
Abstract
The colorectal cancer is the leading contributor of cancer-related mortality. Mammalian target of rapamycin (mTOR), existing in 2 complexes (mTORC1/2), is frequently dysregulated and constitutively activated in colorectal cancers. It represents an important drug target. Here we found that INK-128, the novel ATP-competitive kinase inhibitor of mTOR, blocked both mTORC1 and mTORC2 activation in colorectal cancer cells (both primary and transformed cells). The immunoprecipitation results showed that the assembly of mTORC1 (mTOR-Raptor association) and mTORC2 (mTOR-Rictor-Sin1 association) was also disrupted by INK-128. INK-128 inhibited colorectal cancer cell growth and survival, and induced both apoptotic and non-apoptotic cancer cell death. Further, INK-128 showed no effect on Erk/MAPK activation, while MEK/Erk inhibition by MEK-162 enhanced INK-128-induced cytotoxicity in colorectal cancer cells. Meanwhile, INK-128 downregulated Fascin1 (FSCN1)/E-Cadherin expressions and inhibited HT-29 cell in vitro migration. In vivo, daily INK-128 oral administration inhibited HT-29 xenograft growth in mice, which was further enhanced by MEK-162 administration. Finally, we found that INK-128 sensitized 5-fluorouracil-(5-FU)-mediated anti-HT-29 activity in vivo and in vitro. Thus, our preclinical studies strongly suggest that INK-128 might be investigated for colorectal cancer treatment in clinical trials.
Abbreviations
| Co-IP | = | co-immunoprecipitation |
| ECL | = | enhanced chemiluminescence |
| FSCN10 | = | Fascin1 |
| HRP | = | horseradish peroxidase |
| (mTOR) | = | mammalian target of rapamycin |
| (mTORC1) | = | mTOR complex 1 |
| (mTORC2) | = | mTOR complex 2 |
| (S6K) | = | p70S6K1 |
| PI | = | propidium iodide |
| SD | = | standard deviation (SD) |
Introduction
As one of the leading contributors of cancer-related mortality around the world, the colorectal cancer has drawn significant attentions from both oncologists and cancer biologists.Citation1–3 In the past decades, major improvements have been achieved in surgery and/or chemotherapy therapies for colorectal cancers. However, no significant improvement in overall survival has been accomplished, especially for the advanced/malignant colorectal cancer patients.Citation4 The molecular heterogeneity of colorectal cancers hinders the uniform application of specific molecularly targeted agents.Citation1,2 Mammalian target of rapamycin (mTOR) signaling is frequently dysregulated in colorectal cancers, and is vital for cancer cell progression.Citation5,6 Thus, this pathway is a favorite target for anti-colorectal cancer therapy.Citation5,6
The mTOR kinase forms 2 distinct multi-protein complexes, namely mTOR complex 1 (mTORC1), or the rapamycin-sensitive complex composed of mTOR, Raptor and PRAS40, as well as rapamycin-insensitive mTOR complex 2 (mTORC2), containing mTOR, Rictor, Sin1 and Protor.Citation7-9 MTORC1 phosphorylates its downstream targets p70S6K1 (S6K) and 4E-BP1 to promote protein translation and cell growth.Citation12 On the other hand, mTORC2 phosphorylates Akt at Ser 473 to activate its enzymatic activity.Citation7-9 The first generation of mTOR inhibitors (i.e. rapamycin) only inhibits mTORC1 activity. The second generation of mTOR inhibitors, or the mTOR ATP-competitive kinase inhibitors, interfere both mTORC1 and mTORC2 simultaneously and suppress Akt activation.Citation10
INK-128 is an oral available mTOR ATP-competitive kinase inhibitor which selectively inhibits mTORC1 and mTORC2 with the IC-50 around 1 nM.Citation11 Thus, the compound is much more potent than the well-known mTOR inhibitor rapamycin, not to mention it's inhibitory effect on mTORC2 and Akt.Citation11 In both in vitro and in vivo experiments, INK-128 was shown to effectively suppress several cancer cell growth and to reduce phosphorylation of mTORC1 targets S6K and 4E-BP1, and mTORC2 target Akt (Ser 473).Citation11,12 A phase I clinical trial has been performed to test the safety and pharmacokinetics of INK-128 in advanced solid tumors.Citation12 However, the potential role of INK-128 in colorectal cancers is not fully tested. In the current study, we found that INK-128 blocks mTORC1/2 signaling and inhibits colorectal cancer cell growth both in vivo and in vitro. It also inhibits colorectal cancer cell in vitro migration probably through downregulating fascin1 (FSCN1) and E-Cadherin expressions.
Results
INK-128 inhibits colorectal cancer cell growth
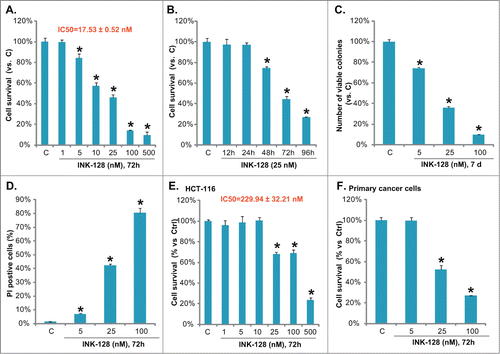
In cultured HT-29 colorectal cancer cells, INK-128 induced a significant decrease of cell survival (indicated by MTT OD), and the effect of INK-128 was both dose- (, with IC 50 = 17.53 ± 0.52 nM) and time-dependent (). Similar results were also observed in another colorectal cancer cell line HCT-116 () and in primary cultured colon cancer cells (). Next we tested the effect of INK-128 on HT-29 cell death, which was tested by the “Clonogenicity” assay and PI staining. As shown in and , INK-128 dose-dependently inhibited the number of survival colonies (also see representative photographs in Fig. S1A), while increasing the PtdIns staining in HT-29 cells. Thus, INK-128 is cytotoxic and inhibits growth of colorectal cancer cells.
Figure 1. INK-128 inhibits colorectal cancer cell growth. HT-29 cells were exposed to the indicated concentration of INK-128 for 72 h (A), or treated with 25 nM of INK-128 for indicated time (B), cell survival was analyzed by MTT assay. HT-29 cells were exposed to indicated concentration of INK-128 (5, 25 and 100 nM), cells were subjected to the “Clonogenicity” assay (7 d after stimulation) and PI staining (72 h after stimulation), number of survival colonies (C) and PtdIns positive cells were recorded (D). HCT-116 cells (E) or the primary cultured colon cancer cells (F) were either left untreated, or exposed to indicated concentration of INK-128 (5, 25 and 100 nM) for 72 h, cell survival was analyzed by MTT assay. Data were expressed as mean ± SD, experiments were repeated 3 times, and similar results were obtained. *P < 0 .05 vs. control group.
INK-128 induces both apoptotic and non-apoptotic death of colorectal cancer cells
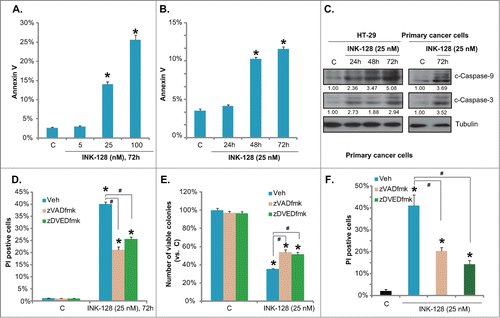
Above results confirmed the cytotoxic effect of INK-128 against colorectal cancer cells. Then we wanted to know if this was due to cell apoptosis. As described in our previous studies, HT-29 cell apoptosis was analyzed by Annexin V staining ( and , also see representative photographs in Fig. S1B), and Western blots assaying apoptosis proteins (). Results showed that INK-128 induced a moderate cell apoptosis in both primary and transformed (HT-29) colorectal cancer cells (-), as the number of Annexin V staining and the expression of cleaved-caspase-3/-9 were increased after INK-128 stimulation in colorectal cancer cells. Meanwhile, 2 apoptosis inhibitors z-VAD-fmk and z-DVED-fmk only inhibited, but not reversed, INK-128-mediated cytotoxicity in HT-29 cells ( and ), and in primary colorectal cancer cells (). The cytotoxicity was analyzed by PI staining and/or the “Clonogenicity” assay (-). Thus, INK-128 induces both apoptotic and non-apoptotic death of colorectal cancer cells
Figure 2. INK-128 induces both apoptotic and non-apoptotic death of colorectal cancer cells. HT-29 cells were either left untreated or exposed to indicated concentration of INK-128 (5, 25 and 100 nM) for 72 h, or treated with 25 nM of INK-128, and cultured for indicated time, cell apoptosis was analyzed by Annexin V FACS (A and B), expressions of cleaved-caspase-3/-9 and tubulin in both HT-29 cells and primary colorectal cancer cells were tested (C). HT-29 or the primary colorectal cancer cells were pretreated with z-VAD-fmk (25 μM) and z-DVED-fmk (25 μM) for 1 hr, followed by INK-128 (25 nM) stimulation, PI positive cells (D and F) and the number of survival colonies (E) were recorded. Data were expressed as mean ± SD, experiments were repeated 3 times, and similar results were obtained. *P < 0 .05 vs. control group. #P < 0 .05.
INK-128 blocks mTORC1 and mTORC2 activation in colorectal cancer cells
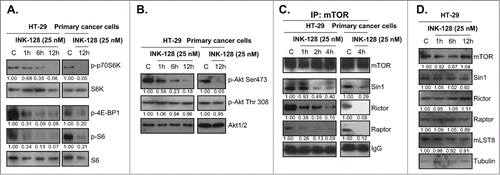
INK-128 is novel dual mTORC1 and mTORC2 inhibitor.Citation11 As discussed early, constantly activated Akt/mTOR signaling contributes to colorectal cancer cell progression,Citation13 we then examined INK-128s effect on Akt/mTOR activation in cultured colorectal cancer cells. Western blots results demonstrated that INK-128 significantly inhibited both mTORC1 and mTORC2 activation in HT-29 and primary colorectal cancer cells ( and ). Note that the activation of mTORC1 was indicated by phospho-S6K (Thr 389), phospho-4E-BP1 (Ser 65) and phospho-S6 (Ser 235/236), while activation of mTORC2 was reflected by Akt Ser 473 phosphorylation ( and ). Akt Thr 308 phosphorylation was not affected by INK-128 (). Significantly, the complex assembly of mTORC1 (mTOR-Raptor association) and mTORC2 (mTOR-Rictor-Sin1 association)Citation8 in HT-29 and primary colorectal cancer cells was disrupted by INK-128 (). While the expressions of mTORC1/2 components including mTOR, Sin1, mLST8, Raptor and Rictor were not affected by INK-128 (). Thus, INK-128 disrupts mTORC1/2 assembly and blocks mTORC1/2 activation in cultured colorectal cancer cells.
Figure 3. INK-128 blocks mTORC1 and mTORC2 activation in colorectal cancer cells. HT-29 or the primary colorectal cancer cells were treated with INK-128 at indicated concentration, regular and/or phospho (P)- levels of Akt, S6, S6K and 4E-BP1 were measured by Western blot (A and B); the mTORC1 assembly (mTOR-raptor association) and mTORC2 assembly (mTOR-Sin1-Rictor association) were tested by Co-IP (C). Expressions of list mTORC1/2 components in HT-29 cells after INK-128 treatment were also examined (D). Experiments were repeated 3 times, similar results were obtained.
Erk inhibition enhances INK-128-induced cytotoxicity in colorectal cancer cells
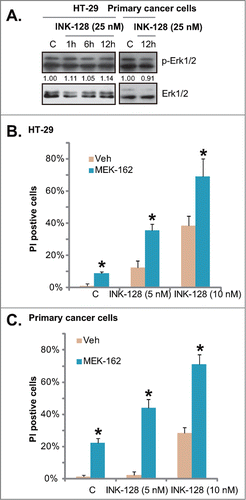
MTORC1 inhibition by rapamycin was found to activate Erk, as part of its negative feedback loop and a resistance factor.Citation14 Thus, we tested the potential role of INK-128 on Erk activation. In both HT-29 and primary colorectal cancer cells, Erk was over-activated at the basal condition, and INK-128 had no significant effect on Erk activation (). Importantly, Erk inhibition by the MEK/Erk inhibitor MEK-162 enhanced INK-128-induced death of colorectal cancer cells, and the 2 showed an addictive effect ( and ). Thus, INK-128 had no effect on Erk activation, but Erk inhibition might increase its cytotoxicity.
Figure 4. Erk inhibition enhances INK-128-induced cytotoxicity in colorectal cancer cells. HT-29 or primary colorectal cancer cells were treated with INK-128 (25 nM) for indicated time, regular and phospho (p)-Erk1/2 were tested (A). HT-29 or primary colorectal cancer cells were treated with INK-128 (5 and 10 nM), or with MEK-162 (25 nM) for 72 h, cell death was tested by PtdIns staining (B and C). Data were expressed as mean ± SD, experiments were repeated 3 times, and similar results were obtained. *P < 0 .05 vs. group without MEK-162 co-administration.
INK-128 inhibits fascin1/E-Cadherin expressions and HT-29 cell migration
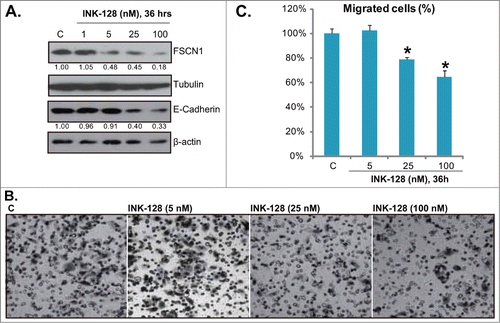
Fascin1 (FSCN1) is an actin bundling protein, which is important for cell migration and proliferation through its functional roles in the formation of cell protrusions.Citation15 Studies including oursCitation16 have suggested that FSCN1 is over-expressed in colorectal cancers, and is associated with cancer cell progression.Citation15 In HT-29 cells, our previous study has found that FSCN1 RNAi-knockdown inhibited HT-29 cell migration and proliferation.Citation16 Significantly, activation of mTORC1 was required for FSCN1 expression, HT-29 cell migration and proliferation. Further, mTORC1 inhibition by mTORC1 inhibitors RAD001 and rapamycin, as well as by activation of its negative regulator AMP-activated protein kinase (AMPK) inhibited FSCN1 expression and HT-29 cell migration.Citation16 Another important protein for cell migration is E-Cadherin, studies have confirmed the role of E-Cadherin in HT-29 cell migration.Citation17,18 It has been suggested that Akt-mTOR activation might be important for E-Cadherin expression.Citation19 Since in this study we have shown that INK-128 blocked mTORC1/2 activation and Akt Ser 473 phosphorylation in HT-29 cells, thus we tested its effect on FSCN1/E-Cadherin expressions and HT-29 cell migration. Results in clearly demonstrated that INK-128 dose-dependently inhibited the expressions of FSCN1/E-Cadherin in HT-29 cells. Further, HT-29 cell in vitro migration, detected by “transwell” assay, was also suppressed by INK-128 ( and ). Note that for the cell migration assay, HT-29 cells were treated with INK-128 for 36 h, when no cytotoxicity was occurred (Data not shown). Also, mitomycin C was always added to block cell proliferation.
Figure 5. INK-128 inhibits fascin1/ E-Cadherin expressions and HT-29 cell migration. HT-29 cells were exposed to indicated concentration of INK-128 (1, 5, 25 or 100 nM) for 36 h, expressions of FSCN1, E-Cadherin, β-actin and tubulin were tested by Western blot (A). Cell migration was assessed by “transwell” assay (B and C). Data were expressed as mean ± SD, experiments were repeated 3 times, and similar results were obtained. *P < 0 .05 vs. control group.
Enhancement of 5-fluorouracil (5-FU)-mediated anti-HT-29 cell activity by INK-128
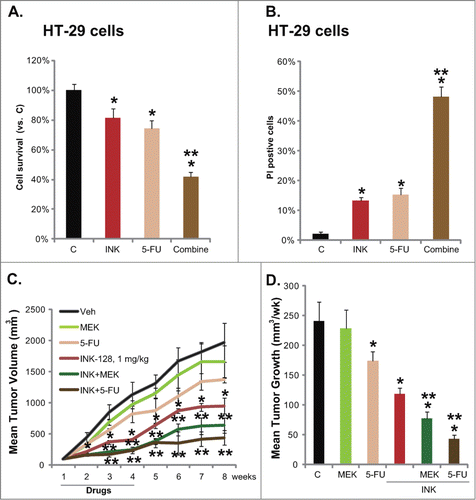
Five-fluorouracil (5-FU)-based adjuvant chemotherapy is widely used for the treatment of colon cancer. However, 5-FU resistance in the course of treatment is a common issue. Therefore, 5-FU sensitization strategies need to be developed.Citation20 We thus tested the potential role of INK-128 in 5-FU-induced cytotoxicity in HT-29 cells. Results in and showed that 5-FU (5 μM) only induced minor viability decrease and death of HT-29 cells. Significantly, co-administration with a low concentration of INK-128 (5 nM) dramatically enhanced 5-FU-induced cytotoxicity ( and ). Further, INK-128 enhanced 5-FU-meditated inhibition on HT-29 growth in mice xenograft ( and ). Note that oral INK-128 (1 mg/kg, p.o. daily) alone inhibited HT-29 xenograft growth ( and ), an effect that was further potentiated by MEK/ERK inhibitor MEK-162 ( and ). These results show that INK-128 sensitizes 5-FU-mediated anti-HT-29 activity in vivo and in vitro.
Figure 6. Enhancement of 5-FU-mediated anti-HT-29 cell activity by INK-128. HT-29 cells were treated with INK-128 (5 nM), or with 5-FU (5 μM) for 72 h, cell viability and cell death were tested by MTT assay (A) and PI staining assay (B), respectively. Mice bearing HT-29 tumors were administrated 3 weeks with: vehicle (“Veh”), INK-128 (1 mg/kg, p.o. daily), 5-FU (30 mg/kg, i.p. twice a week), MEK-162 (2.5 mg/kg, p.o. daily), INK-128 + 5-FU, or INK-128 + MEK-162, n = 8 per group. Mean Tumor Volume (in mm3, recorded every week) and average tumor growth (in mm3/week) were presented (C and D). Data were expressed as mean ± SD, experiments were repeated 3 times, and similar results were obtained. *P < 0 .05 vs. vehicle or “C” group. **P < 0 .05 vs. Five-FU only group.
Discussions
Our groupCitation16 among others have found that the mTOR signaling plays a vital role in controlling cancer cell growth, proliferation and survival, and is frequently dysregulated and over-activated in human colorectal cancers,Citation5,6,21 laying the foundation for the use of mTOR blockers as novel targeted anti-cancer agents. MTOR forms at least 2 structurally and functionally distinct multi-protein complexes including mTORC1 and mTORC2.Citation7,8 These 2 complexes have different subunits composition, downstream substrates and biological effects, which are both important for mTOR's functions.Citation7,8 Here we found that INK-128 disrupted mTOR-Raptor association and mTOR-Rictor-Sin1 association in cultured HT-29 cells (). Furthermore, INK-128 inhibited S6K, S6 and 4E-BP1 phosphorylation, it also inhibited Akt Ser 473 phosphorylation, while leaving Akt Thr-308 phosphorylation unaffected (). Thus, these data indicated that INK-128 inhibited both mTORC1 and mTORC2 assembly and activation in cultured colorectal cancer cells. Our results are similar to what reported by other groups.Citation11,22
Rapamycin, the well known inhibitor of mTOR, inhibits mTOR kinase activity by binding to the FKBP-12 (FK506-binding protein of 12 kDa), which forms a complex with C-terminus of mTOR.Citation23 Thus, it interferes exclusively with the kinase activity of the mTORC1, but not mTORC2. The limited pharmacological properties of rapamycin, such as its poor bioavailability and aqueous insolubility in water, prompted the preparation of synthetic analogs (rapalogs). However, several mechanisms of potential resistance to rapalogs have been identified, of which a negative feed-back loop activation of AktCitation24 and ErkCitation14 have been reported in preclinical and clinical settings.Citation22,25 Thus, ATP-competitive kinase inhibitors of mTOR, or the the second generation mTOR inhibitors, have been developed.Citation22 These kinase inhibitors not only block mTORC1 activation, but also interfere with mTORC2 and Akt activation.Citation12,22 Here we found that INK-128 not only blocked mTORC1 activity, it also decreased Akt activity as it inhibited Akt Ser 473 phosphorylation (). Meanwhile, it had no significant effect on Erk activation (), and Erk inhibition by MEK-162 was found to increase INK-128s cytotoxicity. Thus, these results suggest that INK-128 and possible other ATP-competitive mTOR kinase inhibitors should have higher efficiency than rapamycin or rapalogs against colorectal cancer cells. As a matter of fact, INK-128 efficiently inhibited colorectal cancer cell growth both in vitro ( and ) and in vivo (). In consistent with these findings, a recent study by Li et a., showed that AZD8055, another dual mTORC1 and mTORC2 inhibitor, suppressed head and neck squamous cell carcinoma cell growth in vivo and in vitro, and its efficiency was better that rapamycin.Citation26
MTOR activation is also known important for cell migration. For example, mTORC1 mediates intestinal cell migration and plays a role in intestinal repair.Citation27 Rapamycin has found to inhibit vascular smooth muscle cell migration.Citation28 Liu et al., found that TNFα-induced cell migration also requires mTORC1 activation.Citation29 S6K was co-localized to the actin protein arc locating at the leading edge of migrating cells, directing cell migration.Citation30 Our previous study has shown that mTORC1 activation is important for FSCN1 expression and colorectal cancer migration. Inhibition of mTORC1 by rapamycin, AMPK activation or microRNA-451 suppresses FSCN1 expression and cell migration. In the current study, we found that INK-128 also inhibited FSCN1 expression and colorectal cancer cell in vitro migration. Furthermore, INK-128 induced E-Cadherin downregulation in HT-29 cells, which might also be responsible for migration inhibition.
Chemotherapy is currently used to reduce tumor recurrence and prolong postoperative survival in colorectal cancer patients.Citation20 5-FU remains the most widely-used chemotherapeutic drug for colorectal cancers.Citation20 Its cytotoxicity to tumor cells is associated with the mis-incorporation of fluoronucleotides into RNA and DNA and the inhibition of the nucleotide synthetic enzyme thymidylate synthase.Citation20 The limited response rate for 5-FU as a single agent in advanced colorectal cancer treatment has led to the alternative modulation strategies.Citation20 Studies have shown that mTOR inhibition could increase 5-FU-mediated cytotoxicity.Citation31-33 In the current study, we found that INK-128 sensitized 5-FU-induced anti-HT-29 cell activity both in vivo and in vitro, indicating that INK-128 could be developed as an 5-FU adjuvant.
In summary, our results show that INK-128, the novel ATP-competitive kinase inhibitor of mTOR, blocks both mTORC1 and mTORC2 activation in colorectal cancer cells. INK-128 inhibits colorectal cancer cell growth, survival and migration, and induces both apoptotic and non-apoptotic cancer cell death. Further, it sensitizes 5-FU-mediated anti-HT-29 activity in vivo and in vitro. Our preclinical studies revealed that INK-128 might be investigated for colorectal cancer treatment in clinical trials.
Material and Methods
Cell culture
As previously describedCitation16, colorectal cancer HT-29 and HCT-116 cells were maintained in a RPMI medium (Sigma, St. Louis, MO), with a 10% FBS (Sigma, St. Louis, MO), Penicillin/Streptomycin (1:100, Sigma), and in a CO2 incubator at 37°C.
Primary colon cancer cell isolation and culture
Fresh malignant colonic mucosal specimens from patients were thoroughly washed in PBS containing 100 units/ml penicillin–streptomycin and 2 mM DTT (wash buffer) to remove debris and then minced by scalpel into small pieces into DMEM containing 100 units/ml penicillin–streptomycin. Colon cancer cell pellets were thoroughly washed, then re-pelleted at 400 g for 5min. Single-cell suspensions of malignant colon cancer cells were achieved by re-suspending cells in 0.15% (w/v) collagenase dissolved in DMEM and incubating the suspension at 37°C and 5% CO2. After 1 h, individual cell was pelleted and rinsed twice with DMEM before re-suspending the cell pellets in cell culture medium (DMEM, 15% FBS, 10 mg/ml transferrin, 2 mM glutamine, 1 mM pyruvate, 10 mM HEPES, 100 units/ml penicillin/streptomycin, 0.1 mg/ml gentamicin, 0.2 units/ml insulin, 0.1 mg/ml hydrocortisone, and 2 g/liter fungizone). The study was approved by the institutional review board of all authors’ institutions, and written informed consent was obtained from all individual participated patients. All clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki.
Chemicals and reagents
INK-128 and MEK-162 were obtained from Selleck (Shanghai, China). Five-fluorouracil (5-FU) was purchased from Sigma (St Louis, MO).Anti-FSCN1, Erk1, Akt1, mTOR, Raptor, Sin1, E-Cadherin, β-actin, Rictor and tubulin antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). All other kinase antibodies used in this study were obtained from Cell Signaling Technology (Shanghai, China).
“Transwell” migration assay
As previously reported,Citation16 the “Transwell” assay was performed using modified Corning chambers (12.0-μm pore size, Corning, Lowell, MA). The lower chamber was filled with 600 μl of RPMI medium containing 20% FBS. Cells were harvested with trypsin/EDTA, resuspended to 2.5 × 105 cells/ml and added to the upper chamber. The cells were allowed to migrate in 37°C at 5% CO2 for 36 h. INK-128 was added to the both upper and lower chambers. Non-migratory cells at the upper surface of the membrane were removed and the number of migrant cells attached to the lower surface was monitored and counted under microscopy. The number of migrated cells in at least 8 random views from 4 repeat chambers was counted. Mitomycin C (5 μg/mL) was always to both upper and lower chambers.
Western blots
Cells with indicated treatments were harvested in a buffer containing 20 mM Tris-HCl (pH 7.8); 137 mM NaCl; 15% glycerol; 1% Triton X-100; 20 mM NaF; 10 mM sodium pyrophosphate; 25 mM β-glycerophosphate, and 1 mM phenylmethylsulfonyl fluoride. Protein concentrations were determined by Bio-Rad protein assay (Bio-Rad, Beijing, China). Aliquots of 30–40 μg of lysates were electrophoresed on 10% SDS-PAGE and transferred to PVDF membranes. Western blots were carried out with primary antibodies at 4°C overnight. Appropriate secondary antibodies conjugated to horseradish peroxidase (HRP) were then added for 1.5 h. Antigen-antibody complex was detected by using enhanced chemiluminescence (ECL) reagent (Amersham Biosciences, Piscataway, NJ). All Western blots in this study were subjected to different exposures: from 10 seconds to 10 minutes, the best exposures were selected for data presentation. The blot's intensity was quantified by ImageJ software (NIH) after normalization to the corresponding loading controls. And the quantification number was expressed as fold change vs the band labeled with “1.00.”
The MTT cell survival assay
The cell survival was assessed using the MTT (Sigma) assay. In brief, cells were collected and seeded in 96-well plates at a density of 1 × 10Citation5 cells/well in 200 ml of culture medium (containing 1% FBS). After treatment, 20 μl of MTT (5 mg/ml) solution was added to each well for 4 hrs at 37°C, the cell viability was determined by measuring absorbance at 490 nm using a microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA), OD value of treatment group was normalized to that of untreated control group.
The apoptosis assay
Cell apoptosis was measured by Annexin-V FACS according to the manufacturer's protocol (BD PharMingen). Briefly, after treatment, cells were washed twice with PBS and incubated in 300 μL binding buffer containing 3 μL of Annexin V-FITC and 2 μL of propidium iodide (PtdIns) in the dark for 15 min at room temperature. The stained samples (containing 250,000 cell/sample) were then analyzed on a FACScalibur flow cytometer within 1 hr following the manufacturer's protocol (BD PharMingen).
Cell growth “clonogenicity” assay
After treatment, cancer cells were suspended in 1 mL of DMEM containing 0.25% agar (Sigma), 10% FBS and indicated treatments. The cell suspension was then added on the top of a pre-solidified 100 mm culture dish. The drug-containing medium was switched every 2 d After 7 d of incubation, the remaining survival large colonies (>50 μm in diameter) were manually counted, and the number was normalized to that of control group.
Co-immunoprecipitation (Co-IP) assay
After treatment, cells growing in 10 cm diameter dishes were rinsed once with cold PBS and lysed on ice for 20 min in 1 ml of ice-cold buffer A (40 mM HEPES [pH 7.5], 120 mM NaCl, 1 mM EDTA, 10 mM pyrophosphate, 10 mM glycerophosphate, and EDTA-free protease inhibitors [Roche]) containing 0.3% CHAPS in order to preserve the integrity of the complexes. To the cleared lysates, 2 μg of mTOR (Santa Cruz) antibody was added per 1.2 mg of soluble proteins, and immune complexes were allowed to form by incubating with rotation for 24 hs at 4°C. A 50% slurry (30 μl) of protein A/G-Sepharose was then added, and the incubation continued for 1 h. Immunoprecipitates captured with protein A/G -Sepharose were washed 4 times with CHAPS-containing buffer and analyzed by Western blot as described.
HT-29 xenograft
The in vivo experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC). Male BALB/c nu/nu nude mice at 6 weeks of age were injected subcutaneously with one million HT-29 cells grown in RPMI +10% FBS medium. Drug treatment was initiated 10 d after tumor cell inoculation, when established tumors were around 0.5 cm in diameter. Mice were randomized to following 6 groups (Vehicle control; INK-128, 1 mg/kg, p.o., daily; MEK-162, 2.5 mg/kg, p.o. daily; 5-FU, 30 mg/kg, i.p. twice a week; INK-128 + 5-FU and INK-128 + MEK-162) (n = 8 per group). The drug dose administrated and the duration were based on pilot studies and published literatures. Drugs were given for a total of 3 weeks. Tumor size were measured weekly by the modified ellipsoid formula: (π / 6) × AB2, and A is the longest and B is the shortest perpendicular axis of an assumed ellipsoid corresponding to tumor mass.
Statistical analysis
In each experiment, a minimum of 3 wells/dishes were used. Each experiment was repeated a minimum of 3 times. All data were normalized to control values of each assay and were presented as mean ± standard deviation (SD). Data were analyzed by one-way ANOVA followed by a Scheffe's f-test by using SPSS software (SPSS Inc., Chicago, IL, USA). IC-50 was also calculated by SPSS software. Significance was chosen as P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
972274_Supplementary_Materials.zip
Download Zip (3.1 MB)Funding
This work was supported by the National Natural Science Foundation (nos. 81101223, 81472305), and the Foundation of Xuzhou science and Technology Bureau (no. XM12B046). The founders have no roles in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
References
- Schmoll HJ, Stein A. Colorectal cancer in 2013: Towards improved drugs, combinations and patient selection. Nat Rev Clin Oncol 2014; 11:79-80. PMID:24445520.
- Kuipers EJ, Rosch T, Bretthauer M. Colorectal cancer screening–optimizing current strategies and new directions. Nat Rev Clin Oncol 2013; 10:130-42. PMID:23381005.
- Brouquet A, Nordlinger B. Metastatic colorectal cancer outcome and fatty liver disease. Nat Rev Gastroenterol Hepatol 2013; 10:266-7. PMID:23567218.
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9-29. PMID:24399786.
- Ekstrand AI, Jonsson M, Lindblom A, Borg A, Nilbert M. Frequent alterations of the PI3K/AKT/mTOR pathways in hereditary nonpolyposis colorectal cancer. Fam Cancer 2010; 9:125-9. PMID:19731079.
- Zhang YJ, Dai Q, Sun DF, Xiong H, Tian XQ, Gao FH, et al. mTOR signaling pathway is a target for the treatment of colorectal cancer. Ann Surg Oncol 2009; 16:2617-28. PMID:19517193.
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell 2007; 12:9-22. PMID:17613433.
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 2006; 6:729-34. PMID:16915295.
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93. PMID:22500797.
- Sun SY. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett 2013; 340:1-8. PMID:23792225.
- Garcia-Garcia C, Ibrahim YH, Serra V, Calvo MT, Guzman M, Grueso J, et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin Cancer Res 2012; 18:2603-12. PMID:22407832.
- Garcia-Echeverria C. Allosteric and ATP-competitive kinase inhibitors of mTOR for cancer treatment. Bioorg Med Chem Lett 2010; 20:4308-12. PMID:20561789.
- Freudlsperger C, Burnett JR, Friedman JA, Kannabiran VR, Chen Z, Van Waes C. EGFR-PI3K-AKT-mTOR signaling in head and neck squamous cell carcinomas: attractive targets for molecular-oriented therapy. Expert Opin Ther Targets 2011; 15:63-74. PMID:21110697.
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008; 118:3065-74. PMID:18725988.
- Ma Y, Machesky LM. Fascin1 in carcinomas: Its regulation and prognostic value. Int J Cancer 2014. PMID:25302416.
- Chen MB, Wei MX, Han JY, Wu XY, Li C, Wang J, et al. MicroRNA-451 regulates AMPK/mTORC1 signaling and fascin1 expression in HT-29 colorectal cancer. Cell Signal 2014; 26:102-9. PMID:23899558.
- Hu M, Xia M, Chen X, Lin Z, Xu Y, Ma Y, et al. MicroRNA-141 regulates Smad interacting protein 1 (SIP1) and inhibits migration and invasion of colorectal cancer cells. Dig Dis Sci 2010; 55:2365-72. PMID:19830559.
- Fabre M, Garcia de Herreros A. Phorbol ester-induced scattering of HT-29 human intestinal cancer cells is associated with down-modulation of E-cadherin. J Cell Sci 1993; 106 (Pt 2):513-21. PMID:8282758.
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem 2000; 275:36803-10. PMID:10969078.
- Chung KY, Saltz LB. Adjuvant therapy of colon cancer: current status and future directions. Cancer J 2007; 13:192-7. PMID:17620769.
- Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene 2006; 25:6436-46. PMID:17041628.
- Vilar E, Perez-Garcia J, Tabernero J. Pushing the envelope in the mTOR pathway: the second generation of inhibitors. Mol Cancer Ther 2011; 10:395-403. PMID:21216931.
- Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol 1993; 229:105-24. PMID:7678431.
- O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66:1500-8. PMID:16452206.
- Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget 2011; 2:135-64. PMID:21411864.
- Li Q, Song XM, Ji YY, Jiang H, Xu LG. The dual mTORC1 and mTORC2 inhibitor AZD8055 inhibits head and neck squamous cell carcinoma cell growth in vivo and in vitro. Biochem Biophys Res Commun 2013; 440:701-6. PMID:24103749.
- Rhoads JM, Niu X, Odle J, Graves LM. Role of mTOR signaling in intestinal cell migration. Am J Physiol Gastrointest Liver Physiol 2006; 291:G510-7. PMID:16710051.
- Poon M, Marx SO, Gallo R, Badimon JJ, Taubman MB, Marks AR. Rapamycin inhibits vascular smooth muscle cell migration. J Clin Invest 1996; 98:2277-83. PMID:8941644.
- Liu Y, Cao GF, Xue J, Wan J, Wan Y, Jiang Q, et al. Tumor necrosis factor-alpha (TNF-alpha)-mediated in vitro human retinal pigment epithelial (RPE) cell migration mainly requires Akt/mTOR complex 1 (mTORC1), but not mTOR complex 2 (mTORC2) signaling. Eur J Cell Biol 2012; 91:728-37. PMID:22595285.
- Berven LA, Willard FS, Crouch MF. Role of the p70(S6K) pathway in regulating the actin cytoskeleton and cell migration. Exp Cell Res 2004; 296:183-95. PMID:15149849.
- Bu X, Le C, Jia F, Guo X, Zhang L, Zhang B, et al. Synergistic effect of mTOR inhibitor rapamycin and fluorouracil in inducing apoptosis and cell senescence in hepatocarcinoma cells. Cancer Biol Ther 2008; 7:392-6. PMID:18075305.
- Lee KH, Hur HS, Im SA, Lee J, Kim HP, Yoon YK, et al. RAD001 shows activity against gastric cancer cells and overcomes 5-FU resistance by downregulating thymidylate synthase. Cancer Lett 2010; 299:22-8. PMID:20727673.
- Punt CJ, Boni J, Bruntsch U, Peters M, Thielert C. Phase I and pharmacokinetic study of CCI-779, a novel cytostatic cell-cycle inhibitor, in combination with 5-fluorouracil and leucovorin in patients with advanced solid tumors. Ann Oncol 2003; 14:931-7. PMID:12796032.