
Abstract
Multiple drug resistance (MDR) is a major obstacle to attenuating the effectiveness of chemotherapy to many human malignancies. Proteasome inhibition induces apoptosis in a variety of cancer cells and is recognized as a novel anticancer therapy approach. Despite its success, some multiple myeloma patients are resistant or become refractory to ongoing treatment by bortezomib suggesting that chemoresistant cancer cells may have developed a novel mechanism directed against the proteasome inhibitor. The present study aimed to investigate potential mechanism(s) of attenuation in a MDR cell line, MES-SA/Dx5. We found that compared to the parental human uterus sarcoma cell line MES-SA cells, MES-SA/Dx5 cells highly expressed the ABCB1 was more resistant to MG132 and bortezomib, escaping the proteasome inhibitor-induced apoptosis pathway. The resistance was reversed by co-treatment of MG132 and the ABCB1 inhibitor verapamil. The data indicated that ABCB1 might play a role in the efflux of MG132 from the MES-SA/Dx5 cells to reduce MG132-induced apoptosis. Furthermore, the canonical Wnt pathway was found activated only in the MES-SA/Dx5 cells through active β-catenin and related transactivation activities. Western blot analysis demonstrated that Wnt-targeting genes, including c-Myc and cyclin D1, were upregulated and were relevant in inhibiting the expression of p21 in MES-SA/Dx5 cells. On the other hand, MES-SA cells expressed high levels of p21 and downregulated cyclin D1 and caused cell cycle arrest. Together, our study demonstrated the existence and participation of ABCB1 and the Wnt pathway in an MDR cell line that attenuated proteasome inhibitor-induced apoptosis.
Introduction
Multidrug resistance (MDR) of cancer cells has become a barrier to clinical applications of anticancer agents.Citation1 P-glycoprotein 1 (permeability glycoprotein, abbreviated as P-gp or Pgp), also known as multidrug resistance protein 1 (MDR1) or ATP-binding cassette sub-family B member 1 (ABCB1) or cluster of differentiation 243 (CD243), is a glycoprotein that in humans is encoded by the ABCB1 gene. Since ABCB1 is highly related to MDR in cancer cells, ABCB1 inhibitors have been tested in clinical trials in attempts to circumvent MDR. Unfortunately, such attempts have failed in the phase III trials because of possible involvement of some unknown alternative mechanisms of drug resistance in cancer cells which seemed to be adverse to the pharmacological properties of the ABCB1 inhibitors tested.Citation2 An innovative approach of proteasome inhibition capable of inducing apoptosis in multiple myeloma cells has been reported.Citation3 The proteasome inhibitor, bortezomib (Velcade®, formerly known as PS-341), inhibited the NFκB pathway leading to decreased cell proliferation and induction of apoptosis. Bortezomib obtained FDA approval in 2005 for clinical treatment against relapsed multiple myeloma and mantle cell lymphoma.Citation4,5 Despite successes with this drug, some patients develop resistant or become refractory to ongoing treatment; clinical responses to bortezomib in other hematologic malignancies and solid tumors remain low.Citation6,7 To date, several mechanisms of resistance to proteasome inhibitors have been reported, including upregulation of heat shock proteins,Citation8 increased activity of the aggresome pathway,Citation9 mutations or overexpression of the β5 unit of the proteasome catalytic unitCitation10 and overexpression of ABCB1.Citation11,12
Combined use of an ABCB1 inhibitor and bortezomib was shown to increase anticancer cytotoxicity in Ewing's family tumors cells.Citation11 Furthermore, siRNA knockdown of ABCB1 expression in K562 cells rendered treated cells to be more sensitive to bortezomib cytotoxicity; based on this observation, bortezomib has been considered as a possible ABCB1 substrate.Citation12,13 On the other hand, some studies have indicated that treating multidrug-resistant cancer cells with a proteasome inhibitor can alleviate the effects of ABCB1 protein translation and posttranslational modifications thereby facilitating the accumulation of immature ABCB1, and consequently reducing signs of multidrug resistance.Citation12,14,15
It has also been shown that human DLD1 colon cancer cells induce ABCB1 gene expression through T-cell Factor 4 and the β-catenin transcription factor complex, causing ABCB1 expression levels to increase.Citation16 Furthermore, studies have indicated that a T-cell Factor 4 and β-catenin transcription factor complex bind at a site located at nucleotides -261 to -1813 upstream of the promoter of the ABCB1 gene.Citation17 Thus, ABCB1 is a known downstream target gene of the Wnt pathway. Furthermore, T-cell Factor 4 assists β-catenin translocation from the cytosol to the nucleus to participate in the activation of downstream target genes such as c-Myc, cyclin D1, MMPs.Citation18 Liang et al. proposed that the proto-oncogene c-Myc could inhibit p21 expression, facilitating cell cycle progression.Citation19 Another study demonstrated that MG132 caused glioma cancer cells to remain in the G2 phase.Citation20 Furthermore, p53 and p21 co-expression caused the cell cycle in cancer cells to remain at the G2 phase, preventing the cells from entering mitosis and, thus, arresting cell growth.Citation21,22 Together, these findings suggest that chemoresistant cancer cells may have developed a Wnt pathway-related mechanism directed against the proteasome inhibitor-induced apoptosis.
Hence, the multidrug-resistant cell line, MES-SA/Dx5, established from a poorly differentiated human uterine sarcoma cell line, MES-SA cells, grown in the presence of doxorubicin and were overexpressing ABCB1 but not ABCC1 and ABCG2,Citation23,24,25 was used in this study. Two proteasome inhibitors, namely MG132 and Bortezomib, were used to investigate possible mechanism(s) of drug resistance developed in the multidrug-resistant cancer cells. We found that Wnt-targeting genes, including c-Myc and cyclin D1, were upregulated and were relevant in inhibiting the expression of p21 in MES-SA/Dx5 cells. Moreover, we also found that the canonical Wnt pathway was only activated in ABCB1-overexpressed MES-SA/Dx5 cells through active β-catenin and its related transactivation activities regulating the cell cycle. Together, our study demonstrated the existence and involvement of ABCB1 and that the Wnt pathway in a MDR cell line would attenuate proteasome inhibitor-induced apoptosis.
Results
Cytotoxic effects of proteasome inhibitors on multidrug-resistant cancer cells
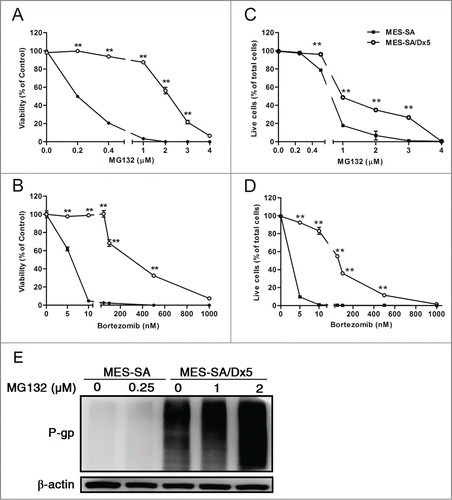
To understand the damage caused by proteasome inhibitors in multidrug-resistant cancer cells, different concentrations of proteasome inhibitors, MG132 and bortezomib, were used to treat MES-SA and MES-SA/Dx5 cancer cells. Results obtained in MTT assays () and live/dead viability assay () were consistent. The survival rate of both cell lines declined with increasing drug concentrations, and the half maximal inhibitory concentrations (IC50) of the 2 cell lines were significantly different. The IC50 of MG132 treatment was 0.2 μM in MES-SA and 2 μM in MES-SA/Dx5 cells (). The IC50 of bortezomib treatment was 6 nM in MES-SA cells and 300 nM in MES-SA/Dx5 cells (), respectively. Thus, these results indicated that compared to data on the MES-SA cells, multidrug-resistant MES-SA/Dx5 cells seemed to possess a higher tolerance against proteasome inhibitors. Furthermore, the MES-SA/Dx5, which overexpressed ABCB1 but not ABCC1 and ABCG2, were selected from MES-SA cell lines by doxorubicin treatment.Citation23,24,25 Western blot analysis showed that ABCB1 expression occurred only in MES-SA/Dx5 cells, and there was significant accumulation of the ABCB1 protein in coincidence with the elevation of MG132 concentration (). Our data, thus, indicate that a direct association of multidrug resistance in MES-SA/Dx5 cells with ABCB1 function to reduce the cytotoxic effects of proteasome inhibitors.
Figure 1. Multidrug-resistant MES-SA/Dx5 cancer cells is more resistant to proteasome inhibitors MG132- and bortezomib-induced cytotoxicity. Cells were incubated with the indicated concentration of (A, C) MG132 or (B, D) bortezomib for 60 h. At the end of the drug treatment, cells were harvested to measure viability using (A, B) the MTT assay, and (C, D) the live/dead viability assay. (E) Expression of ABCB1 of both cell lines after MG132 treatment at the indicated drug concentrations incubated for 60 h. ABCB1 was identified by Western blot analysis. The surviving fractions are expressed as the mean ± SD from 3 independent experiments. **P < 0 .05 indicates the differences between the MG132- or bortezomib-treated cells and relative to the respective untreated controls.
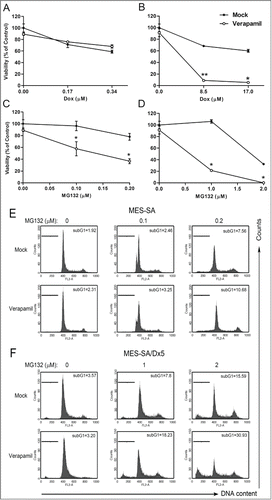
To further verify the correlation between doxorubicin and ABCB1, both cell types were treated with doxorubicin in the presence of an ABCB1 inhibitor, verapamil. MES-SA cells showed similar survival rates regardless of the presence of verapamil (). As for the MES-SA/Dx5 cells, following verapamil treatment, the survival rate was reduced to 60% () indicating that the ABCB1 inhibitor increased anticancer drug cytotoxicity on MES-SA/Dx5 most likely by inhibiting the extracellular efflux and exclusion of doxorubicin by ABCB1. We further investigated correlation between the proteasome inhibitor and ABCB1 by treating both cells with MG132 in the presence of verapamil. Results showed that following 60-hour treatment with MG132 and in the presence of verapamil, the survival rate of the treated MES-SA cells decreased to 30% (), and that of MES-SA/Dx5 to 80% (). Compared to MES-SA cells, the survival rate of MES-SA/Dx5 cells was more greatly affected by verapamil. Furthermore, the DNA contents treated cells were analyzed with propidium iodine dye. sub-G1 apoptotic population of MES-SA cells increased to 3% (). and that of MES-SA/Dx5 to 15% (). Compared to MES-SA cells, the sub-G1 apoptotic population of MES-SA/Dx5 cells was significantly affected by verapamil. These results verified that MES-SA/Dx5 cells relied on ABCB1 to express multidrug resistance to reduce the cytotoxic effects induced by proteasome inhibitors ().
Figure 2. ABCB1 inhibitor resensitizes the multidrug-resistant MES-SA/Dx5 cancer cells to proteasome inhibitor-induced cytotoxicity. (A-B) MES-SA (A) and MES-SA/Dx5 cells (B) were incubated with the indicated concentrations of doxorubicin and 2 μM verapamil for 60 h. (C-D) MES-SA (C) and MES-SA/Dx5 cells (D) were incubated with the indicated concentration of MG132 and 2 μM verapamil for 60 h. At the end of treatment with MG132 or doxorubicin in the presence of the verapamil, cells were harvested for measurement of viability using the MTT assay. (E-F) MES-SA (E) and MES-SA/Dx5 cells (F) were incubated with the indicated concentration of MG132 and 2 μM verapamil for 48 h. At the end of treatment with MG132 in the presence of the verapamil, cells were subjected to cell cycle analysis by FACS after propidium iodide staining, as described in Materials and methods. The sub-G1 population represents apoptotic as well as dead cells. The surviving fractions are expressed as the mean ± SD from 3 independent experiments. *P < 0 .05 and **P < 0 .01 indicate the differences between the ABCB1 inhibitor-treated cells and the respective untreated controls.
Involvement of the apoptosis pathway of proteasome inhibitor in triggering cell death of multidrug-resistant cancer cells
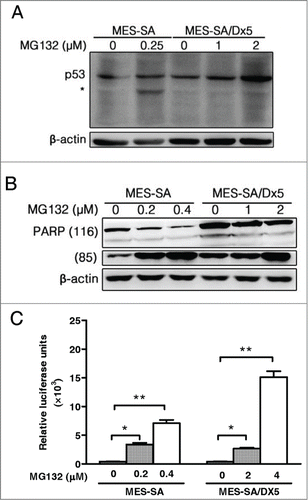
To evaluate possible involvement of the apoptosis pathway in proteasome inhibitor-triggered cell death, the MES-SA and MES-SA/DX5 cancer cells were treated with respective IC50 concentrations of MG132, and the expression of the transcription factor p53, apoptosis enzyme caspases 3 and 7 and the DNA repair enzyme, poly ADP-ribose polymerase (PARP), were analyzed. Western blot results showed that p53 protein expression levels increased along with elevated MG132 concentrations (). An increase in p53 expression levels possibly indicates activation of the caspase family apoptosis enzymes. Among such enzymes, caspase 3 performs a pivotal function in apoptosis signal transduction.Citation26 Caspase-Glo 3/7 assay was used to measure the activities of caspases 3 and 7 in both cell types. Results showed that in both MES-SA and MES-SA/Dx5 cell lines, activities of caspases 3 and 7 increased with increasing MG132 concentrations (). Activation of the intracellular caspase 3 usually involves loss of activity of certain enzymes, such as DNA repair enzyme PARP.Citation27 Results showed that when MG132 concentration was increased, the levels of intact PARP proteins declined gradually while the amounts of cleaved PARPs increased (). Thus, the same apoptotic pathway of MG132 contributed to in triggering cell death in both cancer cell lines and the effects are independent on multidrug resistance.
Figure 3. Proteasome inhibitor-induced apoptosis through increased expression of p53, caspase 3/7 and cleaved PARP in a dose-dependent manner. (A-B) Expression of p53 (A) and PARP (B) in MES-SA and MES-SA/D×5 cells after MG132 treatment for 60 h examined by Western blotting. (C) Caspase 3/7 activities of MES-SA and MES-SA/D×5 cells were detected by the Caspase-Glo 3/7 assay after 60 h MG132 treatment at the indicated dosages. The Caspase 3/7 activities were detected by luminescence and were proportional to its intensity normalized by cell viability. Data are expressed as relative luciferase activity (RLU) as the mean ± SD from 3 independent experiments. *P < 0 .05 and **P < 0 .01 indicate the differences between the MG132-treated cells and the respective untreated controls.
Involvement of the Wnt pathway in regulating drug resistance in MDR cancer cells against proteasome inhibitor
Are there some unknown mechanisms that exist in ABCB1-overexpressed MDR MES-SA/Dx5 cells to attenuate proteasome inhibitors-induced apoptosis? Since ABCB1 is a known downstream target gene of the Wnt pathway, we hypothesized that MES-SA/Dx5 cells upregulated the expression of ABCB1 downstream target genes through the Wnt pathway. Western blot results showed that β-catenin expression levels increased substantially following proteasome inhibition in MES-SA/Dx5 compared to MES-SA cells, especially in the nuclear fraction (). T-cell Factor 4 assists β-catenin translocation from the cytosol to the nucleus to participate in the activation of the downstream target gene such as c-Myc, cyclin D1 and MMPs.Citation18 Results showed no notable difference in T-cell Factor 4 expression levels in the 2 cell lines; most T-cell Factor 4 protein was located in the cytosol and not in the nucleus (). We next evaluated whether T-cell Factor 4 and the β-catenin transcription factor complex could regulate transcription of c-Myc and cyclin D1. The Wnt reporter gene system included TOPflash which contained a wild-type β-catenin/T cell factor-binding elements, whereas FOPflash contained a mutated β-catenin/T cell factor-binding elements for T-cell Factor 4 and the β-catenin transcription factor complex. In MES-SA cells, transfection of either the TOPflash or FOPflash reporter vector both resulted in relatively low T-cell Factor 4 activities () due to low β-catenin expression in this cell line. On the other hand, in MES-SA/Dx5 cells, transfection of the FOPflash negative control group also resulted in low luminescence; however, luminescence intensity following TOPflash transfection was 150 times higher than the intensity in the similarly-transfected MES-SA cells. These results indicated that T-cell Factor 4 and the β-catenin transcription factor complex in the nucleus of MES-SA/Dx5 cells possessed the ability to regulate the transcription of β-catenin/Tcf-4 downstream target genes. Transfection of mutant β-catenin-expressing plasmids into MES-SA/Dx5 cells also resulted in an elevation in luminescence intensity (). MG132 treatment of TOPflash-transfected MES-SA/Dx5 cells also led to increases in luminescence intensity. MES-SA/Dx5 cells transfected with mutant β-catenin and subsequently treated with MG132 resulted in the highest luminescence intensity (). Thus, the results revealed that besides upregulating β-catenin expression in the nucleus, the transcriptional functions of T-cell Factor 4 and the β-catenin transcription factor complex were also upregulated in MES-SA/Dx5 cells to facilitate the activation of ABCB1 downstream target genes.
Figure 4. The Wnt pathway is activated in multidrug-resistant MES-SA/Dx5 cancer cells through active β-catenin and its related transactivation activities against proteasome inhibitor. (A-B) Expression of the Wnt pathway ligand β-catenin (A) and the transcription factor Tcf-4 (B) of nuclear and cytoplasmic fractions in MES-SA and MES-SA/D×5 cell lines after bortezomib treatment for 60 h examined by Western blotting. (C) β-catenin/TCF transcription activities of MES-SA and MES-SA/D×5 cells treated with MG132. The β-catenin/TCF transcription activities of the MG132-treated cells were detected by the TOP/FOP assay. The TOP-Flash or FOP-Flash reporter-transfected MES-SA and MES-SA/D×5 cells were incubated with different concentrations of MG132 for 60 h. MES-SA and MES-SA/D×5 cells transfected with the mutant-type β-catenin were used as controls. At the end of the treatment, cell lysates were harvested and were assayed for luciferase activities. The β-catenin/TCF transcription activities were represented as a ratio of firefly/renilla. (D)The β-catenin/TCF transcription activities or ABCB1 transactivation activity of the CTNNB1 shRNA transfected MES-SA/Dx5 cells was detected by TOP assay or ABCB1 promoter–Luciferase reporter assay, respectively. Data are expressed as relative luciferase activity (RLU) as the mean ± SD from 3 independent experiments. *P < 0 .05 and **P < 0 .01 indicate the differences between the MG132-treated cells or CTNNB1 shRNA transfected cells and the respective untreated controls.
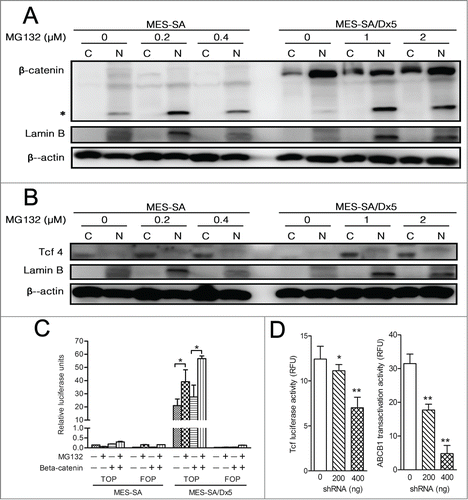
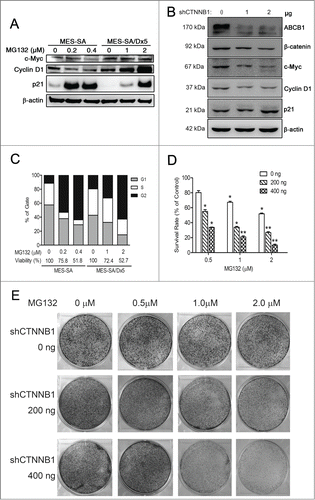
To confirm the regulatory of the expression of ABCB1 downstream target genes through the β-catenin, we deliver the indicated concentration of CTNNB1 shRNA to MES-SA/Dx5 cell, the TOP reporter assay indicated that Tcf transctivation activity were significant reduced in dose-dependent manner of CTNNB1 knockdown MES-SA/Dx5 cell (). Transcriptional control of ABCB1 by the β-catenin/TCF response elements in its promoter site had been proved in the previous study.Citation28 We further to evaluate the ABCB1 expression level and function in CTNNB1 knockdown MES-SA/Dx5 cell. The ABCB1-Luc reporter assay data showed that ABCB1 promoter transactivation activity was reduced in dose-dependent manner of CTNNB1 knockdown MES-SA/Dx5 cell (). Thus, the results revealed that besides upregulating β-catenin expression in the nucleus, the transcriptional functions of T-cell Factor 4 and the β-catenin transcription factor complex were also upregulated in MES-SA/Dx5 cells to facilitate the activation of ABCB1 downstream target genes.
Involvement of cell cycle in regulating drug resistance in MDR cancer cells against proteasome inhibitor
To investigate whether MES-SA/Dx5 cells enhanced resistance against proteasome inhibitors through the Wnt pathway-targeted downstream genes, such as c-Myc and cyclin D1, western blot analysis was performed to assay the levels of these 2 proteins in MES-SA and MES-SA/Dx5 cells. The results showed higher expression levels in both c-Myc and cyclin D1 in these cells (). Following MG132 treatment, c-Myc remained highly expressed in MES-SA/Dx5 cells and displayed no effects to the cells even with increased drug concentrations. The cyclin D1 expression levels increased markedly with increasing MG132 concentrations in the 2 cell lines; however, in higher treatment concentrations of MG132, the cyclin D1 expression level in MES-SA cells reduced rapidly, possibly due to the function of other regulating factors in the cancer cells. To explain the considerable reduction of cyclin D1 levels in MES-SA cells in high MG132 concentrations, we further examined the expression level of the p21 protein, a cell cycle regulator responsible for cyclin D1 regulation. Western blot results showed that following an increase in MG132 concentration, p21 expression levels in MES-SA cells markedly increased (). The results clearly revealed that after cell injury induced by MG132, MES-SA cells expressed high levels of p21 leading to inhibition of cyclin D1 expression. In contrast, the c-Myc expression level was also high, causing p21 expression to become considerably inhibited in MES-SA/Dx5 cells. Furthermore, to confirm the regulatory of the expression of Wnt pathway-targeted downstream genes in MDR cancer cells through the β-catenin, we deliver the CTNNB1 shRNA vector to MES-SA/Dx5 cell. The Western blot data indicated that ABCB1, β-catenin, c-Myc and cyclin D1 were significant decreased in dose-dependent manner of CTNNB1 knockdown MES-SA/Dx5 cell ().
Figure 5. Cell cycle-regulated resistance against proteasome inhibitor in the multidrug-resistant MES-SA/Dx5 cancer cells (A) Expression of Wnt-targeting genes, c-Myc, cyclin D1 and a cell cycle regulator, p21, in MES-SA and MES-SA/Dx5 cells after MG132 treatment for 60 h examined by Western blotting. (B) Expression of ABCB1, β-catenin, c-Myc, cyclin D1 and p21 in CTNNB1 shRNA transient transfected MES-SA/Dx5 cell examined by Western blotting. (C) Cells were incubated with the indicated concentrations of MG132 for 60 h. At the end of the treatment, cells were harvested for measurement of viability using the MTT assay and cell cycle distribution using Flow cytometry/PI staining. (D) Cells were transfected with the indicated concentrations of CTNNB1-shRNA plasmid for 24 h, and incubated with the indicated concentrations of MG132 for 60 h. At the end of the treatment, cells were harvested for measurement for of viability by using (D) the MTT assay and (E) the colony formation assay. The colony formation was scored after 10 d incubation. Data represent mean ± SD of 3 independent experiments. *P < 0 .05 and **P < 0 .01 indicate differences between the MG132-treated cells or CTNNB1 shRNA transfected cells and the respective untreated controls.
Following MG132 treatment, MES-SA cells expressed a large amount of the cell cycle regulator protein p21, which could subsequently regulate cell cycle distribution of the cell population.Citation21 As for MES-SA/Dx5 cells expressing the Wnt pathway protein, whether MG132 affected cell cycle distribution was next investigated by PI staining. Results showed that following an increase in the MG132 concentration, cell cycle distribution in MES-SA cells showed a considerable increase in the G2 phase from 12% to 64%, and a reduction of the S phase from 31% to 7%. However, following MG132 treatment, the cell cycle distribution in MES-SA/Dx5 cells displayed similar results: increase in the G2 phase from 20% to 63% and reduction in the S phase from 38% to 22% (). Thus, even after the damage caused by MG132, MES-SA/Dx5 cells could still sustain normal cell cycle progression and increase cancer cell proliferation.
Possible inhibition of β-catenin leading to resensitizition of MG132 toxicity in drug-resistant MES-SA/Dx5 cells was examined. About 40% decreased cell viability was observed when MES-SA/Dx5 cells were treated with indicated concentration of MG132 in MTT assays () and colony formation assays (), the viability and colony formation ability significantly decreased in a dose-dependent manner of MG132 treated CTNNB1-shRNA transfected MES-SA/Dx5 cells (). Our data, therefore, support the proposition that expression of the Wnt pathway contribute significantly to the mitigation of the cytotoxic damages caused in the MES-SA/Dx5 cancer cells.
Discussion
Several studies have reported using proteasome inhibitors as ABCB1 substrates in their studies.Citation11,12,15,29-32 The drugs MG132 and bortezomib applied in this study both contain benzene rings and have molecular weights of approximately 400 Da, which are fitting as ABCB1 substrates. We noted that doxorubicin resistance-selected MES-SA/Dx5 exhibit 10-fold and 25-fold cross-resistance to the proteasome inhibitors MG132 and bortezomib, respectively. Our studies was consistent with original studies by Sharma et al, 1992 showed that peptide aldehyde structures like MG132 are bona fide substrates for ABCB1,Citation29 but a boronated peptide compound like bortezomib has relative poor ABCB1 substrate affinity resulting in moderate resistance levels (approx. 5-fold). Besides these differences it should also be taken into account that MG132 target multiple catalytic proteasome subunits,Citation29 whereas bortezomib has preferential binding to the β5 unit of the proteasome catalytic unit.Citation10 Moreover, these drugs could also efflux to outside of the cell membrane. It has been shown that siRNA knockdown of ABCB1 expression in K562 cells is more sensitive to bortezomib cytotoxicity and considered bortezomib has been considered as a possible ABCB1 substrate.Citation13 Combined use of an ABCB1 inhibitor and bortezomib increases anticancer cytotoxicity on Ewing's family tumors cellsCitation13 and it is assumed that bortezomib is a possible ABCB1 substrate drug.Citation12,13 The published results are essentially similar with our study in the weakened cytotoxicity of proteasome inhibitors due to the presence of ABCB1 (). Subsequent to adding an ABCB1 inhibitor to MES-SA/Dx5 cells, cell survival rates were reduced, though not as markedly as in the MES-SA cancer cells (). Therefore, MG132 and doxorubicin possible act via different mechanisms in cells. Alternatively, the combined application of MG132 and an ABCB1 inhibitor results in drug interference due to their concerted action.
Following proteasome dysfunction in cancer cells, human DLD1 colon cancer cells were shown to induce ABCB1 gene expression through T-cell Factor 4 and β-catenin transcription factor complex, causing increases in the ABCB1 expression levels.Citation16 Furthermore, studies have indicated that a T-cell Factor 4 and β-catenin transcription factor complex binding site is located upstream of the ABCB1 gene promoter.Citation17 Our study, therefore, targeted T-cell Factor 4 and β-catenin transcription factor complex as well as ABCB1 expression in MES-SA/Dx5 cells. Regardless of MG132 treatment, β-catenin expression increased markedly in the nucleus of MES-SA/Dx5 cancer cells relative to data obtained with MES-SA cells (). Western blot results showed β-catenin bands with higher MW in the cytosol than in the nucleus, possibly due to phosphorylation of β-catenin in the cytosol. Smaller protein bands (marked with asterisks in ) are possibly β-catenin products of caspase reactions. Similar β-catenin smaller-fragment proteins have also been previously reported in a study using MG132 treatment to activate caspases in hepatocellular carcinoma cells.Citation33
Treatment of MES-SA/Dx5 cancer cells with the proteasome inhibitor MG132 induced upregulated transcriptional activities of T-cell Factor 4 and β-catenin transcription factor complex on downstream target genes (). ABCB1 is a known downstream target gene of the Wnt pathway. Hence, data reported in this study have led us to propose that MG132 activation of the Wnt pathway possibly increases ABCB1 expression. This proposition is consistent with the Western blot results indicating elevated ABCB1 expression in conjunction with MG132 concentration (). Moreover, this result also explains why MG132 treatment did not inhibit ABCB1 expression.
Treatment with a proteasome inhibitor not only causes apoptosis in cancer cells, but also results in cell-cycle arrest. The relationship between cell cycle progression and apoptosis is complementary.Citation34 In the apoptosis experiments using 2 cancer cell lines reported here yielded similar results showing increases in p53 expression in response to increases in the concentration of the proteasome inhibitor (). Aside from inducing the activation of cell apoptosis proteins, p53 also indirectly regulates cell cycle progression.Citation21,22 This is primarily achieved through the activation of the cell-cycles regulating p21 protein, thus, achieving the observed effects in cell cycle regulation. The p21 protein is an inhibitor of cyclin dependent kinase (CDK), and is arrested in the cell cycle via CDK-cyclin inhibition sparing time for damaged DNA to repair in the nucleus before continuing the cell cycle processes.
Studies have indicated that MG132 treatment of MES-SA cells caused accumulation of p21 and inhibition of intracellular cyclin D1 (). Cyclin D1 is a cell cycle protein that forms a complex with CDK4 to regulate cell cycle progression from the G1 phase to the S phase. MES-SA/Dx5 cancer cells, however, did not express p21 (), mainly due to activation of the target gene c-Myc downstream of the Wnt pathway. Liang J et al. (2003) proposed that the anticancer gene c-Myc could inhibit p21 expression, facilitating cell cycle progression.Citation19 However, the function of p21 in cell cycle regulation following its accumulation remains to be investigated.
Previous studies demonstrated that MG132 caused glioma cancer cells to stay in the G2 phase.Citation20 Furthermore, p53 and p21 co-expression caused the cell cycle in cancer cells to remain at the G2 phase, preventing them from performing mitosis and, thus, arresting cell growth.Citation21,22 The results of our study () are consistent with the abovementioned conclusions indicating that, in MES-SA cells, the S phase was notably reduced while the G2 phase increased. MES-SA/Dx5 cells, following the same treatment with MG132, displayed a gradual decrease in the S phase but the decrease was more substantial in the G2 phase () due to Wnt pathway activation of c-Myc, consequently inhibiting p21 expression. These results confirm that activated Wnt pathways in MES-SA/Dx5 cells render the cells resistant to the MG132 effects on the cell cycle enabling normal progression of the cell cycle and cancer cell proliferation.
Following MG132 treatment, p21 in MES-SA cells accumulated considerably, causing a substantial increase in the G2 phase and a marked reduction in the S phase (). This indicates that cells remaining in the G2 phase could not proceed into the M phase to perform mitosis. MES-SA/Dx5 cells lacked p21 expression, and while there was an increase in the G2 phase, S phase reduction was relatively limited (). Thus, even after the damage caused by MG132, MES-SA/Dx5 cells could still sustain normal cell cycle progression and increase cell proliferation. Taken together, Wnt pathway expression contributes positively to the mitigation of the cytotoxic damages caused by MG132 in the MES-SA/Dx5 cancer cells.
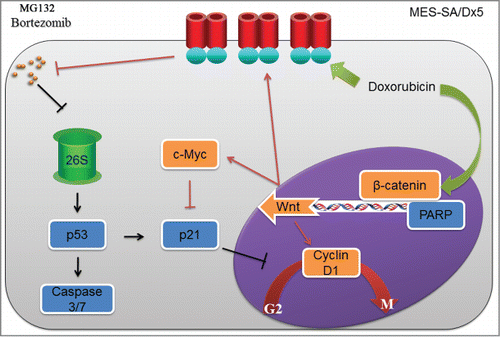
Results from our study have indicated that MG132 treatment caused an increase in the p53 expression levels, increased Caspase 3/7 activities and elevated amounts of fragmented PARP level in MES-SA cancer cells (). Furthermore, p53 regulation on the expression of the downstream p21 gene results in cell-cycle arrest in the G2 phase leading to cessation of MES-SA cell growth in a proposed mechanism depicted in . Though MES-SA/Dx5 and the parent MES-SA cancer cells both resulted in similar apoptosis pathway after MG132 treatment, MES-SA/Dx5 cells were able to transport MG132 out of the cell via ABCB1 expression, resulting in reduced bortezomib cytotoxicity. MES-SA/Dx5 cells then increased the expression of the cancer cell growth factors cyclin D1 and c-Myc through the Wnt pathway, subsequently inhibiting p21 regulation on the cell cycle. This rendered MES-SA/Dx5 cells capable of resisting cell cycle suspension in G2 phase caused by MG132 (), permitting the cells to populate normally, and also alleviating MG132 cytotoxicity. In conclusion, MES-SA/Dx5 cancer cells resisted cytotoxicity of proteasome inhibitors through the functions of ABCB1 and the Wnt pathway.
Figure 6. (A)proposed mechanism of attenuated Proteasome Inhibitor-induced cytotoxicity in the multidrug-resistant MES-SA/Dx5 cancer cells Since MES-SA/Dx5 and the parental MES-SA cancer cells both trigger similar apoptosis pathway after MG132 or bortezomib treatment, MES-SA/Dx5 cells efflux MG132 or bortezomib out of the cell via ABCB1 expression, resulting in reduced MG132 or bortezomib cytotoxicity. MES-SA/Dx5 cells then increase the expression of the cancer-cell growth factors cyclin D1 and c-Myc through the Wnt pathway to subsequently inhibit p21 regulation on the cell cycle. These events render MES-SA/Dx5 cells capable of resisting cell cycle suspension in the G2 phase caused by MG132 or bortezomib, permitting the cancer cells to proliferate normally and consequently alleviating proteasome Inhibitor cytotoxicity.
Materials and Methods
Cell culture
The multiple drug-resistant cell line MES-SA/Dx5 (ATCC, CRL-1977) was established from the human sarcoma cell line MES-SA (ATCC, CRL-1976) in the presence of increasing doxorubicin concentrations.Citation23 All cell lines were continuously maintained in McCoy's 5A medium (Invitrogen, 16600) supplemented with 10% fetal bovine serum (Invitrogen, 10099) and 1% Antimycotics (Invitrogen, 15240) under 95% air and 5% CO2 at 37°C. The MES-SA/Dx5 cell line was maintained in the continuous presence of 1.7 μM doxorubicin (Sigma-Aldrich, D1515).
Western blot analysis
Total cellular proteins were isolated from cell lines by the PRO-PREP™ Protein Extraction Solution (INtRon, 17081). Nuclear and cytoplasmic proteins were isolated from cell lines using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce, 78835). Western blot was performed as described previously.Citation35 Briefly, approximately 40 μg of total proteins was loaded onto each lane and the proteins were separated in SDS-PAGE gels. After electrophoresis, the resolved proteins were transferred to PVDF membrane (EMD Millipore, IPVH00010) and subsequently stained with Ponceau S (Sigma-Aldrich, P7170) to confirm complete protein transfer. The membranes were blocked with 5% nonfat dry milk in PBS-T (0.1% Tween-20 (Sigma-Aldrich, P1379) in PBS, pH7.4) for 1 hour and probed overnight with the following antisera at appropriate dilutions: 1:1000 dilution of anti-ABCB1 (Santa Cruz, sc-13131), 1:500 dilution of anti-p53 (Invitrogen, 081129), 1:500 dilution of anti-PARP (Cell Signaling, 9542), 1:500 dilution of anti-Lamin B1 (Santa Cruz, sc-56145), 1:500 dilution of anti-Tcf 4 (Santa Cruz, sc-13027), 1:500 dilution of anti-c-Myc (Santa Cruz sc-70469), 1:500 dilution of anti-cyclin D1 (Santa Cruz, sc-8396) 1:500 dilution of anti-p21 (Cell Signaling, 2946), a 1:1,000 dilution of the anti-β-catenin (Invitrogen, 138400) and 1:10,000 dilution of an anti-actin (Novus, NN600501) antiserum in PBS–T. The identification of each protein was achieved with the Western Lighting Plus Reagent (Perkin Elmer, NEL103E001EA) using an appropriate alkaline phosphatase-conjugated secondary antibody. The level of each protein in the protein gel blot analysis was detected by the LAS-3000 chemiluminescence detection device (FUJIFILM). To adjust for loading differences, the optical density of each protein was normalized to that of the β-actin band.
shRNA
The CTNNB1 shRNA clone (TRCN0000003845; GCTTGGAATGAGACTGCTGAT) targeted at the human CTNNB1 transcript for β catenin, and the control scramble shRNA clone (CCTAAGGTTAAGTCGCCCTCGCTC) in a TRC001-based shRNA pLKO.1 vector was purchased from the National RNAi Core Facility (Academia Sinica).
Transfection and luciferase assays
MES-SA or MES-SA/Dx5 cells were plated at a density of 1 × 105 cells/well in a 24-well plate. For Tcf transactivation activity assay as described previously,Citation36 cells were co-transfected with 800 ng TOP-Flash, FOP-Flash reporter (Addgene) or the mutated β-catenin overexpresssion plasmid, p-cat T41AS45A.Citation37 and 1 ng phRL-TK (Promega, E2261) premixed with Lipofectamine 2000 (Invitrogen, 11668027) for 6 h. The indicated concentration of bortezomib (Millennium Pharmaceuticals) was then added to the transfected cells and incubated for 60 h. For ABCB1 transactivation activity assay, cells were co-transfected with indicated concentration of CTNNB1 shRNA DNA, 800 ng ABCB1 reporter (Addgene), and 1 ng phRL-TK (Promega, E2261). At the end of the incubation, cells were lysed by Passive Lysis Buffer and were assayed by the Dual-Luciferase Reporter Assay System (Promega, E1910). The luminescence intensity was measured by GLOMAX 20/20 Luminometer (Promega). The firefly luciferase activities were adjusted for background luminescence and normalized to renilla luciferase activities to adjust for transfection efficiencies. The firefly luciferase units were divided by the renilla units to obtain the relative luciferase units (RLU). The experiments were performed in triplicates, and data shown were obtained from 3 independent experiments.
Cell viability MTT assay
MES-SA or MES-SA/Dx5 cells were plated at an approximate density of 1 × 105 cells/well in a 24-well plate. The cells were then treated with the indicated concentration of various drugs in different sets of experiments. The concentrations of verapamil (Sigma-Aldrich, V4629) were used as previously reported.Citation23 After 60 h of drug incubation, the medium was removed and phosphate buffered saline (PBS) was used to wash the cells. Two hundreds microliter of Thiazolyl Blue Tetrazolium (Sigma-Aldrich, M2128) was added to each well and was incubated with the cells at 37°C for 2 h. Subsequently, 400 μl DMSO was added to each well and incubation at 37°C was continued for 20 min. Absorbance of the mixture was read at 540 nm using a Microplate Reader (VersaMax). Cell viability (%) was calculated as the ratio of the surviving cells in each drug-treated experiment set to that of the control.
Colony formation assay
The cells were plated at a density of 5 × 105 cells/well in a 6-well plate and grown for 24 h. After CTNNB1 shRNA transfection, the cells were grown in the replaced fresh medium for 24 h. The cells were then treated with the indicated concentration of MG132 for 48 h. The cells were then replated at a density of 1000 cells/10 cm2 dish and grown for 7 to 8 d until discrete colonies were visualized. After washing with PBS, the colonies were stained with 0.5% crystal violet (Sigma-Aldrich, C3886) and counted. After cell counting, cell number was estimated by dissolving the crystal violet in 70% ethanol (4 ml/well) and then optical density (OD) values was measured at 562 were used as previously reported.Citation38 In each group, cells transfected with the control scramble shRNA were set at 100%. Results (mean ± standard error) represent data from triplicate wells.
Caspase 3/7 assays
MES-SA and MES-SA/Dx5 cells were plated at a density of approximately 8 × 103 cells/well in a 96-well plate. The cells were then treated with the indicated dosages of bortezomib for 60 h. At the end of the drug treatment, 100 μl Caspase-Glo 3/7 Reagent (Promega, G8091) was added to the treated cells in each well. After 30 min of incubation at room temperature, the relative luminescence unit (RLU) was measured by GLOMAX 20/20 Luminometer (Promega) and as an indication of Caspase 3/7 apoptotic activity.
Live/Dead viability assay
In preparation of flow cytometry, the cells were then treated with the indicated concentration of various drugs in different sets of experiments. The concentrations of verapamil (Sigma-Aldrich, V4629) used were as previously reported.Citation23 After 48 h of drug incubation, the cells were collected at the indicated time points. The cells were then stained with Live-Dye™, a cell-permeable green fluorescent dye (Ex/Em = 488/518 nm) and propidium iodine, a cell non-permeable red fluorescent dye (Ex/Em = 488/615), by using the Live-Dead cell staining Kit (Biovision, K501) following the manufacturer's protocol. Stained live and dead cells was assessed with the FACS Calibur flow cytometer and CellQuest Pro software (Becton Dickinson) as previously described.Citation35
Cell cycle analysis by FLOW cytometry
The cell cycle was performed as previously described.Citation39 Brief, MES-SA and MES-SA/Dx5 cells were treated with MG132 for 60 h following which the cells were washed with PBS and fixed in 4% paraformaldehyde. Samples were treated with 10 μg/ml RNase and Triton X-100 at 37°C for 1 h. The cells were then stained with 50 μg/ml propidium iodine (EMD Millipore, 537060). After 20 min of incubation at 4°C in the dark, the cells were analyzed using a FCM scan flow cytometer using the CellQuest software (Becton Dickinson). The percentage of cells in the apoptotic sub-G1 and G1, S-phase and G2-M phase were calculated using the Modfit software (Verity Software House).
Statistical analysis
The surviving fraction and the relative luminescence unit (RLU) were measured in triplicate samples and were expressed as mean ± SD. The Student's t-test or one way ANOVA was used for statistical analysis. P < 0.05 was considered as statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported in part by the Ministry of Science and Technology (Taiwan; NSC 95–2320-B-182–028-MY3, NSC 99–2632-B-182–001-MY3, MOST 103–2320-B-182–021), Chang Gung Memorial Hospital Grant (CMRPD 34012, CMRPD 180133 and CMRPD 1B0473) and Ministry of Education (Taiwan; EMRPD1C0121).
References
- Goldman ID. Transport energetics of the folic acid analogue, methotrexate, in L1210 leukemia cells. Enhanced accumulation by metabolic inhibitors. J Biol Chem 1969; 244: 3779-85; PMID:5805398
- Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007; 446: 749-57; PMID:17429392; http://dx.doi.org/10.1038/nature05630
- Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 2001; 61, 3071-76; PMID:11306489
- Richardson PG, Anderson KC. Bortezomib: a novel therapy approved for multiple myeloma. Clin Adv Hematol Onco 2003; 1: 596-600.
- Bross PF, Kane R, Farrell AT, Abraham S, Benson K, Brower ME, Bradley S, Gobburu JV, Goheer A, Lee SL, et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin Cancer Res 2004; 10: 3954-64; PMID:15217925; http://dx.doi.org/10.1158/1078-0432.CCR-03-0781
- Cortes J, Thomas D, Koller C, Giles F, Estey E, Faderl S, Garcia-Manero G, McConkey D, Ruiz SL, Guerciolini R, et al. Phase I study of bortezomib in refractory or relapsed acute leukemias. Clin Cancer Res 2004; 10: 3371-76; PMID:15161691; http://dx.doi.org/10.1158/1078-0432.CCR-03-0508
- Dispenzieri A, Jacobus S, Vesole DH, Callandar N, Fonseca R, Greipp PR. Primary therapy with single agent bortezomib as induction, maintenance and re-induction in patients with high-risk myeloma: results of the ECOG E2A02 trial. Leukemia 2010; 24: 1406-11; PMID:20535147; http://dx.doi.org/10.1038/leu.2010.129
- Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, Mitsiades C, Mitsiades N, Yasui H, Letai A, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005; 8: 407-19; PMID:16286248; http://dx.doi.org/10.1016/j.ccr.2005.10.013
- Hideshima T, Chauhan D, Richardson P, Anderson KC. Identification and validation of novel therapeutic targets for multiple myeloma. J Clin Oncol 2005; 23: 6345-50; PMID:16155018; http://dx.doi.org/10.1200/JCO.2005.05.024
- Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008; 112: 2489-99; PMID:18565852; http://dx.doi.org/10.1182/blood-2007-08-104950
- Nakamura T, Tanaka K, Matsunobu T, Okada T, Nakatani F, Sakimura R, Hanada M, Iwamoto Y. The mechanism of cross-resistance to proteasome inhibitor bortezomib and overcoming resistance in Ewing's family tumor cells. Int J Oncol 2007; 31: 803-11; PMID:17786311
- O'Connor R, Ooi MG, Meiller J, Jakubikova J, Klippel S, Delmore J, Richardson P, Anderson K, Clynes M, Mitsiades CS, et al. The interaction of bortezomib with multidrug transporters: implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother Pharmacol 2013; 71: 1357-68; PMID:23589314; http://dx.doi.org/10.1007/s00280-013-2136-7
- Rumpold H, Salvador C, Wolf AM, Tilg H, Gastl G, Wolf D. Knockdown of Pgp resensitizes leukemic cells to proteasome inhibitors. Biochem Biophys Res Commun 2007; 361: 549-54; PMID:17662692; http://dx.doi.org/10.1016/j.bbrc.2007.07.049
- Fujita T, Washio K, Takabatake D, Takahashi H, Yoshitomi S, Tsukuda K, Ishibe Y, Ogasawara Y, Doihara H, Shimizu N. Proteasome inhibitors can alter the signaling pathways and attenuate the P-glycoprotein-mediated multidrug resistance. Int J Cancer 2005; 117: 670-82; PMID:15945097; http://dx.doi.org/10.1002/ijc.21063
- Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 2001; 61, 3071-6; PMID:11306489
- Yamada T, Takaoka AS, Naishiro Y, Hayashi R, Maruyama K, Maesawa C, Ochiai A, Hirohashi S. Transactivation of the multidrug resistance 1 gene by T-cell factor 4/beta-catenin complex in early colorectal carcinogenesis. Cancer Res 2000; 60: 4761-6; PMID:10987283
- Labialle S, Gayet L, Marthinet E, Rigal D, Baggetto LG. Transcriptional regulators of the human multidrug resistance 1 gene: recent views. Biochem Pharmacol 2002; 64: 943-8; PMID:12213590; http://dx.doi.org/10.1016/S0006-2952(02)01156-5
- Luu HH, Zhang R, Haydon RC, Rayburn E, Kang Q, Si W, Park JK, Wang H, Peng Y, Jiang W, et al. Wnt/beta-catenin signaling pathway as a novel cancer drug target. Curr Cancer Drug Targets 2004; 4: 653-71; PMID:15578921; http://dx.doi.org/10.2174/1568009043332709
- Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003; 2: 339-45; PMID:12851486; http://dx.doi.org/10.4161/cc.2.4.433
- Wagenknecht B, Hermisson M, Eitel K, Weller M. Proteasome inhibitors induce p53/p21-independent apoptosis in human glioma cells. Cell Physiol Biochem 1999; 9: 117-25; PMID:10494025; http://dx.doi.org/10.1159/000016308
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282: 1497-501; PMID:9822382; http://dx.doi.org/10.1126/science.282.5393.1497
- Kim OH, Lim JH, Woo KJ, Kim YH, Jin IN, Han ST, Park JW, Kwon TK. Influence of p53 and p21Waf1 expression on G2/M phase arrest of colorectal carcinoma HCT116 cells to proteasome inhibitors. Int J Oncol 2004; 24: 935-41; PMID:15010833
- Harker WG, Sikic BI. Multidrug (pleiotropic) resistance in doxorubicin-selected variants of the human sarcoma cell line MES-SA. Cancer Res 1985; 45: 4091-6; PMID:4028002
- Calcagno AM, Ambudkar SV. Molecular mechanisms of drug resistance in single-step and multi-step drug-selected cancer cells. Methods Mol Biol 2010; 596: 77-93; PMID:19949921; http://dx.doi.org/10.1007/978-1-60761-416-6_5
- Calcagno AM, Salcido CD, Gillet JP, Wu CP, Fostel JM, Mumau MD, Gottesman MM, Varticovski L, Ambudkar SV. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J Natl Cancer Inst 2010; 102: 1637-52; PMID:20935265; http://dx.doi.org/10.1093/jnci/djq361
- Lauricella M, D'Anneo A, Giuliano M, Calvaruso G, Emanuele S, Vento R, Tesoriere G. Induction of apoptosis in human osteosarcoma Saos-2 cells by the proteasome inhibitor MG132 and the protective effect of pRb. Cell Death Differ 2003; 10: 930-2; PMID:12868001; http://dx.doi.org/10.1038/sj.cdd.4401251
- Yuan B-Z, Chapman JA, Reynolds SH. Proteasome inhibitor MG132 induces apoptosis and inhibits invasion of human malignant pleural mesothelioma cells. Transl Oncol 2008; 1: 129-14; PMID:18795123; http://dx.doi.org/10.1593/tlo.08133
- Corrêa S, Binato R, Du Rocher B, Castelo-Branco MT, Pizzatti L, Abdelhay E. Wnt/β-catenin pathway regulates ABCB1 transcription in chronic myeloid leukemia. BMC Cancer 2012; 12: 303; http://dx.doi.org/10.1186/1471-2407-12-303
- Sharma RC, Inoue S, Roitelman J, Schimke RT, Simoni RD. Peptide transport by the multidrug resistance pump. J Biol Chem 1992; 267: 5731-4; PMID:1348245
- Minderman H, Zhou Y, O'Loughlin KL, Baer MR. Bortezomib activity and in vitro interactions with anthracyclines and cytarabine in acute myeloid leukemia cells are independent of multidrug resistance mechanisms and p53 status. Cancer Chemother Pharmacol 2007; 60: 245-55; PMID:17096161; http://dx.doi.org/10.1007/s00280-006-0367-6
- Verbrugge SE, Assaraf YG, Dijkmans BA, Scheffer GL, Al M, den Uyl D, Oerlemans R, Chan ET, Kirk CJ, Peters GJ, van der Heijden JW, et al. Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno) proteasome inhibitors inmononuclear blood cells from patients with rheumatoid arthritis. J Pharmacol Exp Ther 2012; 341: 174-82; PMID:22235146; http://dx.doi.org/10.1124/jpet.111.187542
- Zheng B, Zhou R, Gong Y, Yang X, Shan Q. Proteasome inhibitor bortezomib overcomes P-gp-mediated multidrug resistance in resistant leukemic cell lines. Int J Lab Hematol 2012; 34: 237-47; PMID:22145750; http://dx.doi.org/10.1111/j.1751-553X.2011.01384.x
- Cervello M, Giannitrapani L, La Rosa M, Notarbartolo M, Labbozzetta M, Poma P, Montalto G, D'Alessandro N. Induction of apoptosis by the proteasome inhibitor MG132 in human HCC cells: Possible correlation with specific caspase-dependent cleavage of beta-catenin and inhibition of beta-catenin-mediated transactivation. Int J Mol Med 2004; 13, 741-8; PMID:15067380
- Conzen SD, Gottlob K, Kandel ES, Khanduri P, Wagner AJ, O'Leary M, Hay N. Induction of cell cycle progression and acceleration of apoptosis are two separable functions of c-Myc: transrepression correlates with acceleration of apoptosis. Mol Cell Biol 2000; 20: 6008-18; PMID:10913183; http://dx.doi.org/10.1128/MCB.20.16.6008-6018.2000
- Hung TH, Chen CM, Tseng CP, Shen CJ, Wang HL, Choo KB, Chong KY. FZD1 activates protein kinase C delta-mediated drug-resistance in multidrug-resistant MES-SA/Dx5 cancer cells. Int J Biochem Cell Biol 2014; 53: 55-65; PMID:24814288; http://dx.doi.org/10.1016/j.biocel.2014.04.011
- Huang CL, Cheng JC, Kitajima K, Nakano T, Yeh CF, Chong KY, Tseng CP. Disabled-2 is required for mesoderm differentiation of murine embryonic stem cells. J Cell Physiol. 2010; 225: 92-105; PMID:20648627; http://dx.doi.org/10.1002/jcp.22200
- Hsu HT, Liu PC, Ku SY, Jung KC, Hong YR, Kao C, Wang C. Beta-catenin control of T-cell transcription factor 4 (Tcf4) importation from the cytoplasm to the nucleus contributes to Tcf4-mediated transcription in 293 cells. Biochem Biophys Res Commun 2006; 343: 893-8; PMID:16564030; http://dx.doi.org/10.1016/j.bbrc.2006.02.193
- Chong KY, Lai CC, Su CY. Inducible and constitutive HSP70s confer synergistic resistance against metabolic challenges. Biochem Biophys Res Commun. 2013; 430: 774-9; PMID:23206709; http://dx.doi.org/10.1016/j.bbrc.2012.11.072
- Hung TH, Hsu SC, Choo KB, Tseng CP. Chen TC, Lan YW, Huang TT, Lai HC, Chen CM, Chong KY. Wnt5A regulates ABCB1 expression in multidrug-resistant cancer cells through activation of the non-canonical PKA/β-catenin pathway. Oncotarget 2014; 5: 12273-90.