
The cellular response to DNA damage ranges from transient cell cycle arrest to the induction of apoptosis or senescence. It is generally thought that permanent cell cycle withdrawal is initiated in response to severe, irreparable DNA damage. This way, cells can prevent the propagation of a damaged genome. Alternatively, following DNA repair, the cell can shut down the checkpoint and resume the cell cycle, a process known as checkpoint recovery. If a DNA damaged-induced arrest is established in G2 phase, cells need to maintain the expression of cell cycle regulatory proteins, such as Cyclin B1, in order to retain the ability to resume cell cycle progression. On the other hand, degradation of cell cycle regulatory proteins due to the activation of APC/CCdh1 leads to cell cycle exit.
In this edition of Cell Cycle Müllers et al. have investigated the regulation of checkpoint recovery and cell cycle exit, by investigating Cyclin B1 dynamics in response to DNA damage in single cells. Consistent with previous observations,Citation1–3 Müllers et al. find that non-transformed cells respond to DNA damage by translocating Cyclin B1 to the nucleus in a p53- and p21-dependent manner.Citation4 Nuclear translocation of Cyclin B1 is followed by APC/CCdh1 dependent degradation of Cyclin B1. Interestingly, DNA damage-induced nuclear translocation of Cyclin B1 marks the point-of-no-return during the DNA damage response, as cells with nuclear Cyclin B1 fail to induce mitosis upon inhibition of the DNA damage checkpointCitation2,4 (, top). The finding that Cyclin B1 becomes nuclear in response to DNA damage is counterintuitive, since nuclear translocation of Cyclin B1 marks the onset of mitosis in an unperturbed cell cycle. However, it was shown that, following DNA damage, the p21-bound Cyclin B1-Cdk1 complexes in the nucleus are inactive and cannot be dephosphorylated by Cdc25.Citation1,2 Consequently, nuclear p21-bound Cyclin B1-Cdk1 complexes are refractory to reactivation upon silencing of the DNA damage checkpoint, thus marking a point-of-no-return during the DNA damage response. In addition, Müllers et al. now report that transformed U2OS cells, unlike non-transformed cells, do not translocate Cyclin B1 to the nucleus in response to DNA damage.Citation4 Accordingly, these cells maintain recovery competence much longer than do non-transformed cells (, middle).
Figure 1. DNA damage in p53-proficient cells induces nuclear translocation of Cyclin B1 and subsequent APC/CCdh1 activation, leading to cell cycle exit (top). p53-impaired cells fail to translocate Cyclin B1, and maintain a reversible checkpoint (middle). Forced p21-expression or Wip1 inhibition following DNA damage may result in nuclear translocation of Cyclin B1 and induction of cell cycle exit in p53-impaired cells (bottom).
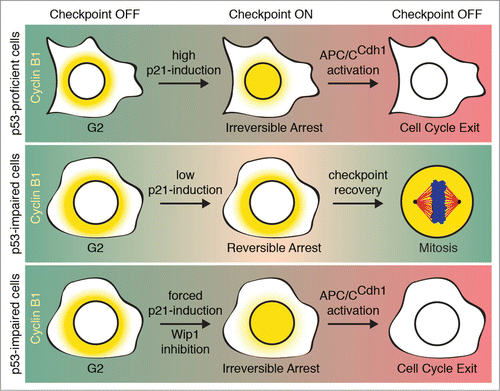
Induction of DNA damage, through irradiation or exposure to DNA damaging chemotherapeutics, is routinely used in the treatment of cancer. This strategy relies on the induction of cell death or cell cycle exit in the damaged cells. However, resistance to these therapies may arise over time due to the survival of a few cells. Given the findings from Müllers et al, it will be interesting to see if restoration of the cell-cycle exit machinery in these transformed cells can increase sensitivity to DNA damaging agents and potentially decrease therapy resistance. In support of this hypothesis is the finding that forced induction of p21 in U2OS cells can result in the nuclear translocation of inactive Cyclin B1,Citation5 indicating that the underlying cell-cycle exit mechanisms are present yet suppressed in these cells (, bottom). Recently described gain-of-function mutations of Wip1 in U2OS and HCT116 cells were shown to contribute to the suppression of p21-induction upon DNA damage,Citation6 providing a possible explanation for the fact that Cyclin B1 does not translocate to the nucleus in these cells after DNA damage. Similar mutations were found in cancer patients, highlighting the clinical relevance of these findings. The recent discovery of a Wip1 inhibitorCitation7 makes it possible to investigate whether inhibition of Wip1 in U2OS and other Wip1-mutated cells can reactivate a DNA damage-induced cell cycle exit mechanism (, bottom), and possibly improve therapeutic outcome in patients.
References
- Charrier-Savournin FB, et al. Mol Biol Cell 2004; 15:3965-76; PMID:15181148; http://dx.doi.org/10.1091/mbc.E03-12-0871
- Krenning Let al. Mol Cell 2014; 55:59-72; PMID:24910099; http://dx.doi.org/10.1016/j.molcel.2014.05.007
- Johmura Y, et al. Mol Cell 2014; 55:73-84; PMID:24910096; http://dx.doi.org/10.1016/j.molcel.2014.05.003
- Müllers E, et al. Cell Cycle 2014; 13:2733-43; http://dx.doi.org/10.4161/15384101.2015.945831
- Smits VA, et al. J Biol Chem 2000; 275:30638-43; PMID:10913154; http://dx.doi.org/10.1074/jbc.M005437200
- Kleiblova P, et al. J Cell Biol 2013; 201:511-21; PMID:23649806; http://dx.doi.org/10.1083/jcb.201210031
- Gilmartin AG, et al. Nat Chem Biol 2014; 10:181-7; PMID:24390428; http://dx.doi.org/10.1038/nchembio.1427