
Dysregulation of the DNA damage response (DDR) creates the genomic instability that promotes tumorigenesis. Depending on the type of DDR defect, it can have prognostic significance and confer sensitivity or resistance to different chemotherapeutic drugs. Exploiting defects in the DDR by inhibiting complementary DDR pathways is an exciting new paradigm in anticancer therapy. The prime example is the exquisite sensitivity of cells with defects in BRCA1 and BRCA2 to PARP inhibitors (PARPi).Citation1 BRCA1 and BRCA2 are key components of homologous recombination DNA repair (HRR) while PARP plays a central role in DNA single strand (SS) break repair (SSBR). While loss of SSBR or HRR alone does not impact significantly on viability that disruption of both SSBR and HRR together is synthetically lethal. Several PARPi are in currently late-stage clinical evaluation, largely in patients with tumors harbouring known (BRCA mutations) or suspected defects in HRR. Since HRR is a multicomponent pathway and a defect in any one component can compromise the whole pathway the search is on for other determinants of sensitivity to PARPi in order to identify patients that may benefit from this novel tumor-specific therapeutic approach.
ATM signals DNA double-strand breaks (DSBs) to cell cycle checkpoints via Chk2 and p53. ATM has been linked to HRR and knockdown of ATM is also synthetically lethal with PARPi.Citation2 Following on from their studies in ATM-defective Mantle cell leukemia,Citation3 and similar studies in ATM-defective (11q deletion) chronic lymphocytic leukemia,Citation4 the group in Calgary now demonstrate the increased sensitivity of gastric carcinoma cells with low levels of ATM protein and p53 dysfunction to the PARPi, olaparib.Citation5 They support this finding with a number of complementary approaches using unmatched cells with differing ATM and p53 status, ATM and p53 knockdown and the use of inhibitors, with converging results. Based on these data one might speculate that PARP, by promoting repair, reduces replication stress and replication-associated DSBs and that p53 and ATM prevent replication stress leading to cell death by activating cell cycle arrest and also promoting repair. Therefore PARPi will preferentially kill cells in which cell cycle checkpoint activation has been compromised by the inactivation of p53 and ATM ().
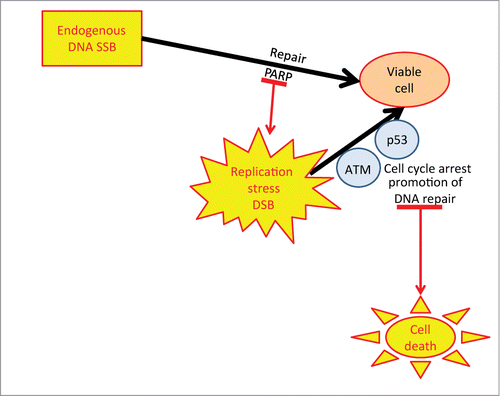
Figure 1. Role of PARP ATM and p53 in cell viability Endogenous DNA damage is repaired by PARP to maintain viability. If PARP is inhibited replication stress and DSB ensue, triggering p53 and ATM to arrest the cell and promote repair of the DSB. In the absence/inhibition of ATM and p53 function the DSB will accumulate and the cell will progress through the cell cycle with broken DNA leading to cell death.
Since gastric cancer is the second most common cause of cancer deaths in the world and many harbour defects in ATM, associated with microsatellite instability,Citation6 these findings suggest that PARPi therapy may benefit a substantial number of patients and that ATM levels may be used as a biomarker to stratify patients to receive a PARPi or conventional therapy. Olaparib, in combination with paclitaxel was recently shown to be of benefit in patients with gastric cancer, particularly those with low ATM levels determined by IHC (NCT01063517), demonstrating the feasibility of the approach.Citation7
Interestingly, in the study by Kubota et al Citation5 levels of ATM protein in the cell line panel did not correlate with mutations in the ATM gene on the COSMIC database, and the olaparib sensitivity correlated with the protein level rather than the genomic data. This may have implications for clinical trials of molecularly targeted agents, such as PARPi, in which the patients are stratified on the basis of genomics rather than protein levels.
References
- Bryant HE, et al. Nature 2005; 434:913-7; PMID:15829966
- McCabe N, et al. Cancer Res 2006; 66:8109-15; PMID:16912188
- Williamson CT, et al. EMBO Mol Med 2012; 4:515-27; PMID:22416035; http://dx.doi.org/10.1002/emmm.201200229
- Weston VJ, et al. Blood 2010; 116:4578-87; PMID:20739657; http://dx.doi.org/10.1182/blood-2010-01-265769
- Kubota E, et al. Cell Cycle 2014; 13(13):2129-37; PMID:24841718; http://dx.doi.org/10.4161/cc.29212
- Kim JW, et al. Int J Cancer 2014; 134:72-80; PMID:23649938; http://dx.doi.org/10.1002/ijc.28245
- Bang YJ, et al. J Clin Oncol 2013; 31