a poor prognosis in glioblastomas. Cancer Manag. Res.12, 769–775 (2020).
- Mao XG , ZhangX , XueXYet al. Brain tumor stem-like cells identified by neural stem cell marker CD15. Transl. Oncol.2(4), 247–257 (2009).
- Lee HJ , ChoeG , JheonS , SungSW , LeeCT , ChungJH. CD24, a novel cancer biomarker, predicting disease-free survival of non-small cell lung carcinomas: a retrospective study of prognostic factor analysis from the viewpoint of forthcoming (seventh) new TNM classification. J. Thorac. Oncol.5(5), 649–657 (2010).
- Shaikh MV , KalaM , NivsarkarM. CD90 a potential cancer stem cell marker and a therapeutic target. Cancer Biomark.16(3), 301–307 (2016).
- Lathia JD , GallagherJ , HeddlestonJMet al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell6(5), 421–432 (2010).
- Zheng X , XieQ , LiS , ZhangW. CXCR4-positive subset of glioma is enriched for cancer stem cells. Oncol. Res.19(12), 555–561 (2011).
- Raveh S , GavertN , Ben-Ze'evA. L1 cell adhesion molecule (L1CAM) in invasive tumors. Cancer Lett.282(2), 137–145 (2009).
- Li Y , WangZ , AjaniJA , SongS. Drug resistance and cancer stem cells. Cell Commun. Signal.19(1), 19 (2021).
- Neradil J , VeselskaR. Nestin as a marker of cancer stem cells. Cancer Sci.106(7), 803–811 (2015).
- Glazer RI , WangXY , YuanH , YinY. Musashi1: a stem cell marker no longer in search of a function. Cell Cycle7(17), 2635–2639 (2008).
- Rasper M , SchaferA , PiontekGet al. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro. Oncol.12(10), 1024–1033 (2010).
- Gupta MK , PolisettyRV , SharmaRet al. Altered transcriptional regulatory proteins in glioblastoma and YBX1 as a potential regulator of tumor invasion. Sci. Rep.9(1), 10986 (2019).
- Godlewski J , NewtonH , ChioccaE , LawlerS. MicroRNAs and glioblastoma; the stem cell connection. Cell Death Different.17(2), 221–228 (2010).
- Pottoo FH , JavedMN , RahmanJU , Abu-IzneidT , KhanFA. Targeted delivery of miRNA based therapeuticals in the clinical management of Glioblastoma Multiforme. Presented at: Seminars in Cancer Biol.69, 391–398 (2021).
- Yeh M , WangYY , YooJYet al. MicroRNA-138 suppresses glioblastoma proliferation through downregulation of CD44. Scientific Reports11(1), 1–11 (2021).
- Galli R , BindaE , OrfanelliUet al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res.64(19), 7011–7021 (2004).
- Joo KM , KimSY , JinXet al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab. Invest.88(8), 808–815 (2008).
- Glumac PM , LebeauAM. The role of CD133 in cancer: a concise review. Clin. Transl. Med.7(1), 18 (2018).
- Ahmed SI , JavedG , LaghariAAet al. CD133 expression in glioblastoma multiforme: a literature review. Cureus10(10), e3439 (2018).
- Galli R , BindaE , OrfanelliUet al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res.64(19), 7011–7021 (2004).
- Behrooz AB , SyahirA. Could we address the interplay between CD133, Wnt/beta-catenin, and TERT signaling pathways as a potential target for glioblastoma therapy?Front. Oncol.11, 642719 (2021).
- Xi G , LiYD , GrahovacGet al. Targeting CD133 improves chemotherapeutic efficacy of recurrent pediatric pilocytic astrocytoma following prolonged chemotherapy. Mol. Cancer16(1), 21 (2017).
- Wei Y , JiangY , ZouFet al. Activation of PI3K/Akt pathway by CD133-p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc. Natl Acad. Sci. USA110(17), 6829–6834 (2013).
- Liu G , YuanX , ZengZet al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer5, 67 (2006).
- Beier D , WischhusenJ , DietmaierWet al. CD133 expression and cancer stem cells predict prognosis in high-grade oligodendroglial tumors. Brain Pathol.18(3), 370–377 (2008).
- Michalczyk K , ZimanM. Nestin structure and predicted function in cellular cytoskeletal organisation. Histol. Histopathol.20(2), 665–671 (2005).
- Matsuda Y , HagioM , IshiwataT. Nestin: a novel angiogenesis marker and possible target for tumor angiogenesis. World J. Gastroenterol.19(1), 42–48 (2013).
- Ehrmann J , KolarZ , MokryJ. Nestin as a diagnostic and prognostic marker: immunohistochemical analysis of its expression in different tumours. J. Clin. Pathol.58(2), 222–223 (2005).
- Tang X , ZuoC , FangPet al. Targeting glioblastoma stem cells: a review on biomarkers, signal pathways and targeted therapy. Front. Oncol.11, 701291 (2021).
- Zhang M , SongT , YangLet al. Nestin and CD133: valuable stem cell-specific markers for determining clinical outcome of glioma patients. J. Exp. Clin. Cancer Res.27(1), 85 (2008).
- Jin X , JinX , JungJE , BeckS , KimH. Cell surface Nestin is a biomarker for glioma stem cells. Biochem. Biophys. Res. Commun.433(4), 496–501 (2013).
- Brown DV , MantamadiotisT. Insights into the next generation of cancer stem cell research. Front. Biosci. (Landmark Ed)19, 1015–1027 (2014).
- Anido J , Sáez-BorderíasA , Gonzàlez-JuncàAet al. TGF-β receptor inhibitors target the CD44high/Id1high glioma-initiating cell population in human glioblastoma. Cancer Cell18(6), 655–668 (2010).
- Vaillant B , BhatK , SulmanEet al. CD44 as a prognostic and predictive marker for GBM. J. Clin. Oncol.29, 2049–2049 (2011).
- Mooney KL , ChoyW , SidhuSet al. The role of CD44 in glioblastoma multiforme. J. Clin. Neurosci.34, 1–5 (2016).
- Penar PL , KhoshyomnS , BhushanA , TrittonTR. Inhibition of epidermal growth factor receptor-associated tyrosine kinase blocks glioblastoma invasion of the brain: experimental studies. Neurosurgery40(1), 141–151 (1997).
- Sato H , KuwashimaN , SakaidaTet al. Epidermal growth factor receptor-transfected bone marrow stromal cells exhibit enhanced migratory response and therapeutic potential against murine brain tumors. Cancer Gene Ther.12(9), 757–768 (2005).
- Gomez KE , WuF , KeysarSBet al. Cancer cell CD44 mediates macrophage/monocyte-driven regulation of head and neck cancer stem cells. Cancer Res.80(19), 4185–4198 (2020).
- Wang HH , LiaoCC , ChowNHet al. Whether CD44 is an applicable marker for glioma stem cells. Am. J. Translational Res.9(11), 4785 (2017).
- Wei KC , HuangCY , ChenPYet al. Evaluation of the prognostic value of CD44 in glioblastoma multiforme. Anticancer Res.30(1), 253–259 (2010).
- Nishikawa M , InoueA , OhnishiTet al. Hypoxia-induced phenotypic transition from highly invasive to less invasive tumors in glioma stem-like cells: significance of CD44 and osteopontin as therapeutic targets in glioblastoma. Translat. Oncol.14(8), 101137 (2021).
- Qiu GZ , JinMZ , DaiJX , SunW , FengJH , JinWL. Reprogramming of the tumor in the hypoxic niche: the emerging concept and associated therapeutic strategies. Trends Pharmacol. Sci.38(8), 669–686 (2017).
- Shi Y , LimSK , LiangQet al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature567(7748), 341–346 (2019).
- Brown DV , FilizG , DanielPMet al. Expression of CD133 and CD44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PLoS ONE12(2), e0172791–e0172791 (2017).
- Wang R , ChadalavadaK , WilshireJet al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature468(7325), 829–833 (2010).
- Calabrese C , PoppletonH , KocakMet al. A perivascular niche for brain tumor stem cells. Cancer Cell11(1), 69–82 (2007).
- Da Ros M , DeGregorio V , IorioALet al. Glioblastoma chemoresistance: the double play by microenvironment and blood-brain barrier. Int. J. Mol. Sci.19(10), 2879 (2018).
- Di Mauro PP , CascanteA , BrugadaP , Gómez-VallejoV , LlopJ , BorrósS. Peptide-functionalized and high drug loaded novel nanoparticles as dual-targeting drug delivery system for modulated and controlled release of paclitaxel to brain glioma. Int. J. Pharmaceutics553(1), 169–185 (2018).
- Portnow J , BadieB , ChenM , LiuA , BlanchardS , SynoldTW. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: potential implications for the current approach to chemoradiation. Clin. Cancer Res.15(22), 7092–7098 (2009).
- Lah TT , NovakM , BreznikB. Brain malignancies: glioblastoma and brain metastases. Semin. Cancer Biol.60, 262–273 (2020).
- Wu W , KlockowJL , ZhangMet al. Glioblastoma multiforme (GBM): an overview of current therapies and mechanisms of resistance. Pharmacol. Res.171, 105780 (2021).
- Xiong L , WangF , QiXie X. Advanced treatment in high-grade gliomas. J. Buon.24(2), 424–430 (2019).
- Abadi B , Ahmadi-ZeidabadiM , DiniL , VergalloC. Stem cell-based therapy treating glioblastoma multiforme. Hematol Oncol Stem Cell Ther14(1), 1–15 (2021).
- Lee CY , OoiIH. Preparation of temozolomide-loaded nanoparticles for glioblastoma multiforme targeting-ideal versus reality. Pharmaceuticals (Basel, Switzerland)9(3), 54 (2016).
- Fang C , WangK , StephenZRet al. Temozolomide nanoparticles for targeted glioblastoma therapy. ACS Appli. Mater. Interf.7(12), 6674–6682 (2015).
- Emamgholizadeh Minaei S , KhoeiS , KhoeeS , KarimiMR. Tri-block copolymer nanoparticles modified with folic acid for temozolomide delivery in glioblastoma. Int. J. Biochem. Cell Biol.108, 72–83 (2019).
- Ramalho MJ , SevinE , GosseletFet al. Receptor-mediated PLGA nanoparticles for glioblastoma multiforme treatment. Int. J. Pharm.545(1–2), 84–92 (2018).
- Wang JB , DongDF , WangMD , GaoK. IDH1 overexpression induced chemotherapy resistance and IDH1 mutation enhanced chemotherapy sensitivity in Glioma cells in vitro and in vivo. Asian Pacific J. Cancer Prevent.15(1), 427–432 (2014).
- Huang J , YuJ , TuL , HuangN , LiH , LuoY. Isocitrate dehydrogenase mutations in glioma: from basic discovery to therapeutics development. Front. Oncol.9, 506 (2019).
- Glaser T , HanI , WuL , ZengX. Targeted nanotechnology in glioblastoma multiforme. Front. Pharmacol.8, 166 (2017).
- Hegi ME , DiserensAC , GorliaTet al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N England J. Med.352(10), 997–1003 (2005).
- Beier D , RöhrlS , PillaiDRet al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res.68(14), 5706–5715 (2008).
- Wang H , CaiS , BaileyBJet al. Combination therapy in a xenograft model of glioblastoma: enhancement of the antitumor activity of temozolomide by an MDM2 antagonist. J. Neurosurg.126(2), 446–459 (2017).
- Berberich A , KesslerT , ThomeCMet al. Targeting Resistance against the MDM2 Inhibitor RG7388 in Glioblastoma Cells by the MEK Inhibitor Trametinib. Clin. Cancer Res.25(1), 253–265 (2019).
- Wang S , ZhaoY , AguilarA , BernardD , YangCY. Targeting the MDM2-p53 protein-protein interaction for new cancer therapy: progress and challenges. Cold Spring Harb. Perspect. Med.7(5), a026245 (2017).
- Williams AB , SchumacherB. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med.6(5), a026070 (2016).
- Jerry DJ , TaoL , YanH. Regulation of cancer stem cells by p53. Breast Cancer Res.10(4), 304 (2008).
- Chien CH , HsuehWT , ChuangJY , ChangKY. Dissecting the mechanism of temozolomide resistance and its association with the regulatory roles of intracellular reactive oxygen species in glioblastoma. Journal of Biomedical Science28(1), 18 (2021).
- Zhao Y , AguilarA , BernardD , WangS. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem.58(3), 1038–1052 (2015).
- Zanjirband M , EdmondsonRJ , LunecJ. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget7(26), 40115–40134 (2016).
- Kussie PH , GorinaS , MarechalVet al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science274(5289), 948–953 (1996).
- Sanchez-Vega F , MinaM , ArmeniaJet al. Oncogenic signaling pathways in the Cancer Genome Atlas. Cell173(2), 321–337.e310 (2018).
- Skalniak L , KocikJ , PolakJet al. Prolonged Idasanutlin (RG7388) treatment leads to the generation of p53-mutated cells. Cancers (Basel)10(11), 396 (2018).
- Chen L , RousseauRF , MiddletonSAet al. Pre-clinical evaluation of the MDM2-p53 antagonist RG7388 alone and in combination with chemotherapy in neuroblastoma. Oncotarget6(12), 10207–10221 (2015).
- Khurana A , ShaferDA. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: perspectives on the therapeutic potential of idasanutlin (RG7388). Onco Targets Ther12, 2903–2910 (2019).
- Dadi N , StanleyM , ShahdaS , O'neilBH , SehdevA. Impact of Nab-Paclitaxel-based Second-line Chemotherapy in Metastatic Pancreatic Cancer. Anticancer Res.37(10), 5533–5539 (2017).
- Branham MT , NadinSB , Vargas-RoigLM , CioccaDR. DNA damage induced by paclitaxel and DNA repair capability of peripheral blood lymphocytes as evaluated by the alkaline comet assay. Mutation Res.560(1), 11–17 (2004).
- Horwitz SB . Taxol (paclitaxel): mechanisms of action. Ann. Oncol.5(Suppl. 6), S3–S6 (1994).
- Surapaneni MS , DasSK , DasNG. Designing Paclitaxel drug delivery systems aimed at improved patient outcomes: current status and challenges. ISRN Pharmacol.2012, 623139 (2012).
- Chi EY , ViriyapakB , KwackHSet al. Regulation of paclitaxel-induced programmed cell death by autophagic induction: a model for cervical cancer. Obstetr. Gynecol. Sci.56(2), 84–92 (2013).
- Dong R , XinL , AixiaL , BaohuiL , FengX. Paclitaxel inhibits growth and proliferation of glioblastoma through MMP-9-meidated p38/JNK signaling pathway. Biomed. Res. (0970-938X)28(17), 7348–7353 (2017).
- Yusuf RZ , DuanZ , LamendolaDE , PensonRT , SeidenMV. Paclitaxel resistance: molecular mechanisms and pharmacologic manipulation. Curr. Cancer Drug Targets3(1), 1–19 (2003).
- Rice A , MichaelisML , GeorgG , LiuY , TurunenB , AudusKL. Overcoming the blood-brain barrier to taxane delivery for neurodegenerative diseases and brain tumors. J, Mol. Neurosci.20(3), 339–343 (2003).
- Yusuf RZ , DuanZ , LamendolaDE , PensonRT , SeidenMV. Paclitaxel resistance: molecular mechanisms and pharmacologic manipulation. Curr. Cancer Drug Targets3(1), 1–19 (2003).
- Scripture CD , FiggWD , SparreboomA. Paclitaxel chemotherapy: from empiricism to a mechanism-based formulation strategy. Ther. Clin. Risk Manag.1(2), 107–114 (2005).
- Adams JD , FloraKP , GoldspielBR , WilsonJW , ArbuckSG , FinleyR. Taxol: a history of pharmaceutical development and current pharmaceutical concerns. J. Natl Cancer Inst. Monogr. (15), 141–147 (1993).
- Campos FC , VictorinoVJ , Martins-PingeMC , CecchiniAL , PanisC , CecchiniR. Systemic toxicity induced by paclitaxel in vivo is associated with the solvent cremophor EL through oxidative stress-driven mechanisms. Food Chem. Toxicol.68, 78–86 (2014).
- Lidar Z , MardorY , JonasTet al. Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: a phase I/II clinical study. J. Neurosurg.100(3), 472–479 (2004).
- Chen L , RousseauRF , MiddletonSAet al. Pre-clinical evaluation of the MDM2-p53 antagonist RG7388 alone and in combination with chemotherapy in neuroblastoma. Oncotarget6(12), 10207 (2015).
- Xu Y , ShenM , LiYet al. The synergic antitumor effects of paclitaxel and temozolomide co-loaded in mPEG-PLGA nanoparticles on glioblastoma cells. Oncotarget7(15), 20890–20901 (2016).
- Sørensen MD , FosmarkS , HellwegeS , BeierD , KristensenBW , BeierCP. Chemoresistance and chemotherapy targeting stem-like cells in malignant glioma. Stem Cell Biology in Neoplasms of the Central Nervous System853, 111–138 (2015).
- Saraswathy M , GongS. Different strategies to overcome multidrug resistance in cancer. Biotechnol. Adv.31(8), 1397–1407 (2013).
- Daneman R , PratA. The blood–brain barrier. Cold Spring Harbor Perspect. Biol.7(1), a020412 (2015).
- Bhowmik A , KhanR , GhoshMK. Blood brain barrier: a challenge for effectual therapy of brain tumors. BioMed Res. Int.2015, 320941 (2015).
- Pardridge WM . Drug transport across the blood–brain barrier. J Cerebral Blood Flow Metabolism32(11), 1959–1972 (2012).
- Barua S , MitragotriS. Challenges associated with penetration of nanoparticles across cell and tissue barriers: a review of current status and future prospects. Nano Today9(2), 223–243 (2014).
- Haseloff RF , DithmerS , WinklerL , WolburgH , BlasigIE. Transmembrane proteins of the tight junctions at the blood–brain barrier: structural and functional aspects. Presented at: Seminars in Cell & Developmental Biology.38, 16–25 (2015).
- Mosquera J , GarcíaI , Liz-MarzánLM. Cellular uptake of nanoparticles versus small molecules: a matter of size. Acc. Chem. Res.51(9), 2305–2313 (2018).
- Abbott NJ , PatabendigeAA , DolmanDE , YusofSR , BegleyDJ. Structure and function of the blood–brain barrier. Neurobiol. Dis.37(1), 13–25 (2010).
- Sindhwani S , SyedAM , NgaiJet al. The entry of nanoparticles into solid tumours. Nat. Mater.19(5), 566–575 (2020).
- Westerhout J , PloegerB , SmeetsJ , DanhofM , DeLange EC. Physiologically based pharmacokinetic modeling to investigate regional brain distribution kinetics in rats. AAPS J.14(3), 543–553 (2012).
- De Lange EC . The mastermind approach to CNS drug therapy: translational prediction of human brain distribution, target site kinetics, and therapeutic effects. Fluids Barriers CNS10(1), 1–16 (2013).
- Bonferoni MC , RossiS , SandriGet al. Nanoemulsions for “Nose-to-Brain” Drug Delivery. Pharmaceutics11(2), 84 (2019).
- Kumar A , PandeyAN , JainSK. Nasal-nanotechnology: revolution for efficient therapeutics delivery. Drug Delivery23(3), 681–693 (2016).
- Mikitsh JL , ChackoAM. Pathways for small molecule delivery to the central nervous system across the blood-brain barrier. Perspecti. Med. Chem.6, 11–24 (2014).
- Yang J , ShiZ , LiuR , WuY , ZhangX. Combined-therapeutic strategies synergistically potentiate glioblastoma multiforme treatment via nanotechnology. Theranostics10(7), 3223 (2020).
- Blanco E , ShenH , FerrariM. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nature Biotechnol.33(9), 941–951 (2015).
- Das SK , DasNG. Surface modification strategies in enhancing systemic delivery performance. In: Systemic Delivery Technologies in Anti-Aging Medicine: Methods and Applications.Springer, 365–392 (2020).
- Veronesi MC , AlhamamiM , MiedemaSB , YunY , Ruiz-CardozoM , VannierMW. Imaging of intranasal drug delivery to the brain. Am. J. Nucl. Med. Mol. Imaging10(1), 1–31 (2020).
- Chavda VP . Chapter 4 - Nanobased Nano Drug Delivery: A Comprehensive Review. In: Applications of Targeted Nano Drugs and Delivery Systems.MohapatraSS, RanjanS, DasguptaN, MishraRK, ThomasS ( Eds). Elsevier, 69–92 (2019).
- Gabizon A , BradburyM , PrabhakarU , ZamboniW , LibuttiS , GrodzinskiP. Cancer nanomedicines: closing the translational gap. Lancet (London, England)384(9961), 2175 (2014).
- Jabir NR , AnwarK , FirozCK , OvesM , KamalMA , TabrezS. An overview on the current status of cancer nanomedicines. Curr. Med. Res. Opin.34(5), 911–921 (2018).
- Tong R , LangerR. Nanomedicines targeting the tumor microenvironment. Cancer J.21(4), 314–321 (2015).
- Kammari R , DasNG , DasSK. Nanoparticulate systems for therapeutic and diagnostic applications. Emerging Nanotechnologies for Diagnostics, Drug Delivery and Medical Devices.105–144 (2017).
- Prabhakar U , MaedaH , JainRKet al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res.73(8), 2412–2417 (2013).
- Zhang L , ChenQ , MaY , SunJ. Microfluidic Methods for Fabrication and Engineering of Nanoparticle Drug Delivery Systems. ACS Appl. Bio Materials3(1), 107–120 (2020).
- Bhatia S . Nanoparticles types, classification, characterization, fabrication methods and drug delivery applications. In: Natural Polymer Drug Delivery Systems.33–93 (2016).
- Mohanraj VJ , ChenY. Nanoparticles - A Review. Tropical J. Pharmaceutical Res.5(1), 561–573 (2007).
- De Oliveira Junior ER , NascimentoTL , SalomaoMA , DaSilva ACG , ValadaresMC , LimaEM. Increased nose-to-brain delivery of melatonin mediated by polycaprolactone nanoparticles for the treatment of glioblastoma. Pharm. Res.36(9), 131 (2019).
- D'mello SR , DasSK , DasNG. Polymeric nanoparticles for small-molecule drugs: biodegradation of polymers and fabrication of nanoparticles. In: Drug Delivery Nanoparticles Formulation and Characterization.CRC Press, 36–54 (2016).
- Zhu J , HaywardRC. Spontaneous generation of amphiphilic block copolymer micelles with multiple morphologies through interfacial instabilities. J. Am. Chem. Soc.130(23), 7496–7502 (2008).
- Taghipour B , YakhchaliM , HaririanI , TamaddonAM , SamaniSM. The effects of technical and compositional variables on the size and release profile of bovine serum albumin from PLGA based particulate systems. Res Pharm Sci9(6), 407–420 (2014).
- Wang Y , LiP , Truong-DinhTran T , ZhangJ , KongL. Manufacturing techniques and surface engineering of polymer based nanoparticles for targeted drug delivery to cancer. Nanomaterials (Basel, Switzerland)6(2), 26 (2016).
- López-Royo T , SebastiánV , Moreno-MartínezLet al. Encapsulation of large-size plasmids in PLGA nanoparticles for gene editing: comparison of three different synthesis methods. Nanomaterials (Basel, Switzerland)11(10), 2723 (2021).
- Martínez-Muñoz O , Ospina-GiraldoL , Mora-HuertasCE. Nanoprecipitation: applications for entrapping active molecules of interest in pharmaceutics. Nano-and Microencapsulation-Techniques and Applications (2020).
- Bilati U , AllémannE , DoelkerE. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. European J. Pharmaceutical Sciences24(1), 67–75 (2005).
- Barichello JM , MorishitaM , TakayamaK , NagaiT. Encapsulation of hydrophilic and lipophilic drugs in PLGA nanoparticles by the nanoprecipitation method. Drug Dev Ind Pharm25(4), 471–476 (1999).
- Smiley SB , YunY , AyyagariPet al. Development of CD133 targeting multi-drug polymer micellar nanoparticles for glioblastoma - in vitro evaluation in glioblastoma stem cells. Pharm. Res.38(6), 1067–1079 (2021).
- Niculescu AG , ChircovC , BîrcăAC , GrumezescuAM. Nanomaterials synthesis through microfluidic methods: an updated overview. Nanomaterials11(4), 864 (2021).
- Khan AR , LiuM , KhanMW , ZhaiG. Progress in brain targeting drug delivery system by nasal route. J. Control. Release268, 364–389 (2017).
- Bobo D , RobinsonKJ , IslamJ , ThurechtKJ , CorrieSR. Nanoparticle-based medicines: a review of fda-approved materials and clinical trials to date. Pharm. Res.33(10), 2373–2387 (2016).
- Chu L , WangA , NiLet al. Nose-to-brain delivery of temozolomide-loaded PLGA nanoparticles functionalized with anti-EPHA3 for glioblastoma targeting. Drug Delivery25(1), 1634–1641 (2018).
- Shilo M , SharonA , BaranesK , MotieiM , LelloucheJPM , PopovtzerR. The effect of nanoparticle size on the probability to cross the blood-brain barrier: an in-vitro endothelial cell model. J. Nanobiotechnol.13(1), 1–7 (2015).
- Zhi K , RajiB , NookalaARet al. PLGA nanoparticle-based formulations to cross the blood–brain barrier for drug delivery: from R&D to cGMP. Pharmaceutics13(4), 500 (2021).
- Husseini GA , PittWG. Micelles and nanoparticles for ultrasonic drug and gene delivery. Adv Drug Deliv Rev60(10), 1137–1152 (2008).
- Majumder N , GDas N , DasSK. Polymeric micelles for anticancer drug delivery. Therap. Del.11(10), 613–635 (2020).
- Nabar GM , MahajanKD , CalhounMAet al. Micelle-templated, poly(lactic-co-glycolic acid) nanoparticles for hydrophobic drug delivery. Int. J. Nanomed.13, 351–366 (2018).
- Gao X , YuT , XuGet al. Enhancing the anti-glioma therapy of doxorubicin by honokiol with biodegradable self-assembling micelles through multiple evaluations. Sci. Rep.7, 43501 (2017).
- Zhan C , GuB , XieC , LiJ , LiuY , LuW. Cyclic RGD conjugated poly(ethylene glycol)-co-poly(lactic acid) micelle enhances paclitaxel anti-glioblastoma effect. J. Control. Release143(1), 136–142 (2010).
- Soo Choi H , LiuW , MisraPet al. Renal clearance of quantum dots. Nature Biotechnol.25(10), 1165–1170 (2007).
- Tapeinos C , BattagliniM , CiofaniG. Advances in the design of solid lipid nanoparticles and nanostructured lipid carriers for targeting brain diseases. J. Control. Release264, 306–332 (2017).
- Thi TTH , SuysEJA , LeeJS , NguyenDH , ParkKD , TruongNP. Lipid-based nanoparticles in the clinic and clinical trials: from cancer nanomedicine to COVID-19 vaccines. Vaccines (Basel)9(4), 359 (2021).
- Grillone A , BattagliniM , MoscatoSet al. Nutlin-loaded magnetic solid lipid nanoparticles for targeted glioblastoma treatment. Nanomedicine (Lond)14(6), 727–752 (2019).
- Scioli Montoto S , MuracaG , RuizME. Solid lipid nanoparticles for drug delivery: pharmacological and biopharmaceutical aspects. Front. Mol. Biosci.7, 587997 (2020).
- Jnaidi R , AlmeidaAJ , GonçalvesLM. Solid lipid nanoparticles and nanostructured lipid carriers as smart drug delivery systems in the treatment of glioblastoma multiforme. Pharmaceutics12(9), 860 (2020).
- Akbarzadeh A , Rezaei-SadabadyR , DavaranSet al. Liposome: classification, preparation, and applications. Nanoscale Res. Lett.8(1), 102 (2013).
- Lakkadwala S , DosSantos Rodrigues B , SunC , SinghJ. Dual functionalized liposomes for efficient co-delivery of anti-cancer chemotherapeutics for the treatment of glioblastoma. J. Control. Release307, 247–260 (2019).
- Shen Y , PiZ , YanFet al. Enhanced delivery of paclitaxel liposomes using focused ultrasound with microbubbles for treating nude mice bearing intracranial glioblastoma xenografts. Int. J. Nanomed.12, 5613–5629 (2017).
- Werengowska-Ciećwierz K , WiśniewskiM , TerzykA , FurmaniakS. The chemistry of bioconjugation in nanoparticles-based drug delivery system. Advances Condensed Matter Physics2015, 1–27 (2015).
- Hermanson GT . Bioconjugate techniques.Academic press (2013).
- van de Watering FCJ , RijpkemaM , PerkL , BrinkmannU , OyenW , BoermanO. Zirconium-89 labeled antibodies: a new tool for molecular imaging in cancer patients. BioMed. Res. Int.2014, 203601 (2014).
- Veronesi M , ZamoraM , BhuiyanM , Obrien-PenneyB , ChenC , VannierM. Use of a clinical pet/ct scanner for whole body biodistribution of intranasal nanoparticles. arXiv preprint arXiv:1704.00691 (2017).
- Bien-Moller S , BalzE , HerzogSet al. Association of glioblastoma multiforme stem cell characteristics, differentiation, and microglia marker genes with patient survival. Stem Cells Int2018, 9628289 (2018).
- Gui K , ZhangX , ChenFet al. Lipid-polymer nanoparticles with CD133 aptamers for targeted delivery of all-trans retinoic acid to osteosarcoma initiating cells. Biomed. Pharmacother.111, 751–764 (2019).
- Shigdar S , QiaoL , ZhouSFet al. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett.330(1), 84–95 (2013).
- Funkhouser J . Reinventing pharma: the theranostic revolution. Curr. Drug Discov.2, 17–19 (2002).
- Yordanova A , EppardE , KurpigSet al. Theranostics in nuclear medicine practice. Onco Targets Ther.10, 4821–4828 (2017).
- Agrawal A , RangarajanV , ShahS , PuranikA , PurandareN. MIBG (metaiodobenzylguanidine) theranostics in pediatric and adult malignancies. Br. J. Radiol.91(1091), 20180103 (2018).
- Filippi L , ChiaravallotiA , SchillaciO , CianniR , BagniO. Theranostic approaches in nuclear medicine: current status and future prospects. Expert Rev. Med. Devices17(4), 331–343 (2020).
- Kelkar SS , ReinekeTM. Theranostics: combining imaging and therapy. Bioconjug. Chem.22(10), 1879–1903 (2011).
- Our World in Data . COVID-19 vaccine doses administered by manufacturer, USA (2022). https://ourworldindata.org/grapher/covid-vaccine-doses-by-manufacturer?country=∼USA