
Abstract
Deregulated metabolism is gaining recognition as a hallmark of cancer cells, and is being explored for therapeutic potential. The Warburg effect is a metabolic phenotype that occurs in 90% of tumors, where glycolysis is favored despite the presence of oxygen. Dichloroacetate (DCA) is a pyruvate dehydrogenase kinase (PDK) inhibitor that can reverse the Warburg effect. PENAO (4-(N-(S-penicillaminylacetyl)amino) phenylarsonous acid) is a novel anti-mitochondrial agent that targets the adenine nucleotide transporter in mitochondria and is currently in clinical trials for solid tumors. We have investigated the targeting of two aspects of metabolism, using DCA to promote mitochondrial activity combined with PENAO to inhibit mitochondrial activity, in breast and other carcinoma cell lines. PENAO was effective at low uM concentrations in luminal (T-47D) and triple negative (MDA-MB-231) breast cancer cells, in normoxia and hypoxia. The cytotoxicity of PENAO was enhanced by DCA by a mechanism involving increased reactive oxygen species in both T-47D and MDA-MB-231 cells, however further investigations found it did not always involve PDK2 inhibition or reduction of the mitochondrial membrane potential, which are the accepted mechanisms for DCA induction of apoptosis. Nevertheless, DCA sensitized all cancer cell lines tested toward apoptosis of PENAO. DCA and PENAO are both currently in clinical trials and targeting cancer metabolism with these drugs may offer options for difficult to treat cancers.
Abbreviations
| ANT | = | adenine nucleotide translocase; DCA, dichloroacetate |
| ETC | = | electron transport chain |
| MMP | = | mitochondrial membrane potential |
| MPTP | = | mitochondrial permeability transition pore |
| NAC | = | N-acetylcysteine |
| PDK | = | pyruvate dehydrogenase kinase |
| PDK2-kd | = | knock down of PDK2 |
| PENAO | = | 4-(N-(S-penicillaminylacetyl)amino) phenylarsonous acid |
| ROS | = | reactive oxygen species |
| siNC | = | negative control siRNA |
| siPDK | = | PDK siRNA |
Introduction
Deregulated metabolism as a phenotype of cancer cells has recently received an immense amount of attention, and is considered an emerging hallmark of cancer.Citation1-Citation5 In the 1920s, Otto Warburg discovered metabolic deregulation in cancer cells from his observation that cancer cells prefer to use glycolysis even in the presence of oxygen.Citation6 The Warburg effect refers to the diversion of pyruvate metabolism from mitochondrial oxidation to lactate production. Inhibition of the pyruvate dehydrogenase kinases (PDKs) in cancer cells with a small-molecule PDK inhibitor such as dichloroacetate (DCA) reverses the Warburg effect by activating PDH and redirecting pyruvate metabolism back into the mitochondria.Citation7,8 DCA has been used in the clinic for lactic acidosis disorders for over 3 decades Citation9,10 and as an anticancer drug, is one of the most promising candidates that targets metabolism,Citation11,12 with recent reports indicating its safety in cancer patients.Citation13-Citation15
DCA has been demonstrated to halt tumor growth in vivo, Citation7,16-18 and in glioblastoma patients could normalize the mitochondria, and induce apoptosis in tumors.Citation15 The induction of apoptosis is said to be via increased production of reactive oxygen species (ROS) due to increased mitochondrial activity, with decreased polarization of the mitochondrial membrane restoring apoptotic processes.Citation7,19 However, not all studies find induction of apoptosis with DCA alone at reasonable drug concentrations. Of more practical interest is the ability of DCA to sensitize cancer cells toward apoptosis and enhance the effects of other cytotoxic agents, however this has also produced varied results. Findings range from reducing the effectiveness of doxorubicin and cisplatin,Citation20 to dramatically enhancing the effects of arsenic trioxide (ATO) Citation21 and cisplatin,Citation22-Citation24 or resulting in little change to temozolomide cytotoxicity.Citation20 DCA has been reported in several studies to be most effective when used in combination with agents that inhibit mitochondrial function.Citation18,21,25,26 Thus, targeting two aspects of metabolism, using DCA to promote mitochondrial activity and then an anti-mitochondrial agent to simultaneously inhibit mitochondrial function, is a rational approach to cancer therapy.
4-(N-(S-penicillaminylacetyl)amino) phenylarsonous acid (PENAO) is a second generation arsenic-based mitochondrial toxin Citation27 that is currently being trialled in a Phase I/II dose escalation study in patients with solid tumors refractory to standard therapy. PENAO targets the adenine nucleotide translocase (ANT) component of the mitochondrial permeability transition pore (MPTP), a central component of the mitochondrial apoptotic machinery.Citation28-Citation31 The trivalent arsenical of PENAO reacts with ANT, crosslinking Cys57 and Cys257 of ANT to induce MPTP opening by increasing the sensitivity of pore opening to Ca2+ levels.Citation32 PENAO is taken up into cells faster and exported by MRP1/2 more slowly than its first-generation counterpart, 4-(N-(S-glutathionylacetyl)amino) phenylarsenoxide (GSAO),Citation33 enabling it to target proliferating tumor cells as well as endothelial cells.Citation27 PENAO has been demonstrated to inhibit proliferation of a range of cancer cell lines in vitro, and is effective in vivo against subcutaneous human BxPC-3 pancreatic carcinoma xenografts.Citation27
The present investigation examines the effectiveness of targeting two aspects of metabolism using the novel combination of DCA and PENAO against a panel of breast and other epithelial cancer cells. The mechanisms by which DCA sensitizes cancer cells toward apoptosis when used in combination with PENAO were examined, highlighting the metabolic complexities of cancer cell responses to DCA.
Results
PENAO selectively reduces cell viability of cancer cells
To examine the effects of PENAO on cancer cells, a panel of cancer cells representing 4 common carcinoma types (breast, colon, pancreas, and prostate) was treated with a range of concentrations of PENAO for 48 hr. PENAO significantly reduced the total viable cell number of cancer cells, but did not effect the non-cancerous MCF-10A cells, where only a 5% reduction in viable cell number was observed (). The IC50 for PENAO-induced reduction in cell viability was in the low micromolar range of ∼5–12.5 μM for all cancer cell lines ( and ) with the exception of the Bx-PC3 cell line which had an IC50 of ∼32 μM (Fig. S1; Table 1). PENAO was also able to reduce viable cell number in hypoxia (0.3% O2) in T-47D, MCF7 and MDA-MB-468 cells, with reduced IC50s for these cells indicating that normoxic levels of oxygen are not required for PENAO cytotoxicity (Fig. S2; Table 1).
Table 1. IC50 values for total viable cell number for PENAO treatment of human epithelial cancer cell lines
Figure 1. DCA potentiates PENAO-induced apoptosis. (A) Total viable cell number (neutral red uptake assay) after 48 hr PENAO treatment of epithelial cancer cell lines (MCF10A – non-cancerous control). (B) Viable cell number after 48 hr PENAO treatment +/− 5 mM DCA (5 μM PENAO for all cells except MCF7 (7.5 μM) and BxPC3 (30 μM)). (C) Percentage of apoptotic cells (Annexin V positive) after 48 hr PENAO treatment (concentrations as in (B)) +/− 5 mM DCA. *P < 0.05 **P < 0.01 ***P < 0.001 vs control. # P < 0.01 vs PENAO.
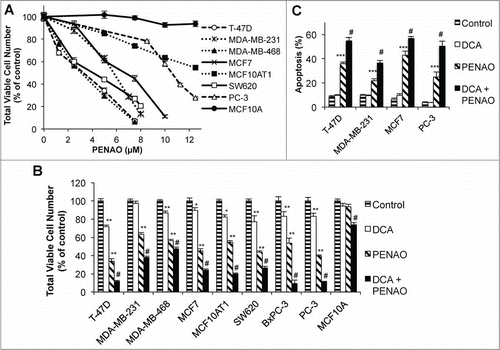
DCA enhances the apoptotic effects of PENAO
The effect of 5 mM DCA on the cytotoxicity of PENAO after 48 hr was then measured (; results for the full range of concentrations of DCA and PENAO tested are in Fig. S1). DCA potentiated the PENAO-induced reduction in viable cells in all cancer cell lines, including the MDA-MB-231 cell line in which DCA alone had no significant effect on viable cell number (). Further investigations were performed on the breast cancer cell lines T-47D, MDA-MB-231 and MCF7, and PC-3 prostate cancer cells to determine whether the increased effect of PENAO by DCA was due to increased induction of apoptosis. In all 4 cell lines, DCA alone did not induce apoptosis, however PENAO was able to induce apoptosis and co-treatment with DCA resulted in a significant increase in the proportion of apoptotic cells compared to PENAO alone (), indicating DCA sensitizes cancer cells toward apoptosis. The effect of PENAO in the MCF10A non-cancerous cells was also increased by DCA, however the cytotoxicity of this drug combination was limited compared to that observed in the cancer cells (), indicating that the cytotoxicity of this drug combination is largely targeted at cancer cells.
DCA enhances apoptosis by increasing ROS production
The mechanism of enhancement of apoptosis was further investigated in T-47D and MDA-MB-231 cells as they were distinct in their response to growth inhibition by DCA alone: T-47D and MDA-MB-231 cells were DCA-sensitive and -insensitive, respectively (, Ref.Citation34); however in both cell lines, PENAO-induced apoptosis was enhanced by DCA treatment ().
DCA increases oxidative stress by redirecting pyruvate into the mitochondria, increasing electron transport chain (ETC) activity and thus generating more ROS.Citation7 The increased oxidative stress could potentially sensitize cancer cells toward apoptosis.Citation35 Treatment with DCA for 24 hr increased ROS by 68 and 65% in T-47D and MDA-MB-231 cells respectively (). As T-47D and MDA-MB-231 cells express predominantly PDK2 (Ki 0.2 mM) and low levels of the other isoforms,Citation34 the effect of knocking down PDK2 (PDK2-kd) on ROS production was investigated. Knockdown of PDK2 with siRNA (∼70–90%, ) increased ROS in a similar manner to 1 mM DCA ( and B) but 1 mM DCA treatment on PDK2-kd T-47D and MDA-MB-231 cells significantly further increased ROS production. This may be due to inhibition of the residual PDK2, or inhibition of PDK1 or PDK4 (Ki 1.0 mM and 0.5 mM respectively) which are expressed at low levels Citation34 or which may have been induced in response to the PDK2-kd. The increase in ROS by DCA could be prevented by co-treatment with the anti-oxidant N-acetylcysteine (NAC, 10 mM) (), thus the effect of NAC on apoptosis was examined. In DCA+PENAO treated MDA-MB-231 cells, co-treatment with 10 mM NAC for 48 hr completely prevented the enhancement of apoptosis (P < 0.001), whereas in T-47D cells, NAC produced only a partial but significant reduction (P < 0.05 vs no NAC) in DCA-PENAO-induced apoptosis (). NAC was also able to reduce apoptosis induced by PENAO alone, almost completely in MDA-MB-231 cells but only partially in T-47D cells (), indicating a contribution of ROS to the cytotoxicity of PENAO. (The thiol group on NAC will not interact directly with PENAO to reduce its cytotoxicity as PENAO interacts only with closely-spaced di-thiol groups, as occur on the ANTCitation32). Thus in both cell lines, oxidative stress was an important mechanism for the enhanced apoptosis of the DCA-PENAO drug combination.
Figure 2. DCA increases ROS. (A) ROS levels after 24 hr DCA treatment in T-47D and MDA-MB-231 cells. (B) ROS levels 24 hr after PDK2-kd +/− 1 mM DCA. (C) Representative protein gel blots of PDK2 protein levels 48 hr post-transfection with siPDK. (D) ROS levels after 24 hr 5 mM DCA treatment +/− 10 mM N-acetylcysteine (NAC). **P < 0.01 vs siPDK2; ***P < 0.001 vs control.
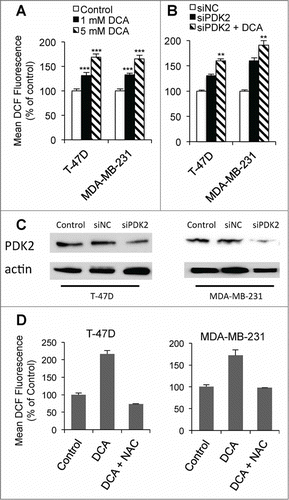
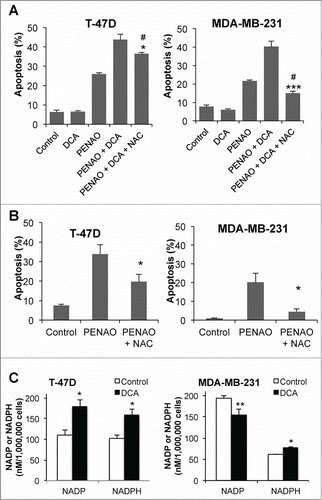
Figure 3. DCA can enhance apoptosis by increasing ROS. (A) Percentage of apoptotic cells after 48 hr DCA-PENAO treatment +/− NAC. # P < 0.01 vs PENAO alone. *P < 0.05 ***P < 0.001 vs PENAO+DCA. (B) Apoptotic cells after 48 hr PENAO +/− NAC. *P < 0.05 vs PENAO. (C) NADP/NADPH levels after 24 hr +/− DCA in T-47D and MDA-MB-231 cells; *p<0.05 **P < 0.01 vs control. Treatments: DCA 5 mM, PENAO 5 μM, NAC 10 mM.
T-47D and MDA-MB-231 cells are distinct in their metabolic response to DCA
Further investigations revealed metabolic differences between the 2 cell lines that may contribute to the different effects of DCA and the relative contribution of ROS. The NADP:NADPH ratio before and after DCA treatment was measured as an indicator of oxidative state (). In both cell lines, the availability of NADPH was increased after 24 hr of treatment with 5 mM DCA, but the context was different. In T-47D cells, the total NADP/NADPH pool was increased by DCA treatment (212 ± 15 vs 337 ± 31, control vs DCA, p = 0.01) but the ratio of NADP:NADPH was unchanged (1.09 ± 0.11 vs 1.13 ± 0.11, control vs DCA, p = 0.4). In contrast, in MDA-MB-231 cells DCA significantly reduced the ratio of NADP:NADPH (3.14 ± 0.08 vs 1.98 ± 0.17, control vs DCA, p = 0.002) while the total NADP/NADPH pool remained the same (236 ± 25 vs 230 ± 25, control vs DCA, p = 0.4). The higher NADP:NADPH ratio of MDA-MB-231 cells indicates they are in a more oxidative state and therefore may be more sensitive to increases in ROS production than T-47D cells.
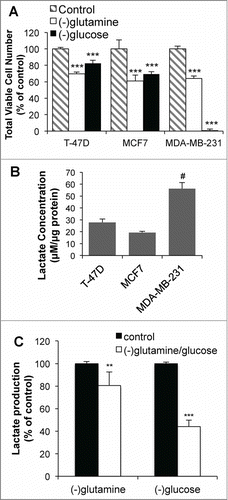
The difference in oxidative state between the 2 cell lines may be due to their distinct metabolic profiles. Glutamine deprivation reduced growth in 3 breast cancer cell lines to a similar extent (). In contrast, while growth of T-47D and MCF7 cells was moderately reduced in glucose-deprived conditions, the deprivation of glucose for 72 hr resulted in complete cell death in MDA-MB-231 cells (). Furthermore, MDA-MB-231 cells produced ∼3-fold more lactate in a 24 hr period compared to the T-47D and MCF7 cells (). The source of lactate production in MDA-MB-231 cells was confirmed to be primarily from glucose, as glucose deprivation reduced lactate production (over 16 hr) by 60% (). These results suggest that metabolic differences between cell lines result in DCA enhancing apoptosis via different mechanisms.
Figure 4. T-47D and MDA-MB-231 cells are metabolically distinct. (A) Viable cell number after 72 hr growth in glucose or glutamine deficient media. (B) Extracellular lactate production over 24 hr for different breast cancer cell lines. (C) Lactate production in MDA-MB-231 cells over 16 hr with glutamine or glucose deprivation. **P < 0.01 ***P < 0.001 vs control; # P < 0.001 vs T-47D and MCF7.
DCA can sensitize cancer cells toward apoptosis via off-target effects
We have previously shown that DCA did not significantly reduce lactate production by MDA-MB-231 cells, correlating with the lack of growth inhibition in these cells.Citation34 Thus we questioned whether the pro-apoptotic effect of DCA was due to PDK2 inhibition by performing siRNA knockdown experiments. Surprisingly, the knockdown of PDK2 in T-47D cells did not enhance the apoptotic effects of PENAO, but did in the MDA-MB-231 cells (). Furthermore, the addition of DCA to T-47D-PDK2-kd cells resulted in a significant enhancement of the apoptotic effect of PENAO, but not in MDA-MB-231-PDK2-kd cells. The findings indicate that DCA can sensitize T-47D cancer cells toward apoptosis via PDK2-independent pathways.
Figure 5. DCA can enhance apoptosis via off-target mechanisms. (A) Apoptosis in T-47D and MDA-MB-231 cells after 48 hr 2 μM PENAO treatment +/− 5 mM DCA in control (siNC) and siPDK2 cells (PDK protein expression 48 hr post-transfection with siPDK2 are shown in ). (B) Noxa and Puma expression after 24 hr 5 mM DCA treatment. (C and D) Mitochondrial membrane potential (JC-1 red indicates presence of MMP) in (C) T-47D cells and (D) MDA-MB-231 cells after 3 and 24 hr treatment with 0.2–5 mM DCA. **P < 0.01 vs PENAO + siNC; *** P < 0.001 vs control.
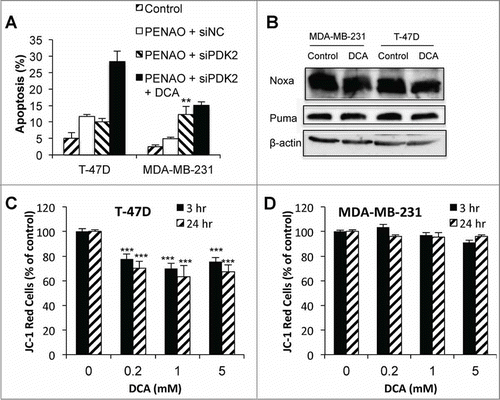
Alternative mechanisms of DCA enhancement of apoptosis were investigated. A decrease in the expression of the anti-apoptotic protein Bcl-2 has been reported with DCA treatment in PC-3 prostate cancer cells,Citation36 however, we have previously found that Bcl-2 expression increased slightly (1.2-fold) with DCA treatment in T-47D breast cancer cells.Citation21 In the current study, we found that DCA treatment did not significantly increase the levels of pro-apoptotic proteins, Noxa and Puma (). Mitochondrial changes may be involved in DCA sensitization of cancer cells toward apoptosis. It has been previously reported that DCA can depolarize the mitochondrial membrane potential (MMP),Citation7,25 which has been postulated to sensitize cancer cells toward apoptosis.Citation37 Using JC-1 staining to measure the MMP, it was found that JC-1 red staining decreased from as early as 3 hr post-DCA treatment in T-47D cells () indicating depolarization, but was unchanged in MDA-MB-231 cells, even after 24 hr of DCA treatment (). Thus DCA-enhanced apoptosis in T-47D cells may be due to changes in MMP.
Discussion
Despite the hype about targeting the metabolic phenotype of cancer cells for therapy, little progress has been made in the clinic to realize this potential. DCA and PENAO are both compounds currently in clinical trials in the treatment of cancer that target different aspects of the cancer metabolic phenotype.Citation13-15,32 Here we demonstrate that PENAO, a novel anti-mitochondrial agent, is effective at low micromolar concentrations against a range of breast cancer cell lines, representing luminal (T-47D and MCF7) and basal (MDA-MB-231 and MDA-MB-468) subtypes of breast cancer, and carcinomas from other tissues (). Previously only a limited number of epithelial cancers had been tested for sensitivity to PENAO.Citation27 Results from the present study indicate that PENAO has promising effects against cancer in two difficult to treat circumstances - triple negative breast cancer (MDA-MB-231 and MDA-MB-468) and hypoxia (). Significant regions of solid tumors are hypoxic, and are generally resistant to traditional therapies and associated with metastases and poor outcomes.Citation38-Citation41 Similarly, triple negative breast cancers, which account for approximately 20% of breast cancer, cannot be targeted with anti-estrogens or Herceptin and have a poorer prognosis than other breast cancer subtypes. Thus, PENAO offers a new treatment option for these tumors.
When combined with DCA, the cytotoxicity of PENAO was enhanced 1.5 – 4-fold in these cell lines ( and ). Thus targeting of two aspects of metabolism in this manner, using DCA to redirect pyruvate metabolism to the mitochondria while using PENAO to inhibit mitochondrial function, is an effective and rational approach to treating cancer. This combination was effective against cancer cells and relatively non-toxic against the non-cancerous MCF10A cells of this study (). A similar distinction between glioblastoma cells and non-cancerous human lung fibroblast cell line MRC-5 and normal human astrocytes has been observed,Citation42 suggesting that a therapeutic window between normal and cancer cells exists, and that this drug combination has potential in the clinic against a number of cancer types.
The success of the principle of combining DCA with a mitochondrial inhibitor in breast cancer cells has been demonstrated previously in vitro with the clinical agent arsenic trioxide, which was shown to inhibit complex IV of the ETC.Citation21 This dual-targeting strategy is also supported by studies showing DCA to be more effective against cancer cells with mitochondrial defects, such as rho(0) MCF7 cells,Citation25 mitochondrial ND2 mutant cells Citation26 and oligomycin-treated HCT116 colon cancer cells.Citation18 However, the precise mechanism by which DCA enhances cytotoxicity is not always clear. Elevated ROS production by DCA has been demonstrated by several investigators, an effect that is attributed to increased ETC activity following the redirection of pyruvate metabolism from lactate production into mitochondrial oxidation.Citation7,21,25,43,44 DCA can enhance the cytotoxicity of pro-oxidant drugs such as elesclomol and sulindac, and the involvement of elevated ROS in apoptosis mediated by DCA was demonstrated by reducing apoptosis with antioxidants such as NAC.Citation43,44 We have also demonstrated that the mechanism of apoptosis in the DCA / PENAO combination treatment clearly involved the increased generation of ROS ( and ), however, the extent to which ROS were responsible, according to intervention with NAC, varied between cell lines.
In T-47D cells where the increase in ROS by DCA did not fully account for the sensitization to apoptosis (), we found significant depolarization of the mitochondrial membrane potential. Bonnet et al hypothesized that when DCA inhibits the PDKs, the increased ROS damage complex I, the most sensitive complex of the ETC to ROS, leading to an inability to efflux protons resulting in depolarization.Citation7 This mechanism is dependent on mitochondrial ROS levels changing, whereas NAC and the DCF readout used in this study reflect cytosolic ROS, thus compartmental differences in ROS levels may explain the incomplete protection by NAC. However, other mechanisms which could be altered by DCA may also contribute to cancer cell hyperpolarization (discussed by Sutendra et al. Citation11), thus the exact mechanism by which DCA depolarizes the mitochondria is uncertain.
Regardless of the mechanism, mitochondrial depolarisation is not essential for the enhancement of apoptosis, as depolarization did not occur in MDA-MB-231 cells but apoptosis was still enhanced ( and ). Further, while the growth inhibitory effects of DCA in T-47D cells correlated well with inhibition of PDK2 activity or PDK2-kd by siRNA,Citation34 the enhanced cytotoxicity of PENAO could not be achieved with siRNA PDK2-kd in T-47D cells (). These results clearly indicate that the anti-proliferative and pro-apoptotic effects of DCA are distinct processes, and that mechanisms of enhanced apoptosis, other than PDK2 inhibition, increased ROS from the ETC and MMP depolarisation, exist.
Were the action of DCA simply due to actions on the MMP, then it should be a universal enhancer of apoptosis through the alteration of this fundamental apoptotic machinery. However, the effect of DCA in combination with other drugs ranges from reduced cytotoxicity of doxorubicin and cisplatin in vitro on a range of pediatric cancer cell lines,Citation20 to no alteration of the effects of temozolomide,Citation20 to synergizing with cisplatin in HeLa cervical cancer cells.Citation23 Our own studies indicate that DCA does not enhance the effects of doxorubicin in our breast cancer panel in a 48 hr cytotoxicity test (unpublished), indicating that DCA is not a universal enhancer of apoptosis.
Conclusion
DCA is a compound that can reverse the widespread phenomenon of aerobic glycolysis. While the accepted mechanism of DCA would suggest that it could be a generic non-toxic enhancer of apoptosis, this is not the case. Combining DCA with mitochondrial inhibitors is a rational strategy for targeting cancer metabolism and shows promise. Further work is needed to fully understand how best to utilize DCA effectively for the treatment of cancer.
Materials and Methods
Cell culture
Human cancer cell lines were obtained from (year obtained in brackets): Breast cancer cell lines: Dr Anna DeFazio, Westmead Millenium Institute, Sydney, Australia provided T-47D (2003), MCF-10AT1 (2005) and MCF-10A (2005, non-cancerous); Prof Chris Parish, Australian National University, Canberra, Australia provided MDA-MB-468 (2003) MCF7 (2003) and MDA-MB-231 (2003); June Hornby, Australian National University, provided PC-3 prostate cancer (2000), SW620 colon cancer (2000); Dr Pierre Dilda Lowy Cancer Center, UNSW, Australia provided Bx-PC3 pancreatic cancer (2011). The cell lines have appearances consistent with published morphologies but have not been authenticated recently. All cells were grown at 37°C, 5% CO2 in RPMI media supplemented with 10% fetal bovine serum, 10 mM HEPES and 2 g/L NaHCO3 with the exception of MCF-10A and MCF-10AT1, which were grown in DMEM/F-12 medium, 5% (MCF-10A) or 2.5% (MCF-10AT1) horse serum, 0.01% EGF, 0.28 IU/ml insulin, 0.01% cholera toxin and 0.5μg/ml hydrocortisone. For hypoxic conditions, cells were grown in a sealed humidified gas chamber (Billups-Rothenberg, Del Mar, CA, USA). Oxygen was flushed out with 95% nitrogen, 5% CO2 at 20 L/min for 4 min and gas inside flasks/plates was allowed to equilibrate for 3 hr. The chamber was then re-flushed as necessary to obtain O2 levels of 0.2–0.3%.
In vitro viability and apoptosis assays
Neutral red staining was used to determine total viable cell number after 48 hr of treatment, as described previously.Citation21 Apoptosis was quantified by flow cytometry (BD LSR1 cell sorter) using FITC-labeled Annexin V (Molecular Probes, Invitrogen Co, CA, USA) and propidium iodide staining as previously described.Citation21
Immunoblotting
Cells at ∼70% confluence were incubated in either normoxia or hypoxia for 18 hr. Cells were lysed with RIPA buffer (150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0), sonicated (low power for 5 s) and centrifuged (13000 x g, 15 min, 4°C). Supernatant protein content was measured using BCA Assay Kit (Pierce, Rockford, IL, USA). Proteins (30 μg/sample) were separated via reducing 10% SDS-PAGE and standard western blotting procedures Citation45 were used to detect proteins of interest with the following primary antibodies: PDK2 (Cat# AP7039b, Abgent, San Diego, CA, USA), β-actin (Abcam), Noxa (OP180, Calbiochem) and Puma (ab9643, Abcam).
Extracellular lactate
Cells were seeded into 12-well plates (1×105 cells/well). After 24 hr, media was collected for lactate measurement as previously described.Citation16 Lactate production at 24 hr was standardized to protein content to account for changes in cell number.
PDK gene silencing with RNAi
Cells were transfected with 10 nM PDK siRNA (siPDK) or negative control siRNA (siNC) using Lipofectamine RNAiMAX (Invitrogen Lifesciences) following the manufacturer's protocol. After 48 hr, knockdown efficiency was determined by immunoblotting (≥70% knockdown achieved), and drug treatment commenced. The total number of viable cells was measured 48 hr later.
Measuring mitochondrial membrane potential
Cells were seeded into 12-well plates at 100,000/well and treated for desired period, then incubated with 0.2 μM JC-1 for 30 min. JC-1 accumulates in cells when a mitochondrial membrane potential (MMP) is present (resting cells are hyperpolarized) and forms aggregates which emit red fluorescence. Upon depolarization (i.e. collapse of the potential), JC-1 does not aggregate but instead is present in the monomeric form that emits green fluorescence. Thus upon depolarization a shift from red to green fluorescence is observed. Cells were with analyzed with FACS (BD LSR1 cell sorter).
Measuring NADP/NADPH
Cells were washed with PBS, harvested then NADP and NADPH levels were detected using a NADP+/NADPH detection kit (Abnova #KA1663) according to the manufacturer's protocol.
ROS measurements
Cells were seeded into 12-well plates at 100,000/well and treated for desired period, then incubated for 1 hr at 37°C with 10 μM 2,7-dichlorodihydrofluorescein (DCF)-diacetate (Molecular Probes, Invitrogen Co, CA, USA). DCF-diacetate is cell permeable and is cleaved by esterases and then oxidized into the fluorescent DCF by ROS, thus fluorescence is proportional to intracellular ROS levels. After incubation, cells were harvested, washed with PBS and then resuspended in PBS and analyzed by FACS (BD LSR1 cell sorter).
Statistical analysis
Differences between groups were analyzed with one-way ANOVA, comparing all treatment groups with each other using the Bonferonni post-hoc test in GraphPad Prism software. Each independent experiment was performed with at least triplicate samples per treatment group. All results are expressed as mean ± SEM of replicate values from at least 2 independent experiments. Statistical significance was considered P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
955992_Supplementary_Materials.zip
Download Zip (288.1 KB)Funding
This project was funded primarily by grant #1008861, awarded through the Priority-driven Collaborative Cancer Research Scheme and co-funded by the National Breast Cancer Foundation and the Australian Government through Cancer Australia. Other support came from grants to ACB from Cancer Council ACT (#585409) and NHMRC CDA Fellowship (#366787).
Supplemental Material
Supplemental data for this article can be accessed on thepublisher's website.
References
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11:85-95; PMID:21258394; http://dx.doi.org/10.1038/nrc2981
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011; 476:346-50; PMID:21760589; http://dx.doi.org/10.1038/nature10350
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012; 488:656-9; PMID:22763442; http://dx.doi.org/10.1038/nature11323
- Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, Huang P, Sawyer SK, Fuerth B, Faubert B, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev 2011; 25:1041-51; PMID:21576264; http://dx.doi.org/10.1101/gad.1987211
- Warburg O. On the origin of cancer cells. Science 1956; 123:309-14; PMID:13298683; http://dx.doi.org/10.1126/science.123.3191.309
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007; 11:37-51; PMID:17222789; http://dx.doi.org/10.1016/j.ccr.2006.10.020
- McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem 2008; 283:22700-8; PMID:18541534; http://dx.doi.org/10.1074/jbc.M801765200
- Stacpoole PW, Greene YJ. Dichloroacetate. Diabetes Care 1992; 15:785-91; PMID:1600837; http://dx.doi.org/10.2337/diacare.15.6.785
- Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Treatment of lactic acidosis with dichloroacetate. N Engl J Med 1983; 309:390-6; PMID:6877297; http://dx.doi.org/10.1056/NEJM198308183090702
- Sutendra G, Michelakis ED. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol 2013; 3:38; PMID:23471124; http://dx.doi.org/10.3389/fonc.2013.00038
- Garber K. Oncology's energetic pipeline. Nat Biotechnol 2010; 28:888-91; PMID:20829819; http://dx.doi.org/10.1038/nbt0910-888
- Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs 2013; 32:452-64; PMID:24297161; http://link.springer.com/article/10.1007%2Fs10637-013-0047-4
- Garon EB, Christofk HR, Hosmer W, Britten CD, Bahng A, Crabtree MJ, Hong CS, Kamranpour N, Pitts S, Kabbinavar F, et al. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J Cancer Res Clin Oncol 2014; 140:443-52; PMID:24442098; http://dx.doi.org/10.1007/s00432-014-1583-9
- Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Translational Med 2010; 2:31ra4; http://dx.doi.org/10.1126/scitranslmed.3000677
- Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 2010; 120:253-60; PMID:19543830; http://dx.doi.org/10.1007/s10549-009-0435-9
- Chen Y, Cairns R, Papandreou I, Koong A, Denko NC. Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PLoS One 2009; 4:e7033; PMID:19753307; http://dx.doi.org/10.1371/journal.pone.0007033
- Sanchez-Arago M, Chamorro M, Cuezva JM. Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis 2010; 31:567-76; PMID:20080835; http://dx.doi.org/10.1093/carcin/bgq012
- Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, McMurtry MS, Michelakis ED. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 2013; 32:1638-50; PMID:22614004; http://dx.doi.org/10.1038/onc.2012.198
- Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol 2010; 67:647-55; PMID:20502900; http://dx.doi.org/10.1007/s00280-010-1361-6
- Sun RC, Board PG, Blackburn AC. Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells. Mol Cancer 2011; 10:142; PMID:22093145; http://dx.doi.org/10.1186/1476-4598-10-142
- Xiao H, Yan L, Zhang Y, Qi R, Li W, Wang R, Liu S, Huang Y, Li Y, Jing X. A dual-targeting hybrid platinum(IV) prodrug for enhancing efficacy. Chem Commun (Camb) 2012; 48:10730-2; PMID:23012698; http://dx.doi.org/10.1039/c2cc34297a
- Xie J, Wang BS, Yu DH, Lu Q, Ma J, Qi H, Fang C, Chen HZ. Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells. Int J Oncol 2011; 38:409-17; PMID:21132264
- Xue X, You S, Zhang Q, Wu Y, Zou GZ, Wang PC, Zhao YL, Xu Y, Jia L, Zhang X, et al. Mitaplatin increases sensitivity of tumor cells to cisplatin by inducing mitochondrial dysfunction. Mol Pharm 2012; 9:634-44; PMID:22289032; http://dx.doi.org/10.1021/mp200571k
- Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer 2010; 127:2510-9; PMID:20533281; http://dx.doi.org/10.1002/ijc.25499
- Sun W, Zhou S, Chang SS, McFate T, Verma A, Califano JA. Mitochondrial mutations contribute to HIF1alpha accumulation via increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma. Clin Cancer Res 2009; 15:476-84; PMID:19147752; http://dx.doi.org/10.1158/1078-0432.CCR-08-0930
- Dilda PJ, Decollogne S, Weerakoon L, Norris MD, Haber M, Allen JD, Hogg PJ. Optimization of the antitumor efficacy of a synthetic mitochondrial toxin by increasing the residence time in the cytosol. J Med Chem 2009; 52:6209-16; PMID:19788237; http://dx.doi.org/10.1021/jm9008339
- Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science 1998; 281:1322-6; PMID:9735050; http://dx.doi.org/10.1126/science.281.5381.1322
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35:495-516; PMID:17562483; http://dx.doi.org/10.1080/01926230701320337
- Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora's box opens. Nat Rev Mol Cell Biol 2001; 2:67-71; PMID:11413468; http://dx.doi.org/10.1038/35048073
- Zamzami N, Marchetti P, Castedo M, Hirsch T, Susin SA, Masse B, Kroemer G. Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett 1996; 384:53-7; PMID:8797802; http://dx.doi.org/10.1016/0014-5793(96)00280-3
- Park D, Chiu J, Perrone GG, Dilda PJ, Hogg PJ. The tumour metabolism inhibitors GSAO and PENAO react with cysteines 57 and 257 of mitochondrial adenine nucleotide translocase. Cancer Cell Int 2012; 12:11; PMID:22448968; http://dx.doi.org/10.1186/1475-2867-12-11
- Don AS, Kisker O, Dilda P, Donoghue N, Zhao X, Decollogne S, Creighton B, Flynn E, Folkman J, Hogg PJ. A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell 2003; 3:497-509; PMID:12781367; http://dx.doi.org/10.1016/S1535-6108(03)00109-0
- Gang BP, Rooke M, Mishra S, Dahlstrom JE, Blackburn AC. Sensitivity to Dichloroacetate is Determined by PDK Expression. Proceedings of the 104th Annual Meeting of the American Association for Cancer Research; Apr 6-10; Washington, DC. Cancer Research 2013; 73(8): Supplement 1; http://dx.doi.org/10.1158/1538-7445.AM2013-5421
- Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 2009; 46:821-31; PMID:19265700; http://dx.doi.org/10.1016/j.yjmcc.2009.02.021
- Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008; 68:1223-31; PMID:18465755; http://dx.doi.org/10.1002/pros.20788
- Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 2008; 99:989; PMID:18766181; http://dx.doi.org/10.1038/sj.bjc.6604554
- Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol 2004; 14:198-206; PMID:15254862; http://dx.doi.org/10.1016/j.semradonc.2004.04.008
- Brizel DM, Scully SP, Harrelson JM, Layfield LJ, Bean JM, Prosnitz LR, Dewhirst MW. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res 1996; 56:941-3; PMID:8640781
- Fyles A, Milosevic M, Hedley D, Pintilie M, Levin W, Manchul L, Hill RP. Tumor hypoxia has independent predictor impact only in patients with node-negative cervix cancer. J Clin Oncol 2002; 20:680-7; PMID:11821448; http://dx.doi.org/10.1200/JCO.20.3.680
- Nordsmark M, Overgaard J. Tumor hypoxia is independent of hemoglobin and prognostic for loco-regional tumor control after primary radiotherapy in advanced head and neck cancer. Acta Oncol 2004; 43:396-403; PMID:15303502; http://dx.doi.org/10.1080/02841860410026189
- Shen H, Decollogne S, Dilda PJ, Chung S, Luk P, Hogg PJ, McDonald KL. PENAO, a Novel Mitochondria-Targeted Agent, Synergizes with Dichloroacetate to Target Aberrant Glucose Metabolism in Glioblastoma. 4th Quadrennial Meeting of the World Federation of Neuro-Oncology; November 21st-24th; San Francisco, CA. Neuro-Oncology 2013; (S3):58; WOS:000327456200243.
- Ayyanathan K, Kesaraju S, Dawson-Scully K, Weissbach H. Combination of sulindac and dichloroacetate kills cancer cells via oxidative damage. PLoS One 2012; 7:e39949; PMID:22866174; http://dx.doi.org/10.1371/journal.pone.0039949
- Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, et al. Inactivation of the HIF-1alphaPDK3 signaling axis drives melanoma toward mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res 2012; 72:5035-47; PMID:22865452; http://dx.doi.org/10.1158/0008-5472.CAN-12-0979
- Lim CE, Matthaei KI, Blackburn AC, Davis RP, Dahlstrom JE, Koina ME, Anders MW, Board PG. Mice deficient in glutathione transferase zetamaleylacetoacetate isomerase exhibit a range of pathological changes and elevated expression of alpha, mu, and pi class glutathione transferases. Am J Pathol 2004; 165:679-93; PMID:15277241; http://dx.doi.org/10.1016/S0002-9440(10)63332-9