
Abstract
Gemcitabine based treatment is currently a standard first line treatment for patients with advanced pancreatic cancer, however overall survival remains poor, and few options are available for patients that fail gemcitabine based therapy. To identify potential molecular targets in gemcitabine refractory pancreatic cancer, we developed a series of gemcitabine resistant (GR) cell lines. Initial drug exposure selected for an early resistant phenotype that was independent of drug metabolic pathways. Prolonged drug selection pressure after 16 weeks, led to an induction of cytidine deaminase (CDA) and enhanced drug detoxification. Cross resistance profiles demonstrate approximately 100-fold cross resistance to the pyrimidine nucleoside cytarabine, but no resistance to the same in class agents, azacytidine and decitabine. GR cell lines demonstrated a dose dependent collateral hypersensitivity to class I and II histone deacetylase (HDAC) inhibitors and decreased expression of 3 different global heterochromatin marks, as detected by H4K20me3, H3K9me3 and H3K27me3. Cell morphology of the drug resistant cell lines demonstrated a fibroblastic type appearance with loss of cell-cell junctions and an altered microarray expression pattern, using Gene Ontology (GO) annotation, consistent with progression to an invasive phenotype. Of particular note, the gemcitabine resistant cell lines displayed up to a 15 fold increase in invasive potential that directly correlates with the level of gemcitabine resistance. These findings suggest a mechanistic relationship between chemoresistance and metastatic potential in pancreatic carcinoma and provide evidence for molecular pathways that may be exploited to develop therapeutic strategies for refractory pancreatic cancer.
Abbreviations
| HDAC | = | histone deacetylase |
| qRT-PCR | = | Quantitative reverse transcription-polymerase chain reaction |
| SAHA | = | suberoylanilide hydroxamic acid |
| CDA | = | cytidine deaminase |
Introduction
Pancreatic cancer is the fourth leading cause of cancer death in the United States with a 5 year survival rate of less than 5% and a median survival of approximately 6-8 months from the time of diagnosis.Citation1,2 The standard of care for patients with localized disease is surgical resection followed by adjuvant treatment with the nucleoside analog, gemcitabine. However, the majority of these individuals will develop recurrent disease that is resistant to further therapy. Only 15-20% of patients are eligible for resection due to the presence of locally advanced or metastatic tumor at diagnosis, suggesting early systemic progression of the disease. Treatment options for individuals with advanced or recurrent disease include gemcitabine, gemcitabine combinations or fluoropyrimidine based combinations such as FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan and oxaliplatin.) These combination therapies generally provide only modest improvements in survival, and the majority of patients die with refractory disease. Understanding the mechanisms of drug resistance to therapies such as gemcitabine is a key factor for improving pancreatic cancer prognosis.
Drug resistance can be either de novo (innate) or acquired, and multiple mechanisms of gemcitabine resistance have been identified.Citation3-6 Gemcitabine is a deoxycytidine (dCyd) analog that requires active cell membrane transport. Once inside the cell, the difluorinated pro-drug undergoes mono-, di-, and tri-phosphorylation before incorporation into DNA, where it causes masked chain termination. Some experimental models have identified acquired resistance to gemcitabine associated with reduced expression or activity of enzymes required for gemcitabine metabolism including hENT1, deoxycytidine kinase (dCK), and ribonucleotide reductase (RR) or increased activity of the detoxifying enzyme, cytidine deaminase (CDA).Citation3 And although low basal expression of hENT1 has been correlated with poor clinical response in newly diagnosed patients,Citation7,8 the relevance of the gemcitabine metabolic pathway in patients with relapsed and refractory disease is not clear, and options for patients with gemcitabine resistant disease remain limited.
Genetic and genomic analysis of pancreatic cancer has demonstrated extensive inter- and intra-tumoral heterogeneity influenced by dense desmoplastic stroma.Citation9-11 In addition to the cancer stem cell (CSC) model, in which a subset of cells display the tumor initiating and chemoresistant phenotype, recent studies have identified 3 transcriptionally defined subtypes of pancreatic adenocarcinoma with different biological characteristics and subtype specific drug responses.Citation12,13 Further heterogeneity can be imposed by extrinsic factors such as the microenvironment,Citation14 which can provide physical protection or transduction of survival signals. These observations demonstrate the need to identify mechanisms of adaptive resistance that support the emergence of the resistant phenotype.
In the present study, a series of gemcitabine-resistant cell lines is used to investigate the mechanisms by which adaptive resistance occurs during pancreatic tumor progression. Characterization of low level (GR300) resistant cells demonstrated no significant changes in gemcitabine metabolic pathways, however prolonged drug exposure leads to cytidine deaminase (CDA) overexpression. The gemcitabine resistant derivatives significantly change other phenotypic features as indicated by a fibroblastic cell morphology, and an invasive phenotype that is increased with increasing drug concentration. Remarkably, the GR cells display dose dependent hypersensitivity to histone deacetylase (HDAC) inhibitors and loss of heterochromatin markers. Taken together, the data suggests that the drug resistant phenotype that emerges is associated with epigenetic reprogramming toward more aggressive disease. Targeting the phenotypic switch could be exploited to enable new therapeutic options for patients with advanced pancreatic adenocarcinoma.
Results
Gemcitabine resistance is a stable phenotype in vitro and in vivo
To investigate mechanisms of gemcitabine resistance, the MiaPaCa-2 (MP) cell line was selected for survival under continuous exposure to gemcitabine. Selection was initiated at 20 nM gemcitabine, which corresponds to the approximate IC50 as determined by 72 hr MTT assay (). After 16 weeks of continuous incubation with increasing concentrations of gemcitabine, a drug resistant population emerged under continuous exposure to 90 nM gemcitabine. Resistance gradually increased with increasing cytotoxic drug selection pressure, and the phenotype was considered stable when cells grown in the presence of 200 nM gemcitabine maintained resistance after being removed from drug for over 16 passages. Increasing drug selection pressure resulted in the establishment of populations that proliferated in the continuous presence of 300 nm (GR300), 800 nM (GR800), or 2 μM (GR2000) gemcitabine. As shown in , the GR derivatives demonstrate a threshold effect with no significant growth inhibition at concentrations of gemcitabine less than approximately 1 μM. Thymidine incorporation demonstrated a non-significant trend of slightly increased proliferative rate in the GR cell lines compared to the parental cells, which was not affected by the presence or absence of gemcitabine (data not shown).
Figure 1. In vitro and in vivo growth inhibition of gemcitabine resistant cell lines. (A) Colorimetric 3-day MTT assay was used to determine the growth inhibition of MiaPaCa (MP) cells and 3 gemcitabine-resistant (GR) derivatives. Cells were incubated for 72 hours with increasing concentrations of gemcitabine. Metabolism of tetrazolium dye to formazan was used to assay cell viability. (B) Subcutaneous tumors were established in SCID mice followed by treatment with gemcitabine (180 mg/kg) q 4 days. Treatment was initiated when the mean tumor volume was 50 mm3, and tumor volume was measured every 3 days. The study was terminated when tumor burden exceeded 20% of total body weight, as per IACUC regulations.
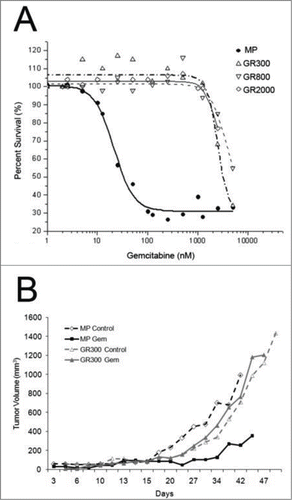
Many studies have demonstrated that in vitro chemoresistance often is not recapitulated by the in vivo phenotype in experimental systems and vice versa.Citation15 To determine if the GR cell lines would maintain the gemcitabine resistant phenotype in vivo, subcutaneous tumors were established in SCID mice. Animals were stratified into equivalent treatment groups, and treatment was initiated on day 12 when mean tumor volume was approximately 50 mm3. Growth of the parental MP tumors was significantly inhibited by 180 mg/kg gemcitabine, while the GR tumors failed to respond to treatment ().
Initial gemcitabine resistance is not associated with alterations in drug metabolism
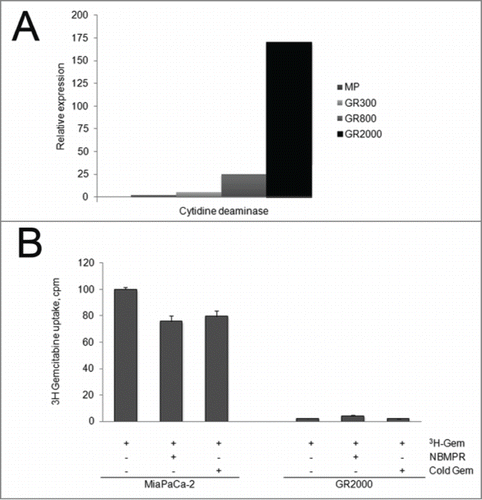
Previous studies have identified increased expression of the gemcitabine metabolic enzyme cytidine deaminase (CDA) as a mechanism of acquired resistance to cytidine nucleosides.Citation3 As shown in , qRT-PCR demonstrated no change in the expression of CDA in the low level resistant (GR300) cells. With prolonged exposure to increasing drug concentrations, there is a significant induction of CDA mRNA in the GR800 and GR2000 cells with 25 and 150 fold increase, respectively, compared to the parental MP cell line. In correlation with the highly induced CDA expression, [3H]-gemcitabine drug accumulation was dramatically reduced in the GR2000 cells compared to the parental MP cells (). The small molecule inhibitor of hENTs S-(4-Nitrobenzyl)-6-thioinosine (NBMPR) was used as a control for nucleoside transport of [3H]-gemcitabine.
Figure 2. Cytidine deaminase expression and gemcitabine accumulation. (A) RNA was extracted from serially selected GR cell lines, and qRT-PCR was used to determine the relative expression of cytidine deaminase mRNA. The 18 S ribosome was used to as an internal standard. (B) Parental sensitive (MP) or resistant cells (GR2000) were incubated with 3H-gemcitabine in the presence or absence of NBMPR or non-radioactive competitor. Cellular accumulation as quantitated by scintillation counting of whole cell lysates, and normalized to cell number. Data are expressed as cpm, Mean + SEM, n = 3. ANOVA was used to determine significance.
Cross resistance and collateral sensitivity of the gemcitabine resistant phenotype
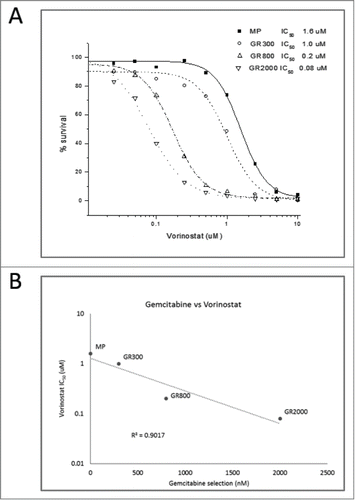
Growth inhibition assays were used to quantitate cross-resistance and collateral sensitivity to additional therapeutic agents. Cross resistance profiles of GR300 demonstrate approximately 100-fold resistance to the pyrimidine nucleoside cytarabine, but no resistance to the same-in-class agents, azacytidine and decitabine (). Similarly, the GR300 cell line displays 20-fold and 10-fold cross resistance to the purine nucleosides clofarabine and cladribine, respectively, but no differential response to fludarabine. No significant difference in response was seen with DNA damaging agents such as doxorubicin and irinotecan, or microtubule targeting agents including vinorelbine, vincristine, and paclitaxel. Interestingly, the gemcitabine resistant derivatives demonstrated a dose dependent hypersensitivity to the Class I HDAC inhibitor suberoylanilide hydroxamic acid (SAHA/Vorinostat) (IC50: MP 1.6 uM; GR300 1.0 uM; GR800 0.2 uM; GR2000 0.08 uM) (). This collateral hypersensitivity extended to additional Class I HDAC inhibitors including MS-275 (Entinostat) and Trichostatin A, but not to the Class IIa selective agent MC1568 (data not shown). Also striking is the observation that hypersensitivity to the HDAC inhibitors is a progressive phenotype that is increased by incubation with higher concentrations of gemcitabine, with a clear inverse correlation (). Combination treatment with gemcitabine plus HDAC inhibitors had no effect on gemcitabine response in the GR cells under any dosing schema investigated, including pre-treatment (1, 10 or 100 nm, 1 hr, 24 hrs, or 3 days), concurrent treatment, or addition of the HDAC inhibitor after gemcitabine (data not shown).
Table 1. Resistance profile comparison of parental (MP) cells with gemcitabine resistant (GR300) cells. Values are IC50 as determined by 72 hr MTT assay
Figure 3. Collateral hypersensitivity of GR cells to Vorinostat. (A) Colorimetric 3-day MTT assay indicated an increased sensitivity to Vorinostat in GR cell lines (MP<GR300<GR800<GR2000). (B) Vorinostat IC50 vs gemcitabine concentration used for continuous maintenance of GR cells.
Gene expression changes in GR cells
To elucidate changes in gene expression that may contribute to the initial gemcitabine resistant/vorinostat sensitive phenotype, the Affymetrix U133A 2.0 gene chip was used to compare the parental MP cells to the early resistant GR300. Of the gene probes examined, 479 genes were differentially expressed in the GR300 cells compared to the parental MP cells. Three hundred-thirty five genes were significantly increased, while one hundred 144 were significantly decreased. The complete gene set is available as supplemental data. In agreement with functional assays () there were no significant differences in the expression of gene products associated with gemcitabine metabolism either by microarray analysis or validation qRT-PCR (). With increasing drug concentration over extended time, changes in gemcitabine metabolic genes began to emerge. For example, microarray analysis and validation qRT-PCR demonstrated a slight (non-significant) change in the expression of the detoxifying enzyme, cytidine deaminase (CDA) in the GR300 derivative, whereas CDA was amplified to nearly 150 fold in the GR2000 derivative compared to the parental MP cell line.
Table 2. Fold change in mRNA expression in GR200 and GR2000 cell lines compared to parental MP cell line
To identify functional processes that might contribute to the drug resistant phenotype, gene enrichment analysis based on Gene Ontology (GO) annotation was applied to the microarray expression data.Citation16,17 In this analysis, genes are ranked according to differential expression between the drug sensitive cells vs the drug resistant cells. GO annotation was used to identify genes that are over-represented within 3 domains, Biological Process, Cellular Component, or Molecular Function. The most significant Biological Process identified was lipid transport (p = 1.5 e-05) followed by cell adhesion, and cell differentiation (). Cell-cell junctions and the extracellular space were identified as significantly changed Cellular Components in the GR cells; oxidoreductase activity and calmodulin binding were significantly changed in the Molecular Function domain. Enrichment of genes associated with nucleoside metabolism (GO:0009116), programmed cell death pathways (GO:0008219), or epithelial to mesenchymal transition (EMT) (GO:0001837), were not present.
Table 3. Gene Ontology domains identified as significantly changed in GR200 cells compared to parental MP cells
GR cells acquire fibroblastic morphology and an invasive phenotype
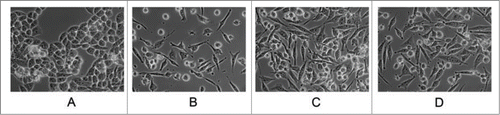
In accordance with the gene expression analysis, one of the most striking features of the gemcitabine resistant cell lines was the change in cellular morphology (). While the parental MP cells display a highly colonized morphology with tight junctions and a cobblestone appearance, the GR300, GR800, and GR2000 cell lines exhibit a spindle-like appearance with loss of cell-cell contact. These morphological features are frequently associated with a fibroblast-like or EMT-like phenotype. However no significant differences were seen in well-established markers of the classic EMT-like phenotype including N-cadherin, E-cadherin, vimentin, or β-catenin (Sup. ). Similarly, no changes were noted in known EMT-associated transcriptional factors such as SNAIL, Twist, ZEB1 or NOTCH signaling elements (data not shown).
Figure 4. Morphology of MP and GR cells by phase contrast microscopy. MiaPaCa-2 cells were selected for increased levels of gemcitabine resistance. Gemcitabine-resistant (GR) cells were derived using incrementally increased levels of gemcitabine (A) MP; (B) GR300; (C) GR800; (D) GR2000.
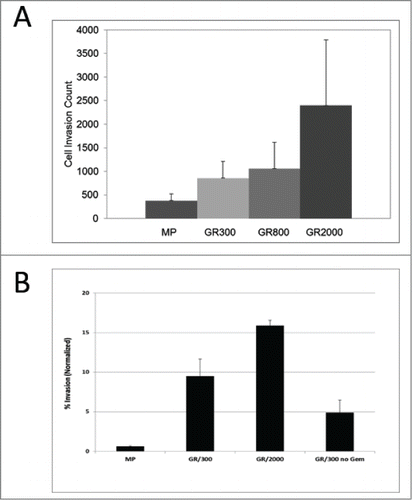
To investigate the functional effects of the fibroblastic morphology, MP and GR cells were assayed for their abilities to invade through extracellular matrix (ECM). Cells were added to Matrigel coated invasion chambers in serum free media with complete media in the lower chamber, and allowed to invade through the ECM for 24 hours. The GR cells were significantly more invasive than the parental MP cells, with an increase in the number of cells recovered from the underside of the Matrigel coated insert that was directly proportional to the level of gemcitabine resistance (MP<GR300<GR800<GR2000) (). In the initial characterization of the GR cells, the stability of the drug resistant phenotype was established by growing cells in the absence of drug selection pressure for up to 16 weeks. GR cells grown in the absence of drug maintained the resistant phenotype (data not shown). To determine if co-selection for the invasive phenotype was a similarly stable characteristic, GR300 cells removed from gemcitabine for 16 weeks were assayed for invasive capacity. As shown in , in the absence of gemcitabine selection pressure, the invasive phenotype of the GR300 cells begins to revert, nearing the level of the parental phenotype.
Figure 5. Matrigel invasion. (A) Cells maintained under continuous gemcitabine selection pressure were analyzed for invasive capacity. Cells were allowed to invade through a Matrigel coated membrane, stained with Wright-Giemsa stain and counted by light microscopy. Data represent 5 independent experiments. (B) Parental MP, GR300 cells maintained under continuous gemcitabine selection GR300 cells that had been removed from gemcitabine for 12 weeks. Cells were allowed to invade through a Matrigel coated membrane, and cells remaining on top of the membrane were removed. Cells in the lower chamber were stained with Calcein-AM, and fluorescence intensity was quantitated. Data are normalized to a Calcein-AM standard curve prepared for each cell type. Data represent 3 independent multi-well plate experiments.
The GR phenotype is associated with changes in chromatin structure
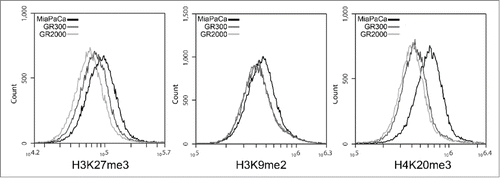
The absence of gene expression signatures associated with the EMT-like phenotype or altered nucleoside metabolism suggested that an epigenetic phenomenon may be responsible for the complex GR cell phenotype. This hypothesis is further supported by the reversal of the invasive capacity, and the GR hypersensitivity to HDAC inhibitors. To investigate changes in the global chromatin structure, 3 markers of heterochromatin were examined in the MP and GR cells. H4K20me3 is an evolutionarily conserved mark of pericentric heterochromatin that is frequently localized to repressed transcriptional start sites. H3K9me3 and H3K27me3 are methyl marks associated with both constitutive and facultative heterochromatin, and are enriched at sites of DNA repair in heterochromatin. All 3 heterochromatin markers were reduced In the GR cells compared to the parental MP cells (). H4K20me3 demonstrated the greatest difference, with 27% reduction in the fluorescence intensity of the GR300 cells and 32% reduction in the GR2000. Similar dose dependent changes were seen in H3K27me3, and to a lesser extent, H3K9me2, demonstrating a progressive loss of heterochromatin structure with increasing drug selection pressure.
Figure 6. Heterochromatin markers. MP, GR300, and GR2000 cells were fixed in formaldehyde and permeabilized with Triton X-100 for staining with methylation specific antibodies, and analysis by flow cytometry. Peak Fluorescence intensity (PFI) values are as follows: H3K27me3: MP 10.0e5; GR300 8.3e5; GR2000 7.3e5. H3K9me2: MP 4.85e5; GR300 4.53e5; GR2000 4.49e5. H4K20me3: MP 6.5e5; GR300 4.6e5; GR2000 4.4e5.
Discussion
In spite of recent advances in systemic treatment of pancreatic adenocarcinoma (gemcitabine based or fluoropyrimidine based combinations) survival remains poor. Many clinical trials have failed to show a survival advantage for most combination therapies over gemcitabine mono-therapy, and there is a need for preclinical investigation of the molecular mechanisms involved in pancreatic cancer development and chemotherapeutic drug resistance. In this study, we present a model of gemcitabine resistance that demonstrates novel features of tumor progression: invasive potential, and collateral sensitivity to HDAC inhibitors.
One of the more frequent observations in previously published models of acquired drug resistance is the emergence of an EMT-like phenotype.Citation16,18-20 Other similar studies have proposed that the gemcitabine resistant population represents pancreatic cancer stem cells.Citation21 In our system, the GR cells display an EMT-like morphology, however, neither gene expression profiling nor analysis of well characterized protein markers provided evidence of classic EMT phenotype, or of cancer stem cells. This is in contrast to the L3.6pl gemcitabine resistant cell line, which was reported to acquire a classic EMT phenotype with activation of Notch signaling.Citation18,19 Subsequent reports from the same investigators identified miRNAs as potential regulatory elements in the drug resistant/invasive phenotype.Citation22 Similarly, Arumugam et al used global transcriptome analysis to identify characteristics associated with intrinsic drug resistance in 10 commonly used pancreatic cancer cell lines.Citation23 Rank ordering based on in vitro IC50 demonstrated a clear correlation of drug resistance with ZEB-1-dependent EMT. In the current study, gene ontology analysis identified significant changes in a large number of gene products that may contribute to the morphological features with an EMT-like phenotype, however there was no evidence for an EMT-genotype signature in the GR cells,Citation24 suggesting that drug response and metastatic potential may be regulated by an epigenetic mechanism such as chromatin structure or miRNA.Citation25
The phenotypic and molecular variability between the published gemcitabine resistant models may be reflective of tumor heterogeneity. Additional phenotypic plasticity may be imposed by the microenvironment both in in vitro model systems and in patient populations. One of the more interesting trends to emerge from gene ontology analysis of the GR cells was the apparent shift from functional groups that mediate laminin/basement membrane adhesion to components that contribute to collagen binding and cell-cell adhesions. Significant decreases were seen in the extracellular matrix proteins LamininA2, B4 and B1, and the hyaluronan and proteoglycan linker protein HAPLN1. In contrast, there were significant increases in subunits of collagens 6, and 13, and desmosome associated proteins such as Desmocollin, Desmoglein2, and Nectin-3. This shift in gene expression associated with the gemcitabine resistant and invasive phenotype may provide clues to the underlying mechanism of the desmoplastic and fibrotic clinical presentation of pancreatic adenocarcinoma. The drug resistant phenotype that emerges under different microenvironmental conditions may be an important tool in designing future therapeutic options.
Our findings identify a direct correlation between differential response to cytotoxic drugs and invasive potential. Cells that tolerate the highest concentration of gemcitabine display the greatest invasive potential. The phenomenon of co-selection for drug resistance and invasion is not limited to gemcitabine resistant pancreatic cells, as other investigators have reported an invasive phenotype associated with breast carcinoma cell lines selected for resistance to paclitaxel or doxorubicin.Citation26 More recently, Wu et al utilized a microfluidic platform to demonstrate biased cell migration toward higher drug concentrations, and increased proliferation of cells in the highest concentration of doxorubicin.Citation27 Taken together, these observations support the hypothesis that the cellular stress response can provide a temporal survival signal that will allow the emergence of a drug resistant population through epigenetic reprogramming.
Similarly, the most invasive cells demonstrate the greatest hypersensitivity to HDAC inhibitors. The clinical implications of these observations are twofold: first of all, in a subset of patients, prolonged treatment with anti-metabolites, such as in an adjuvant or post-surgical maintenance setting may be detrimental overall by promoting tumor progression and metastasis. More importantly, if the invasive, drug resistant phenotype is an induced reprogramming, as opposed to the selection of a pre-existing sub-population, then sequential treatment with different agents in a particular order may be more effective than concurrent administration of the same agents.
Although the underlying mechanism for the observed co-selection of gemcitabine resistance and invasive potential is not fully understood, our findings suggest that in this model, drug resistance and invasion are genotypically independent characteristics. This hypothesis is supported by several lines of evidence. First of all, the emergence of gemcitabine resistance demonstrates a threshold effect in which once the initial resistance is achieved, there is no significant increase in the IC50 with prolonged drug exposure. In contrast, the invasive phenotype displays a dose dependent increase. Secondly, the gemcitabine resistant phenotype is a highly stable characteristic that is maintained in the absence of selection pressure, whereas the invasive phenotype is transient and reverts to levels similar to the parental cells within 12 weeks.
Finally, our data support the findings of Sharma et al in which they suggest a metastable drug-tolerant state that is mediated by alterations in histone lysine methylation or acetylation.Citation28 Using PC9 non-small cell lung cancer cells, the investigators identified a subpopulation of drug-tolerant persistent cells with global chromatin alterations. The drug-tolerant population demonstrated an elevated expression of the histone demethylase KDM5A, and a reduction in the overall level of H3K4 methylation as well as an increased response to HDAC inhibitors. Similarly, in our system, the heterochromatin markers H3K27me3, H3K9me2, and H4K20me3 are reduced or lost in the GR cells in a dose dependent manner that parallels the hypersensitivity to HDAC inhibitors. The interdependence of histone methylation and acetylation in the DNA damage response may provide an important pre-emptive approach to therapy for patients with advanced pancreatic adenocarcinoma.
Materials and Methods
Cell lines and treatments
MiaPaCa-2 (MP) cells were obtained from the American Type Culture Collection (CRL-1420) and maintained in RPMI 1640 media (Mediatech Inc, cat no. 10-040-CV), with 10% fetal bovine serum (FBS) and 100 mM L-glutamine (Mediatech Inc.., cat no. 25-005-Cl). Multiple gemcitabine resistance clones, designated GR, were selected by incubating the parental MiaPaCa-2 cell line with increasing concentrations of gemcitabine. Continuous selection pressure via increasing doses of gemcitabine, resulted in GR300, GR800 and GR2000 derivatives, corresponding to the concentration of gemcitabine in which they were maintained in continuous culture; 300 nM, 800 nM and 2000 nM, respectively. The identity of all cell lines was confirmed by autosomal short tandem repeat (STR) analysis (University of Arizona Genomics Core) and screened for mycoplasma prior to use.
Chemicals
Gemcitabine was obtained from Chemie-tek (Cat no. 122111-03-9). All other chemicals were obtained from Sigma unless otherwise noted. For immunofluorescence staining: β-catenin (Cell Signaling, cat no. 9562), E-cadherin (Cell Signaling, cat no. 3195), N-cadherin (D-4) (Santa Cruz Biotechnology, cat no.8424), Vimentin (Cell Signaling, cat no. 3932), Alexafluor-488 conjugates (Life Technologies, cat no. A-21206, A-21202), Alexafluor-546 conjugate (cat no. A10040), Alexafluor-594 conjugate (cat no. A21203). [3H]gemcitabine was obtained from Moravek Biochemicals (Brea, CA).
Growth Inhibition Assay
MTT dye metabolism was used to assay cell viability as previously described.Citation29 All cross resistance assays were done both in the presence and absence of gemcitabine at the concentration used for maintenance. The IC50 is defined as the concentration of drug required to inhibit cell growth by 50% compared to untreated controls.
In Vivo tumor growth inhibition
Male SCID mice (approximately 6 weeks old) were provided by a breeding colony maintained by the University of Arizona Animal Care Facility. Mice were housed according to guidelines on the American Association for Laboratory Animal Care under protocols approved by the University of Arizona Institutional Animal Care and Use Committee (IACUC). Xenograft tumors were established by subcutaneous injection of 10×106 MP cells or 7.5×106 GR300 cells embedded in Matrigel (BD Biosciences). Animals were stratified into equal treatment groups when tumors were approximately 50 mm3. Gemcitabine (180 mg/kg) or vehicle control (0.9% saline) was administered for 3 cycles on days 1, 5, 9 with one week rest period. Tumor volumes were measured and animals were evaluated for general well-being every 3 days. The study was terminated when tumor volume exceeded 20% of total body weight.
Gene expression analysis
Total RNA for microarray analysis was prepared using the Qiagen RNeasy kit (cat no. 74104). RNA quantity and sample integrity was confirmed using an Agilent Bioanalyzer. All RNA samples scored an RNA integrity number (RIN) of 10. Triplicate RNA samples from MP and GR200 cells were assayed using Affymetrix U133A 2.0 Array Human Genome Gene chip (cat no. 900471) and analyzed on the Agilent/Affymetrix 2500A scanner. This gene chips included 22,000 probe sets covering 14,500 human genes. GR cells were treated for 24 h with 200 nM of gemcitabine prior to RNA isolation. Raw data was summarized to transcript-level signal and normalized using the GC-RMA algorithm as implemented in GeneSpring v7.0 (Silicon Genetics). Microarray expression data were analyzed using R programming environment (R Development Core Team, 2010).
Quantitative real-time PCR
Quantitative PCR (qRT-PCR) was done on an ABI 7700 (Life Technologies). RNA was isolated using Trizol (Life Technologies, cat no. 15596-018) and cDNA prepared using a BioRad iScript cDNA kit (170-8891). Amplification was done with Solaris PCR master mix (Thermo Scientific, cat no. AB-4351) and pre-designed PCR primers and probes (Thermo Scientific). Quantitation was done using the comparative CT method based on a standard curve for each gene analyzed. Data was normalized using the 18S rRNA housekeeping gene.
3H-Gemcitabine accumulation assay
Cells were incubated for 2 h with 50 nM of 3H-gemcitabine (11.0 Ci/mmol), harvested, and total cellular accumulation of 3H-gemcitabine measured. One hundred nM of 4-nitrobenzyl-6-thioinosine (NBMPR) or 250 nM of cold gemcitabine was added 30 m prior to the addition of 3H-gemcitabine. Data are expressed as counts per minute (cpm). Data shown is mean ± SEM (n = 3).
Matrigel invasion Assay
Matrigel invasion was assayed following the manufacturer's instructions (BD Biosciences, cat no. 354480). Cells were plated at a seeding density of 50,000 cells in serum-free media, onto 8 μM PET membranes coated with matrigel. Cells were allowed to migrate through the matrigel for 24 hours. After 24 hours, non-invading cells were removed from the top of the insert, and cells on the underside of the membrane were either fixed in ice cold methanol and stained with Wright-Giemsa for direct counting, or incubated with Calcein AM for 1 hr prior to analysis by fluorescent plate reader. Data shown are the mean of 5 independent assays.
Immunofluorescence staining
For immunofluorescence analysis, MP or GR cells were plated on ECM coated coverslips until approximately 70% confluent, rinsed with PBS, and fixed in 4% formaldehyde. Cells were permeabilized using 0.1% Triton X-100 in PBS containing 1% BSA. Cells were incubated with primary antibodies diluted according to manufacturer guidelines in PBS containing 1% BSA. After incubation, coverslips were washed with PBS and incubated with secondary Alexafluor conjugates and DAPI. Coverslips were dried and mounted on slides using ProLong Gold antifade (Life Technologies, cat no. P36930). For histone methylation assay, cells were harvested with trypsin/EDTA, resuspended in ice cold PBS, and fixed and stained using the same protocol, and counterstained with 7-AAD prior to analysis by flow cytometry.Citation30
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary_Materials.zip
Download Zip (3.4 MB)Acknowledgments
Thanks to Petr Novak and Jaime Gard for data analysis and figure preparation.
Funding
This work was supported, in whole or in part, by NIH CA017094, NIH CA159406 and NIH CA109552. We also acknowledge the UACC Experimental Mouse and Genomics shared services supported by NIH/NCI grant CA023074.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Stathis A, Moore MJ. Advanced pancreatic carcinoma:current treatment and future challenges. Nat Rev Clin Oncol 2010; 7:163-72; http://dx.doi.org/10.1038/nrclinonc.2009.236
- Yachida S, Iacobuzio-Donahue CA. Evolution and dynamics of pancreatic cancer progression. Oncogene 2013; 32:5253-60; PMID:23416985; http://dx.doi.org/10.1038/onc.2013.29
- Damaraju VL, Damaraju S, Young JD, Baldwin SA, Mackey J, Sawyer MB, Cass CE. Nucleoside anticancer drugs:the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003; 22:7524-36; PMID:14576856; http://dx.doi.org/10.1038/sj.onc.1206952
- Giroux V, Iovanna J, Dagorn JC. Probing the human kinome for kinases involved in pancreatic cancer cell survival and gemcitabine resistance. FASEB J 2006; 20:1982-91; PMID:17012250; http://dx.doi.org/10.1096/fj.06-6239com
- Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Inhibition of SRC tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells. Clin Cancer Res 2004; 10:2307-18; PMID:15073106; http://dx.doi.org/10.1158/1078-0432.CCR-1183-3
- Liau SS, Whang E. HMGA1 is a molecular determinant of chemoresistance to gemcitabine in pancreatic adenocarcinoma. Clin Cancer Res 2008; 14:1470-7; PMID:18316571; http://dx.doi.org/10.1158/1078-0432.CCR-07-1450
- Farrell JJ, Bae K, Wong J, Guha C, Dicker AP, Elsaleh H. Cytidine deaminase single-nucleotide polymorphism is predictive of toxicity from gemcitabine in patients with pancreatic cancer:RTOG 9704. Pharmacogenomics J 2012; 12:395-403
- Greenhalf W, Ghaneh P, Neoptolemos JP, Palmer DH, Cox TF, Lamb RF, Garner E, Campbell F, Mackey JR, Costello E, et al. Pancreatic cancer hENT1 expression and survival from gemcitabine in patients from the ESPAC-3 trial. J Natl Cancer Inst 2014; 106:djt347; PMID:24301456; http://dx.doi.org/10.1093/jnci/djt347
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008; 321:1801-6; PMID:18772397; http://dx.doi.org/10.1126/science.1164368
- Penchev VR, Rasheed ZA, Maitra A, Matsui W. Heterogeneity and targeting of pancreatic cancer stem cells. Clin Cancer Res 2012; 18:4277-84; PMID:22896694; http://dx.doi.org/10.1158/1078-0432.CCR-11-3112
- Chang DK, Grimmond SM, Biankin AV. Pancreatic cancer genomics. Curr Opin Genet Dev 2014; 24:74-81; http://dx.doi.org/10.1016/j.gde.2013.12.001
- Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011; 17:500-3
- Cui Y, Brosnan JA, Blackford AL, Sur S, Hruban RH, Kinzler KW, Vogelstein B, Maitra A, Diaz LA Jr, Iacobuzio-Donahue CA, et al. Genetically defined subsets of human pancreatic cancer show unique in vitro chemosensitivity. Clin Cancer Res 2012; 18:6519-30; PMID:22753594; http://dx.doi.org/10.1158/1078-0432.CCR-12-0827
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009; 324:1457-61; PMID:19460966; http://dx.doi.org/10.1126/science.1171362
- Teicher BA, Herman TS, Holden SA, Wang YY, Pfeffer MR, Crawford JW, Frei E 3rd. Tumor resistance to alkylating agents conferred by mechanisms operative only in vivo. Science 1990; 247:1457-61; PMID:2108497; http://dx.doi.org/10.1126/science.2108497
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25-9
- Alexa A, Rahnenfuhrer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006; 22:1600-7; PMID:16606683; http://dx.doi.org/10.1093/bioinformatics/btl140
- Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol 2007; 14:3629-37; http://dx.doi.org/10.1245/s10434-007-9583-5
- Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res 2009; 69:2400-7; PMID:19276344; http://dx.doi.org/10.1158/0008-5472.CAN-08-4312
- Gungor C, Zander H, Effenberger KE, Vashist YK, Kalinina T, Izbicki JR, Yekebas E, Bockhorn M. Notch signaling activated by replication stress-induced expression of midkine drives epithelial-mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res 2011; 71:5009-19; PMID:21632553; http://dx.doi.org/10.1158/0008-5472.CAN-11-0036
- Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007; 1:313-23; PMID:18371365; http://dx.doi.org/10.1016/j.stem.2007.06.002
- Li Y, VandenBoom TG, Kong D, Wang Z, Ali S, Philip PA, Sarkar FH. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res 2009; 69:6704-12; PMID:19654291; http://dx.doi.org/10.1158/0008-5472.CAN-09-1298
- Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res 2009; 69:5820-8; PMID:19584296; http://dx.doi.org/10.1158/0008-5472.CAN-08-2819
- Groger CJ, Grubinger M, Waldhor T, Vierlinger K, Mikulits W. Meta-analysis of gene expression signatures defining the epithelial to mesenchymal transition during cancer progression. PLoS One 2012; 7:e51136
- Bera A, VenkataSubbaRao K, Manoharan MS, Hill P, Freeman JW. A miRNA Signature of Chemoresistant Mesenchymal Phenotype Identifies Novel Molecular Targets Associated with Advanced Pancreatic Cancer. PLoS One 2014; 9:e106343; http://dx.doi.org/10.1371/journal.pone.0106343
- Glynn SA, Gammell P, Heenan M, O’Connor R, Liang Y, Keenan J, Clynes M. A new superinvasive in vitro phenotype induced by selection of human breast carcinoma cells with the chemotherapeutic drugs paclitaxel and doxorubicin. Br J Cancer 2004; 91:1800-7; PMID:15505620; http://dx.doi.org/10.1038/sj.bjc.6602221
- Wu A, Loutherback K, Lambert G, Estevez-Salmeron L, Tlsty TD, Austin RH, Sturm JC. Cell motility and drug gradients in the emergence of resistance to chemotherapy. Proc Natl Acad Sci U S A 2013; 110:16103-8; http://dx.doi.org/10.1073/pnas.1314385110
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010; 141:69-80; PMID:20371346; http://dx.doi.org/10.1016/j.cell.2010.02.027
- Congdon LM, Pourpak A, Escalante AM, Dorr RT, Landowski TH. Proteasomal inhibition stabilizes topoisomerase IIalpha protein and reverses resistance to the topoisomerase II poison ethonafide (AMP-53, 6-ethoxyazonafide). Biochem.Pharmacol 2008; 75:883-90
- Watson M, Chow S, Barsyte D, Arrowsmith C, Shankey TV, Minden M, Hedley D. The study of epigenetic mechanisms based on the analysis of histone modification patterns by flow cytometry. Cytometry A 2014; 85:78-87; PMID:24038859; http://dx.doi.org/10.1002/cyto.a.22344