
Abstract
Whereas many components regulating the progression from S phase through G2 phase into mitosis have been identified, the mechanism by which these components control this critical cell cycle progression is still not fully elucidated. Cyclin A/Cdk2 has been shown to regulate the timing of Cyclin B/Cdk1 activation and progression into mitosis although the mechanism by which this occurs is only poorly understood. Here we show that depletion of Cyclin A or inhibition of Cdk2 during late S/early G2 phase maintains the G2 phase arrest by reducing Cdh1 transcript and protein levels, thereby stabilizing Claspin and maintaining elevated levels of activated Chk1 which contributes to the G2 phase observed. Interestingly, the Cyclin A/Cdk2 regulated APC/CCdh1 activity is selective for only a subset of Cdh1 targets including Claspin. Thus, a normal role for Cyclin A/Cdk2 during early G2 phase is to increase the level of Cdh1 which destabilises Claspin which in turn down regulates Chk1 activation to allow progression into mitosis. This mechanism links S phase exit with G2 phase transit into mitosis, provides a novel insight into the roles of Cyclin A/Cdk2 in G2 phase progression, and identifies a novel role for APC/CCdh1 in late S/G2 phase cell cycle progression.
Abbreviations
| Cdk | = | cyclin dependent kinase |
| Cdki | = | cyclin dependent kinase inhibitor |
| Chk1 | = | checkpoint kinase 1 |
| APC/C | = | anaphase promoting complex/cyclosome |
Introduction
Cell cycle progression is regulated by the ordered activation of Cyclin/Cdk complexes.Citation1 Cyclins are the regulatory subunits of these protein kinase complexes and are essential for activation and substrate specificity of these complexes. Cdk1 is the only essential mammalian Cdk and it can compensate for deletion of other individual Cdks in all but a few tissue-specific functions.Citation2,3 Despite this redundancy, individual Cdks have important functions in the somatic cell cycle. Cdk2 is the most closely related to Cdk1 and has critical roles in initiation of DNA replication when complexed with Cyclin E, and work from a number of groups have shown that Cyclin A/Cdk2 has roles in S phase progression and, perhaps more critically, in G2 phase it regulates the timing of Cyclin B/Cdk1 activation and entry into mitosis.Citation4-8
Several mechanisms by which Cyclin A/Cdk2 controls the subsequent activation of Cyclin B/Cdk1 and progression into mitosis have been proposed, including regulating nuclear envelope breakdown and Cyclin B/Cdk1 accumulation in the nucleus,Citation7 and regulation of the negative regulator of Cyclin B/Cdk1 activity Wee1.Citation5,9 We have previously reported that Cyclin A/Cdk2 also co-ordinates the activation of centrosomal and nuclear pools of Cyclin B/Cdk1.Citation4 This has also been reported to be regulated by the checkpoint kinase Chk1.Citation10 Active Chk1 is present in cells in the absence of DNA damage during interphase,Citation10-12 and inhibition or depletion of Chk1 during an unperturbed G2 phase results in premature activation of Cyclin B/Cdk1 and entry into mitosis, indicating a role for Chk1 in regulating the normal timing of entry into mitosis during normal cell cycle progression.Citation12,13 Cyclin A/Cdk has also been suggested to regulate Chk1 activity directly through the phosphorylaiton of inhibitory sites.Citation14
Chk1 is activated by the apical checkpoint signaling kinase ATR, and this activation is critically dependent on association with an adaptor protein, Claspin.Citation15,16 ATR activates Chk1 by phosphorylation of specific serine residues (Ser317 and Ser345) which have overlapping functions.Citation17,18 Claspin appears to be primarily regulated at the level of its protein stability. The E3 ubiquitin ligase complex APC/C (anaphase promoting complex/cyclosome) regulates exit from mitosis and establishes a stable G1 phase through the ubiquitin-mediated degradation of a suite of protein substrates. APC/C has two substrate recognition subunits, Cdc20 and Cdh1, and although they have some substrate overlap, APC/CCdc20 initiates mitotic exit, while APC/CCdh1 completes this process and establishes G1 phase.Citation19,20 Claspin stability is regulated by βTrCP in mitosis through Plk1-dependent phosphorylation of a phosphodegron,Citation21 and in G1 phase by APC/CCdh1-dependent ubiquitination of Claspin.Citation22,23 Cyclin A/Cdk2 has been reported to bind and phosphorylates Cdh1/Fzr1 to maintain the APC/C in an inactive form during S and G2 phase.Citation24,25
Here we have investigated the mechanism by which Cyclin A/Cdk2 controls the timing of Cyclin B/Cdk1 activation and entry into mitosis in the unperturbed cell cycle, assessing the effect of Cyclin A depletion on known Cyclin A/Cdk interacting proteins with potential G2 phase regulatory functions, Cdh1 and Chk1. We identify an unexpected role for Cyclin A/Cdk2 in regulating Cdh1 levels and thereby APC/CCdh1-regulated Claspin levels and Chk1 activation during normal G2 phase progression.
Results
Cyclin A depletion decreased Cdh1 levels
To assess the effect of Cyclin A depletion on G2 functions, synchronised HeLa cells cultures were used. Depletion of Cyclin A using 2 independent siRNA previously shown to be highly selective for Cyclin ACitation4,26was found to reduce G2 phase Cdh1 levels by 80% (). This effect was mimicked by Cdk2, but not Cdk1 inhibition. The reduction in Cdh1 levels was also observed in asynchronously growing cells, although this was less than observed in synchronised G2 phase cells (), suggesting that the effect is cell cycle phase specific. Cyclin A depletion similarly reduced Cdh1 levels in asynchronous U2OS cells (). The effect of Cyclin A siRNA on Cdh1 levels was rescued by over expressing a Cyclin A-Cherry fusion construct, either wild type or a mutant that was more resistant to Cyclin A siRNA#1,Citation26 in HEK293T cells. Overexpression of both Cyclin A-Cherry fusions alone increased Cdh1 levels, and rescued the reduction in Cdh1 protein levels with Cyclin A siRNA#1 transfection which depleted the endogenous Cyclin A levels (Fig. S1A). These data demonstrated that Cyclin A/Cdk2 was responsible for the maintenance of Cdh1 levels in G2 phase cells.
Figure 1. Cdh1 levels are reduced with Cyclin A depletion. (A) HeLa cells were transfected with nonsense control (NS), cyclin A siRNA #1 and #3 or Cdh1 siRNA, then synchronised with overnight thymidine arrest. Untransfected cells were treated with inhibitors of Cdk2 (Cdk2i), Cdk1 (Cdk1i) or DMSO control (Con) at 4 h after release. Cells were harvested 7 h after release from the synchrony arrest, lysed and immunoblotted for cyclin A, Cdh1 and α-tubulin as a loading control. (B) The bar graph shows quantitation of Cdh1 levels from at least 3 similar independent experiments. (C) Asynchronously growing HeLa cells were transfected with Cyclin A siRNA #1 and #3, a nonsense control (NS) or a lipofectamine treated only control (Con). Cells were harvested at 24 h. The bar graph show quantitation of Cdh1 levels from at least three independent experiments. (D) Asynchronously growing U2OS cells were transfected with the indicated siRNA and harvested 24 h after transfection and analyzed as in B. This is representative of three independent experiments. (E) HeLa cells transfected with either nonsense (NS), Cyclin A siRNA (#1), Cdh1 siRNA, or combinations indicated. Cells were synchronised by thymidine overnight then released into fresh media. At 5 h after release cells were followed by time lapse microscopy, with cells scored for entry into mitosis. Over 200 cells were counted in each case. Inset shows the level of Cyclin A and Cdh1 depletion in this experiment.
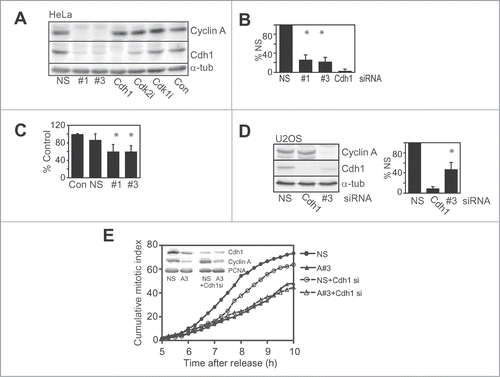
The contribution of the reduced levels of Cdh1 to the G2 phase delay resulting from Cyclin A/Cdk2 depletion/inhibition was assessed in Cdh1 siRNA depleted HeLa cells using time lapse microscopy. Direct siRNA depletion of Cdh1 delayed progression of synchronised cells into mitosis. The extent of the delay appeared dependent on the level of depletion. Depletion of >90% Cdh1 resulted in a delay similar to Cyclin A depletion (Fig. S1B), whereas suboptimal Cdh1siRNA treatment which reduced Cdh1 in to a similar level to that found with Cyclin A depletion, resulted in a shorter delay of entry into mitosis (). Co-depletion of Cdh1 and Cyclin A produced the same delay as Cyclin A alone indicating the effects were via the same mechanism. These data demonstrated that the decreased level of Cdh1 with Cyclin A depletion was a significant contributor to the G2 phase delay observed, but does not alone account for the delay.
The mechanism by which Cyclin A/Cdk2 regulated Cdh1 levels in G2 phase was examined. The reduced level of Cdh1 with Cyclin A/Cdk2 depletion/inhibition was not rescued by inhibition of proteasome activity with MG132, indicating that the reduction was not via proteasome-mediated degradation (). MG132 treatment had no effect on Cdh1 levels in G2 phase cells, but it did increase the levels of Claspin, the APC/C inhibitor Emi1, and Cyclin A to a more modest extent. Interestingly, the Plk1 inhibitor BI-2536 had no effect on Cdh1 levels suggesting that the G1 phase Plk1-dependent destabilisation of Cdh1 did not operate in G2 phase.Citation27 Similarly, the stability of a mutant form of Cdh1 unable to bind Cyclin A was essentially identical to the wild type Cdh1 in G2 phase HEK and HeLa cells (Fig. S2), demonstrating that Cyclin A/Cdk2 did not affect the stability of Cdh1 in G2 phase.
Figure 2. Cyclin A depletion reduces Cdh1 transcript levels. (A) Cells were transfected and synchronised as in A. At 4 h post release cells were treated with or without 20 μM of MG132, Plk1 inhibitor (Plk1i) or Cdk2 inhibitor (Cdk2i) for 3 h. All samples were then collected 7 h post release. Samples were lysed and immunoblotted for the indicated proteins. This is representative of duplicate experiments. (B) HeLa cells were transfected and synchronised as in A, although using only nonsense (NS) and Cyclin A siRNA#1. At 7 h post release when cells were in G2 phase, 10 μg/ml cyclohexamide was added, and washout out 3 h later. Cells were sampled at the indicated times after cyclohexamide addition and immunoblotted for Cdh1, and PCNA as a loading control. (C) Asynchronously growing HeLa cells were transfected with a nonsense control (NS), Cyclin A siRNA #1 or #3, or Cdh1 siRNA. Cells were synchronised with thymidine arrest overnight and released, harvested at 7 h post release when they were in G2 phase and their Cdh1 mRNA content assessed by qRT-PCR. The data are the mean and SEM of four independent experiments and expressed relative to the nonsense control. The level of Cdh1 was quantitated relative to the nonsense control.
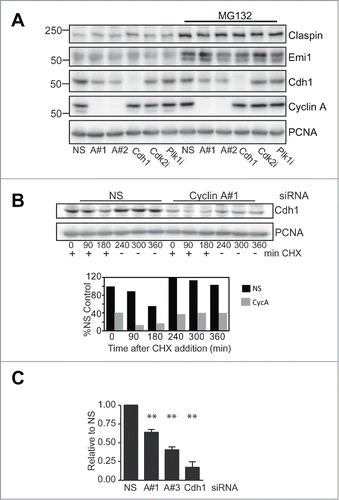
The effect of Cyclin A depletion on de novo synthesis of Cdh1 protein was investigated using a cyclohexamide block release. Incubation with cyclohexamide for 3 h was sufficient to reduce the protein level 50% in the nonsense treated control, and this was rapidly rescued with washout of the cyclohexamide (). Although the level of Cdh1 was <40% of the control in the Cyclin A depleted samples, it was also reduced to 50% of its starting level and rapidly rescued with cyclohexamide washout indicating that there was no overall effect on the translational controls.
The level of Cdh1 mRNA was assessed by qPCR in G2 phase synchronised HeLa cells using two separate probes. There was a 40–50% reduction in Cdh1mRNA levels with two separate Cyclin A siRNAs, corresponding to the reduction in Cdh1 protein and translation observed (), indicating that Cyclin A/Cdk regulates the level of Cdh1 transcript in G2 phase, and thereby the Cdh1 translation and protein levels.
Cyclin A depletion increases the stability of claspin
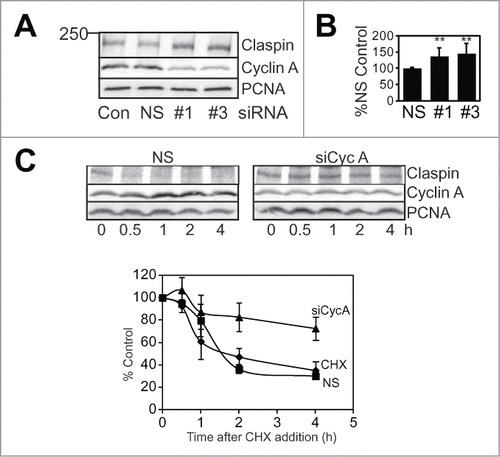
The reduced level of Cdh1 resulting from Cyclin A depletion would be expected to affect the stability of APC/CCdh1 substrate proteins. We observed a consistent 50% increase in Claspin levels with both Cyclin A siRNAs (). G2 phase Claspin levels are regulated by proteasome-mediated degradation, demonstrated by the increased Claspin levels in MG132 treated G2 phase cells (), suggesting that Cyclin A/Cdk2 may regulate Claspin stability. When Claspin protein stability was assessed, Cyclin A depletion increased the half life of Claspin from 1.5 h to over 4 h in synchronised G2 phase cell ().
Figure 3. Cyclin A depletion increases Claspin stability. (A) Asynchronously growing HeLa cells were transfected with a lipofectamine treated only control (Con), nonsense (NS) or Cyclin A siRNAs (#1 and #3). 24 h post transfection cells were collected, lysed and immunoblotted for Claspin, Cyclin A and PCNA. (B) The level of Claspin after treatment relative to nonsense treated control from four independent experiments is shown. The double asterix indicates P < 0.01. (C) HeLa cells were transfected with either nonsense (NS) or Cyclin A siRNA (#3) then synchronised, cyclohexamide (CHX; 10 μg/ml) was added to G2 phase cells and harvested at the indicated time after CHX addition. Lysates were immunoblotted for Claspin, and PCNA as a loading control. The data is from three independent experiments.
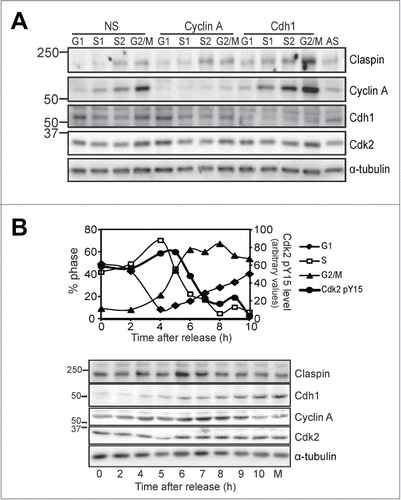
The effect of Cyclin A depletion and Cdk2 inhibition on Cdh1 and Claspin levels appear to be maximal in G2 phase synchronised cells. To ensure that this was not an artefact of the synchrony procedure, cultures that had been transfected with either nonsense, or siRNA to deplete Cyclin A or Cdh1 were sorted by their DNA content by flow cytometry into G1, early and late S phase (S1, S2) and G2/M phase (Fig. S3). Immunoblotting these fractions showed the expected elevated levels of Cdh1 in G1 and G2/M fractions, whereas there appeared to be an accumulation above the control G1 level with Cyclin A depletion and loss of the G2/M phase accumulation. Claspin levels appeared highest in the late S phase fraction in the nonsense control, and there was a modest accumulation of Claspin in the Cyclin A depleted G2/M fraction (). Interestingly, the Cdh1 depleted cells showed a strong accumulation of Claspin in all fractions compared with nonsense controls, and accumulation of Cyclin A, a known APC/CCdh1 substrate.Citation28 This temporal association was found in synchronised cells progressing through S into G2/M phase. There were elevated levels of Claspin during S phase that appeared to be maximal at 6 h after synchrony release and then reduced as cells progressed into mitosis (). This correlated with cells with increase in Cdh1 levels observed from 5 h and the decrease in the inactive Tyr15 phosphorylated Cdk2 associated with Cyclin A (), corresponding to the Cdc25B-dependent G2 phase activation of Cyclin A/Cdk2.Citation29 Together, these data provide strong evidence that Cyclin A/Cdk2 regulates Claspin levels through Cdh1-dependent proteasomal degradation during late S/G2 phase.
Figure 4. Cyclin A regulates S/G2 phase Cdh1 and Claspin levels.(A) HeLa cells were transfected with ether nonsense (NS), Cyclin A (#1) or Cdh1 siRNA for 24 h then fixed, stained for their DNA content with propidium iodide and sorted into four fractions, G1, early (S1), late S phase (S2), and G2/M phase. The histograms are presented in Figure S5. Fractions were immunoblotted for the indicated proteins. An asynchronous culture is shown as a control (AS). These data are representative of replicate experiments. (B) Thymidine synchronised HeLa cells were harvested at the indicated times after release. Samples were analyzed by FACS for their cell cycle distribution, or lysed and immunoblotted for the indicated proteins. Lysates were also immunoprecipitated for Cyclin A and the immunoprecipitates immunoblotted for the presence of the inactive phosphoTyr15 Cdk2. The level of this inactive form was measured by densitometry and shown on the same graph as the cell cycle distribution (Cdk2 pY15). Mitotic shake-off sample (M) was run as a control. α-Tubulin was used as a loading control.
Cyclin A/Cdk2 regulates only a subset of G2 phase Cdh1 substrates
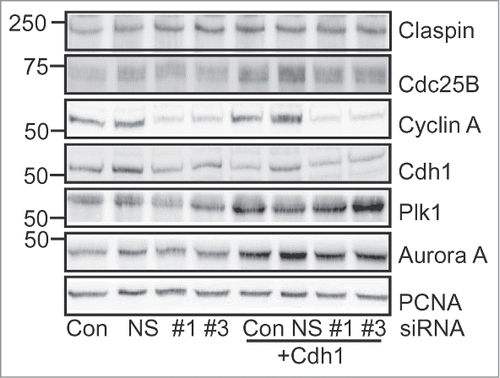
The reduction in Cdh1 levels and corresponding increase in the stability of Claspin with Cyclin A depletion suggested that Claspin is an APC/CCdh1 target in G2 phase. It would also be expected that the levels of other APC/CCdh1 targets such as Plk1 and Aurora ACitation28,30 would increase. Surprisingly, no change in the level of Plk1 or Aurora A was observed with Cyclin A depletion, whereas Cdh1 siRNA-mediated depletion increased the levels of Claspin, Plk1 and Aurora A two-fold (). It also increased the levels of another critical G2/M regulator Cdc25B. Although not a known Cdh1 target, Cdc25B is transcriptionally regulated by the Cdh1 target FoxM1, and increased FoxM1 level is likely to account for the increase observed with Cdh1 depletion.Citation31 Co-depletion of Cyclin A and Cdh1 did not significantly affect the levels of these proteins beyond Cdh1 depletion alone. The difference in the effects of Cyclin A depletion and direct Cdh1 siRNA mediated Cdh1 depletion did not appear to be reliant on the level of Cdh1 depletion. In the experiment shown (), similar levels of Cdh1 were achieved with both Cdh1 and Cyclin A siRNA.
Figure 5. Cyclin A/cdk2 regulates a subset of Cdh1 targets. HeLa cells were transfected with either Lipofectamine alone (Con), nonsense (NS), or Cyclin A siRNAs (#1, #3) without and with co-transfection with Cdh1 siRNA. Cells were synchronised with thymidine and harvested at 7 h after release when they were in G2 phase, lysed and immunoblotted for the indicated proteins.
Cyclin A depletion maintains Chk1 activation through reduced Cdh1 stabilizing Claspin
The regulation of Cdh1-Claspin by Cyclin A/Cdk2 in G2 phase suggested that this may maintain Chk1 activity thereby inhibiting Cyclin B/Cdk1 activation and entry into mitosis. We have previously demonstrated that Chk1 activity regulates normal G2/M phase progression.Citation12 Cyclin A depletion modestly increased the level of activated Ser317 phosphorylated Chk1 (pChk1 Ser317) in thymidine synchronised G2 phase HeLa cells with two independent siRNA to Cyclin A (#1, #3; ). Inhibition of Cdk2 similarly produced increased levels of activated Chk1and G2 phase delayCitation4 (Fig. S4A and B). The increase in activated Chk1 was not a consequence of increased DNA damage caused by Cyclin A siRNA treatment as measured by assessing the levels of γH2AX, a marker of DNA damage (Fig. S4C). Chk1 immunoprecipitates from lysates of either nonsense or Cyclin A siRNA treated G2 phase cells, or G2 phase cells treated with the Cdk2 inhibitor also revealed increased activated Chk1 (). A similar increase in Chk1 phosphorylated at Ser296, marker of active Chk1, was observed with Cyclin A depletion with each siRNA (Fig. S4D). No changes in the levels of activated p38MAPK and Chk2, two other known G2/M regulatory kinasesCitation32,33 were observed with Cyclin A depletion (data not shown).
Figure 6. Chk1 depletion attenuates the Cyclin A dependent G2 phase delay. (A) HeLa cells were transfected with nonsense control (NS), cyclin A siRNAs (#1, #3) or Cdh1 siRNA, then synchronised with overnight thymidine arrest. Cells were harvested 7 h after release from the synchrony arrest, lysed and immunoblotted for cyclin A, Cdh1, pChk1 Ser317 and α-tubulin as a loading control. (B) The bar graph shows quantitation of the increase in pChk1 levels with two independent siRNA, #1 and #3, relative to the nonsense siRNA from at least three independent experiments. The double asterix indicate P < 0.01. (C) HeLa cells treated with either nonsense or Cyclin A siRNA #3 where synchronised then harvested at 7 h post release in G2 phase. A parallel synchronised G2 phase sample treated with Cdk2 inhibitor was also prepared. Lysates from these samples were immunoprecipitated with Chk1 antibody, and the immunoprecipitates and lysates immunoblotted with the indicated antibodies. (D) Asynchronous HeLa cells were either mock transfected (Con) or transfected with nonsense (NS), Cyclin A siRNA (#1), Chk1 siRNA (Chk1) or Cyclin A and Chk1 siRNA. Cells were harvested after 24 h and analyzed for their DNA content by FACS. The data is the G2/M population from three independent experiments. Error bars represent standard error. (E) Samples from the experiment shown in D were immunoblotted Cyclin A, Chk1, pCdk1 Tyr15, and α-tubulin as a loading control. (F and G) HeLa cells treated with either nonsense (NS), Cyclin A (#1), #1 and Chk1 siRNA or #1 and 2.5 μM Chk1 inhibitor (Chk1i) were thymidine synchronised and followed by time lapse microscopy 6 hours after synchrony release. Cells were scored for entry into mitosis, over 200 cells were counted in each case. This result is a typical of three independent experiments.
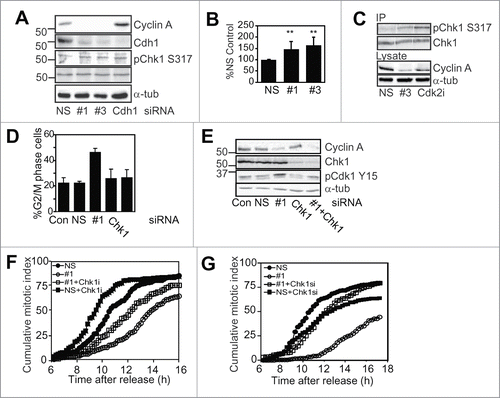
Cyclin A depletion resulted in an accumulation of G2/M phase cells and inactive pCdk1 Tyr15, a marker of G2 phase as reported previously,Citation4 and this was rescued by co-depletion of Chk1 (; Fig. S5). This was an incomplete rescue, demonstrated by the kinetics of mitotic entry assessed using time lapse microscopy. Neither inhibition using a small molecule inhibitor of Chk1 nor depletion Chk1 was capable of completely rescuing the G2 phase delay induced by Cyclin A depletion in synchronised HeLa cells (). Chk1 inhibition also attenuated the G2 phase delay imposed by Cdk2 inhibitor treatment (Fig. S4B).
Cyclin A/Cdk2 has been reported to phosphorylate Chk1 on the inhibitory Ser286 and Ser301 residues also phosphorylated by Cdk1.Citation14,34 Examination of G2 phase HeLa cells depleted of Cyclin A or treated with Cdk2 inhibitor revealed reduced levels of Chk1 Ser301 phosphorylation compared to the control, but this was also completely inhibited by the Cdk1 specific inhibitor Ro-3306, indicating that Cyclin A/Cdk2 regulates this inhibitory phosphorylation indirectly by regulating the timing of Cyclin B/Cdk1 activation (Fig. S6).
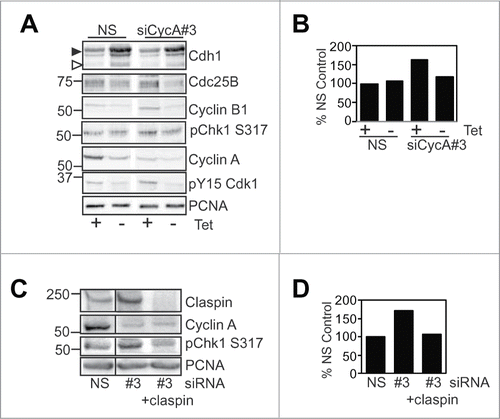
Induced Cdh1 expression in thymidine synchronised G2 phase Tet-Off Myc-Cdh1 U2OS cells promoted a G2 phase delay as reported previously,Citation28 and was able to reverse the increased phosphorylation of Chk1 Ser317 observed with Cyclin A depletion (). However, this did not rescue the G2 phase delay as the increased Cdh1 reduced the levels of critical mitotic regulators such as Cyclin B1 and Cdc25B. Induced Cdh1 expression also reduced Claspin levels (Fig. S7A). Likewise, co-depletion of Claspin and Cyclin A blocked the increase in activation Chk1 observed with Cyclin A depletion alone as expected (), but was unable to rescue the G2 phase delay induced by the Cyclin A depletion. This was because Claspin depletion delayed progression through S phase characterised by elevated S phase levels of Cdc25B, Wee1 and pCdk1Tyr15, although it did reduce the level of activated Chk1 (Fig. S7B and C). This demonstrates that inappropriate increases in Cdh1 level and consequent loss of Claspin during S phase are detrimental to progression from S phase to mitosis. It also demonstrates that in the absence of normal G2 phase Cyclin A/Cdk2-dependent regulation of Cdh1 levels, the inability to destabilise Claspin at the end of S phase results in maintenance of Chk1 activity, thereby inhibiting Cyclin B/Cdk1 activation and entry into mitosis.
Figure 7. Cdh1 over expression and Claspin depletion block Chk1 activation. (A and B) U2OS cell line with tetracycline repressed Myc-Cdh1 were transfected with either nonsense (NS) or Cyclin A siRNAs (#3), synchronised with overnight thymidine arrest, then at release either induced for Cdh1 (-Tet) or not (+Tet). 24 h post induction samples were collected, lysed and immunoblotted for the indicated proteins, PCNA was a loading control. The induced Cdh1 is indicated with the filled arrowhead, and endogenous with the open arrowhead. The level of pChk1 S317 was quantitated and expressed a% NS + tetracycline control. (C and D) HeLa cells were transfected with either nonsense (NS), Cyclin A (A#3), Claspin or Cyclin A and Claspin siRNA. Cells were harvested 24 h post transfection and immunoblotted for the indicated proteins. The level of pChk1 S317 was quantitated and expressed a% NS control.
Discussion
Here we have examined the mechanism by which Cyclin A/Cdk2 regulates normal G2/M phase progression, and found in three cell lines that Cyclin A depletion reduces Cdh1 levels selectively in late S/G2 phase cells. Recently, Cyclin A/Cdk2 acting through Plk1 and βTrCP was shown to regulate the stability of Cdh1 in G1 phase,Citation27 and Cyclin A depletion did appear to increase the level of G1 phase Cdh1 in our FACS sorted cell cycle fractions, supporting this model. Inhibition of Plk1 had no effect on Cdh1 levels in G2 phase, and the stability of a mutant Cdh1 unable to bind Cyclin A was also unaffected, indicating that the Cyclin A/Cdk2-Plk1-βTrCP dependent regulation of Cdh1 stability was restricted to G1 phase. We found a reduction in the level of Cdh1 transcript with Cyclin A depletion that mirrored the reduction in Cdh1 translation and protein levels. Cdh1 transcript accumulates during G2 phase,Citation35 and it is this accumulation from the G1/S phase level of Cdh1 transcript that is regulated by Cyclin A/Cdk2. It would appear difficult to reconcile the data here with previously published work suggesting that Cyclin A/Cdk2 inhibited APC/CCdh1 activity in S/G2 phase.Citation24,25 It may be that these studies relate to the G1 phase Cyclin A/Cdk2-Plk1-βTrCP dependent mechanism where loss of Cyclin A/Cdk2 activity would result in the accumulation of Cdh1 and APC/CCdh1 activity.
The mechanism by which Cyclin A/Cdk2 regulates Cdh1 mRNA levels is at present unknown. Cyclin A/Cdk2 directly regulates the activity of B-Myb,Citation36,37 and this has been shown to bind to the promoter of Cdh1.Citation38,39 Interestingly, another Cyclin A/Cdk2 regulated transcription factor, FoxM1, has been found to be co-recruited to promoters of mitotic genes with B-Myb,Citation38 suggesting that the observed effect is due to defective transcriptional regulation of mitotic genes including Cdh1 by Cyclin A/Cdk2, but this will require further investigation.
The surprising finding was Cyclin A/Cdk2 depletion/inhibition-mediated Cdh1 reduction affected only a limited subset of APC/CCdh1 targets. Direct depletion of Cdh1 regulated all of the APC/CCdh1 substrates examined, Claspin, Plk1 and Aurora A. It also demonstrated that Cdh1 maintained the mitotic substrates Plk1 and Aurora A in a steady state by low level APC/CCdh1 activity in G2 phase. This contrasted with depletion of Cyclin A and the associated reduction in Cdh1, which does not affect APC/CCdh1 activity toward the mitotic substrates Plk1 and Aurora A. The basis for the difference of the effect of Cyclin A-mediated Cdh1 depletion and direct Cdh1 siRNA mediated depletion is unclear. It is unlikely to be a difference in the level of Cdh1 depletion achieved, as suboptimal Cdh1 siRNA depletion was sufficient to increase the levels of all substrates examined.
Claspin levels fluctuate through the cell cycle, being maximal in S/G2 phase then reducing into mitosis, with their lowest levels in early G1 phase.Citation21,40 Claspin stability in mitosis is regulated by βTrCP through Plk1-dependent phosphorylation of a phosphodegron,Citation21 and this low level is maintained in G1 phase by APC/CCdh1.Citation23 Our data points to Claspin stability being regulated by Cdh1in late S/G2 phase. The initial decrease in Claspin from its peak levels is temporally correlated with the increase in Cdh1 levels in late S/G2 phase,Citation41 whereas other reports have focused on the M/G1 phase role of Cdh1Citation23,42 failing to observed the S/G2 phase reported here. The lack of additive effect of Cyclin A and Cdh1 co-depletion over depletion of either alone on Claspin stability implies that Cyclin A/Cdk2 and Cdh1 operate in the same pathway to regulate Claspin stability, and the increased Claspin level in the Cyclin A depleted G2 phase cells is directly attributable to the reduced Cdh1 levels. Cdh1 has been shown to regulate Claspin in G2 phase checkpoint arrested cells, although in this case the activity of Cdh1 is countered by USP28 to maintain Claspin levels.Citation22
The increased Claspin level in the Cyclin A/Cdk2 depleted/inhibited G2 phase cells was responsible for a modest increase in activated Chk1, and a similar modest level of activated Chk1 found in normal G2 phase cells was responsible for delaying progression into mitosis.Citation12 Cyclin A/Cdk2 regulation of Cdh1-Claspin-Chk1 activation contributes to the G2 phase delay observed in Cyclin A/Cdk2 depleted/inhibited cells. Previous studies have also identified Wee1 and Emi1 as Cyclin A/Cdk2 targets,Citation5,9,43 and these also contribute to the Cyclin A/Cdk2-dependent regulation of G2 phase progression, although these are distinct from of Cdh1-regulated mechanism reported here. Cyclin A/Cdk1/2 also regulates FoxM1 activity which regulates the expression of critical mitotic genes such as Plk1, Cyclins B1 and Cdc25B.Citation44,45 We did not observe any changes in the levels of these FoxM1 substrates with Cyclin A depletion, suggesting that whereas chronic Cyclin A/Cdk2 inhibition used in the previous studies can regulate FoxM1-dependent transcription of critical mitotic regulators, an acute response to Cyclin A/Cdk2 inhibition is mediated through regulation of Cdh1 levels.
The regulation of Cdh1 levels and targeting of late S/G2 phase APC/CCdh1 substrates, including Claspin, appears to be a significant contributor to the mechanism by which Cyclin A/Cdk2 regulates progression through G2 phase into mitosis. The elevated levels of Claspin in turn maintain Chk1 activation delaying entry into mitosis. We found that the level of Claspin which is maximal in late S/G2 phase,Citation21,40 correlates with the nadir of Cdh1 in S phase.Citation41 The accumulation of Cdh1 during late S/G2 phase is temporally correlated with the activation of Cyclin A/Cdk2 at occurs during this transition,Citation29 and this appears sufficient to destabilise a subset of APC/CCdh1 substrates including Claspin, but not affecting mitotic substrates of this complex. The reduction in Claspin levels in late S/G2 phase correlates with the decline in activated Chk1 reported previously that is responsible for delaying entry into mitosis during the unperturbed cell cycle.Citation12 Together with the Cyclin A/Cdk2-dependent regulation of Wee1, Emi1 and FoxM1,Citation5,9,Citation43-45 the regulation of Cdh1 levels in late S/G2 phase provide the mechanistic basis for Cyclin A/Cdk2 regulation of Cyclin B/Cdk1 activation and entry into mitosis.
Materials and Methods
Cell Culture- HeLa, U2OS, and U2OS cell line with tetracycline repressed Myc-Cdh1 (a gift of J. LukasCitation28) and HEK293T cells were cultured at 37°C in a 5% CO2-humidified atmosphere in Dulbecco's modified Eagle's medium (Gibco, CA, USA), supplemented with 10% Serum Supreme (BioWhittaker) and 3 mM HEPES. Cells were synchronised with a single thymidine block as described previously.Citation46 For Cdh1 induction, samples were washed 3 times with warmed PBS for 10 minutes then media was reapplied to cells without addition of tetracycline. Inhibition of Cdk2 was achieved using 2 μM PHA533533.Citation47 Cdk1 activity was selectively inhibited using 9 μM Ro-3306 (Calbiochem).
SiRNA-mediated ablation of Cyclin A (2 separate Cyclin A siRNAs #1 and #3), and Chk1 were as reported previously.Citation4,12 Rescue of Cyclin A siRNA depletion was performed by overexpressing with Cherry fusion Cyclin A constructs as reported previously.Citation26 Cdh1 and Claspin siRNAs were On Target Plus Smartpools or individual siRNA duplexes purchased from Dharmacon. Transfections were carried out in 6-well plates using Lipofectamine 2000 (Invitrogen) as per the manufacturer's instructions.
Time lapse microscopy- Time lapse experiments were performed with cells seeded into 6 well plates, transfected, synchronised and images taken at indicated times using a Zeiss Live Cell Observer with a 37°C incubator and 5% CO2 hood. Images were captured with a 20X objective on a Zeiss AxioCam MRm camera using AxioVision software. From the image stacks, time of mitosis entry and exit for each cell was recorded, and percent cumulative mitosis of the entire field was determined. Entry into mitosis was marked by the appearance of the rounded mitotic morphology as reported previously.Citation4
Cell cycle analysis- For DNA content analysis, floating and attached cells were collected, fixed in ice-cold 70% ethanol and stored at −20°C. Cells were stained and analyzed on a FACSCalibur system (BD Biosciences) using Cell Quest (BD Biosciences) and ModFit (Verity Software, Topsham, ME) data analysis software as described previously.Citation29
Fixed Cell Sorting- HeLa and U2OS cells were transfected with siRNA, and both floating and attached cells (1.5 × 107) were harvested at 16 h after transfection, fixed in −20°C 70% ethanol and stored at −20°C. For sorting, cells were washed twice with phosphate buffer saline (PBS) contained 0.1% Triton X-100 (Sigma) and stained in PBS with 2 μg/ml propidium iodide (PI) and 500 μg/mL RNase A (Invitrogen).Citation48 The staining suspension was subsequently filtered through 37 micron gauze and collected into FACS tubes. All the samples were sorted into 4 contiguous windows of cell cycle based on PtdIns fluorescence by using MoFlo Astrios cell sorter (Beckman Coulter). 60,000 cells from each sorted population were solubilised with 1 x SDS-PAGE sample buffer and boiled for 10 minutes before western blotting. Data analysis was performed on Kaluza (Beckman Coulter), where the phase distribution of cell cycle in each sample was calculated by obtaining the sorted number of cells in gated regions.
Immunoblotting- Cells were lysed in buffer (250 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, 20 mM Tris, pH 8) supplemented with 5 μg/ml aprotinin, 5 μg/ml pepstatin, 5 μg/ml leupeptin, 0.5 mM phenylmethylsulfonyl fluoride, 25 mM NaF, 25 mM β-glyerophosphate and 0.1 mM sodium orthovanadate. Samples (20 μg of protein) were resolved on 10% SDS-polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes. Membranes were probed with pChk1 Ser317 (Bethyl and Cell Signaling), pCdk1 Tyr15, pChk1 Ser296, pChk1 Ser345, pMEK1 Thr286 and Claspin (Cell Signaling), Cyclin A, and Cdc25B (Santa Cruz), Chk1, Cdh1 and Plk1 (Abcam), PCNA (DAKO), pChk1 Ser301Citation34 and Cyclin B1Citation49 antibodies and detected by enhanced chemiluminescence detection.
qPCR analysis- Total RNA from different treatments of HeLa cells was obtained by using NucleoSpin RNAII isolation kit (Macherey-Nagel). 1 μg of extracted RNA was used for reverse cDNA synthesis using Superscript III First-Strand Synthesis System for RT-PCR in the presence of both random hexamers and Oligo dT20, as per manufacturer's instruction (Life technologies). Quantification of mRNA levels of certain genes was performed using QuantiFast Probe PCR Kits (Qiagen) according to the manufacturer's instruction. The experiment was designed in triplicate for each sample and 2% of synthesized cDNA was used as template for 10 μl reactions in 384-wells plate on a ViiA 7 Real-Time PCR System (Applied Biosystems, Life technologies). Taqman primers for FZR1 (CDH1) (ID: Hs00393592_m1) and RPL13A (Hs04194366_g1) were purchased from Applied Biosystems, Life technologies. Relative expression of Cdh1 was determined by ΔΔCT method, where CT was when detected fluorescence raised above the background fluorescence, with RPL13A as internal controls. Final quantification of Cdh1 expression was normalized to non-treated HeLa cell samples.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
949111_Supplementary_Material.zip
Download Zip (259.8 KB)Supplemental_Materials
Download Zip (64 KB)Acknowledgments
The author thanks J. Lukas for gifts of the Cdh1 inducible U2OS cell line and Myc-Cdh1 wild type and mutant plasmids, G. McArthur for the PHA 533533 Cdk2 inhibitor, and A. Burgess and P. Duijf for his critical reading of this manuscript.
Funding
This work was supported by funding from the National Health and Medical Research Council of Australia and Cancer Council Queensland Fund. HB was supported by a Lions Research Fellowship, W.J.L is supported by a NHMRC CJ Martin Fellowship, and BG is supported with a NHMRC Senior Research Fellowship.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res 2000; 60:3689-95; PMID:10919634
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9:153-66; PMID:19238148; http://dx.doi.org/10.1038/nrc2602
- Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009; 28:2925-39; PMID:19561645; http://dx.doi.org/10.1038/onc.2009.170
- De Boer L, Oakes V, Beamish H, Giles N, Stevens F, Somodevilla-Torres M, Desouza C, Gabrielli B. Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene 2008; 27:4261-8; PMID:18372919; http://dx.doi.org/10.1038/onc.2008.74
- Fung TK, Ma HT, Poon RY. Specialized roles of the two mitotic cyclins in somatic cells: cyclin A as an activator of M phase-promoting factor. Mol Biol Cell 2007; 18:1861-73; PMID:17344473; http://dx.doi.org/10.1091/mbc.E06-12-1092
- Furuno N, den Elzen N, Pines J. Human cyclin A is required for mitosis until mid prophase. J Cell Biol 1999; 147:295-306; PMID:10525536; http://dx.doi.org/10.1083/jcb.147.2.295
- Gong D, Pomerening JR, Myers JW, Gustavsson C, Jones JT, Hahn AT, Meyer T, Ferrell JEJr. Cyclin A2 regulates nuclear-envelope breakdown and the nuclear accumulation of cyclin B1. Curr Biol 2007; 17:85-91; PMID:17208191; http://dx.doi.org/10.1016/j.cub.2006.11.066
- Mitra J, Enders GH. Cyclin A/Cdk2 complexes regulate activation of Cdk1 and Cdc25 phosphatases in human cells. Oncogene 2004; 23:3361-7; PMID:14767478; http://dx.doi.org/10.1038/sj.onc.1207446
- Li C, Andrake M, Dunbrack R, Enders GH. A bifunctional regulatory element in human somatic Wee1 mediates cyclin A/Cdk2 binding and Crm1-dependent nuclear export. Mol Cell Biol 2010; 30:116-30; PMID:19858290; http://dx.doi.org/10.1128/MCB.01876-08
- Kramer A, Mailand N, Lukas C, Syljuasen RG, Wilkinson CJ, Nigg EA, Bartek J, Lukas J. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat Cell Biol 2004; 6:884-91; PMID:15311285
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8:37-45; PMID:16327781
- Brooks K, Oakes V, Edwards B, Ranall M, Leo P, Pavey S, Pinder A, Beamish H, Mukhopadhyay P, Lambie D, et al. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene 2013; 32:788-96; PMID:22391562; http://dx.doi.org/10.1038/onc.2012.72
- Niida H, Tsuge S, Katsuno Y, Konishi A, Takeda N, Nakanishi M. Depletion of Chk1 leads to premature activation of Cdc2-cyclin B and mitotic catastrophe. J Biol Chem 2005; 280:39246-52; PMID:16159883; http://dx.doi.org/10.1074/jbc.M505009200
- Enomoto M, Goto H, Tomono Y, Kasahara K, Tsujimura K, Kiyono T, Inagaki M. Novel positive feedback loop between Cdk1 and Chk1 in the nucleus during G2/M transition. J Biol Chem 2009; 284:34223-30; PMID:19837665; http://dx.doi.org/10.1074/jbc.C109.051540
- Freire R, van Vugt MA, Mamely I, Medema RH. Claspin: timing the cell cycle arrest when the genome is damaged. Cell Cycle 2006; 5:2831-4; PMID:17172868; http://dx.doi.org/10.4161/cc.5.24.3559
- Lindsey-Boltz LA, Sercin O, Choi JH, Sancar A. Reconstitution of human claspin-mediated phosphorylation of Chk1 by the ATR (ataxia telangiectasia-mutated and rad3-related) checkpoint kinase. J Biol Chem 2009; 284:33107-14; PMID:19828454; http://dx.doi.org/10.1074/jbc.M109.064485
- Leung-Pineda V, Ryan CE, Piwnica-Worms H. Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol Cell Biol 2006; 26:7529-38; PMID:17015476; http://dx.doi.org/10.1128/MCB.00447-06
- Wilsker D, Petermann E, Helleday T, Bunz F. Essential function of Chk1 can be uncoupled from DNA damage checkpoint and replication control. Proc Natl Acad Sci U S A 2008; 105:20752-7; PMID:19091954; http://dx.doi.org/10.1073/pnas.0806917106
- Bassermann F, Eichner R, Pagano M. Biochim Biophys Acta 2014; 1843:150-62; PMID:3466868; http://dx.doi.org/10.1016/j.bbamcr.2013.02.028
- Manchado E, Eguren M, Malumbres M. The anaphase-promoting complex/cyclosome (APC/C): cell-cycle-dependent and -independent functions. Biochem Soc Trans 2010; 38:65-71; PMID:20074037; http://dx.doi.org/10.1042/BST0380065
- Mailand N, Bekker-Jensen S, Bartek J, Lukas J. Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell 2006; 23:307-18; PMID:16885021; http://dx.doi.org/10.1016/j.molcel.2006.06.016
- Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 2008; 134:256-67; PMID:18662541; http://dx.doi.org/10.1016/j.cell.2008.05.043
- Faustrup H, Bekker-Jensen S, Bartek J, Lukas J, Mailand N. USP7 counteracts SCFbetaTrCP- but not APCCdh1-mediated proteolysis of Claspin. J Cell Biol 2009; 184:13-9; PMID:19124652; http://dx.doi.org/10.1083/jcb.200807137
- Mitra J, Enders GH, Azizkhan-Clifford J, Lengel KL. Dual regulation of the anaphase promoting complex in human cells by cyclin A-Cdk2 and cyclin A-Cdk1 complexes. Cell Cycle 2006; 5:661-6; PMID:16582612; http://dx.doi.org/10.4161/cc.5.6.2604
- Sorensen CS, Lukas C, Kramer ER, Peters JM, Bartek J, Lukas J. A conserved cyclin-binding domain determines functional interplay between anaphase-promoting complex-Cdh1 and cyclin A-Cdk2 during cell cycle progression. Mol Cell Biol 2001; 21:3692-703; PMID:11340163; http://dx.doi.org/10.1128/MCB.21.11.3692-3703.2001
- Beamish H, de Boer L, Giles N, Stevens F, Oakes V, Gabrielli B. Cyclin A/cdk2 regulates adenomatous polyposis coli-dependent mitotic spindle anchoring. J Biol Chem 2009; 284:29015-3; PMID:19703905; http://dx.doi.org/10.1074/jbc.M109.042820
- Fukushima H, Ogura K, Wan L, Lu Y, Li V, Gao D, Liu P, Lau AW, Wu T, Kirschner MW, et al. SCF-mediated Cdh1 degradation defines a negative feedback system that coordinates cell-cycle progression. Cell Rep 2013; 4:803-16; PMID:23972993; http://dx.doi.org/10.1016/j.celrep.2013.07.031
- Sorensen CS, Lukas C, Kramer ER, Peters JM, Bartek J, Lukas J. Nonperiodic activity of the human anaphase-promoting complex-Cdh1 ubiquitin ligase results in continuous DNA synthesis uncoupled from mitosis. Mol Cell Biol 2000; 20:7613-23; PMID:11003657; http://dx.doi.org/10.1128/MCB.20.20.7613-7623.2000
- Goldstone S, Pavey S, Forrest A, Sinnamon J, Gabrielli B. Cdc25-dependent activation of cyclin A/cdk2 is blocked in G2 phase arrested cells independently of ATM/ATR. Oncogene 2001; 20:921-32; PMID:11314027; http://dx.doi.org/10.1038/sj.onc.1204177
- van Leuken R, Clijsters L, van Zon W, Lim D, Yao X, Wolthuis RM, Yaffe MB, Medema RH, van Vugt MA. Polo-like kinase-1 controls Aurora A destruction by activating APC/C-Cdh1. PLoS ONE 2009; 4:e5282; PMID:19390576; http://dx.doi.org/10.1371/journal.pone.0005282
- Laoukili J, Alvarez-Fernandez M, Stahl M, Medema RH. FoxM1 is degraded at mitotic exit in a Cdh1-dependent manner. Cell Cycle 2008; 7:2720-6; PMID:18758239; http://dx.doi.org/10.4161/cc.7.17.6580
- Lee K, Kenny AE, Rieder CL. P38 mitogen-activated protein kinase activity is required during mitosis for timely satisfaction of the mitotic checkpoint but not for the fidelity of chromosome segregation. Mol Biol Cell 2010; 21:2150-60; PMID:20462950; http://dx.doi.org/10.1091/mbc.E10-02-0125
- Castedo M, Perfettini JL, Roumier T, Yakushijin K, Horne D, Medema R, Kroemer G. The cell cycle checkpoint kinase Chk2 is a negative regulator of mitotic catastrophe. Oncogene 2004; 23:4353-61; PMID:15048074; http://dx.doi.org/10.1038/sj.onc.1207573
- Ikegami Y, Goto H, Kiyono T, Enomoto M, Kasahara K, Tomono Y, Tozawa K, Morita A, Kohri K, Inagaki M. Chk1 phosphorylation at Ser286 and Ser301 occurs with both stalled DNA replication and damage checkpoint stimulation. Biochem Biophys Res Commun 2008; 377:1227-31; PMID:18983824; http://dx.doi.org/10.1016/j.bbrc.2008.10.119
- Inbal N, Listovsky T, Brandeis M. The mammalian Fizzy and Fizzy-related genes are regulated at the transcriptional and post-transcriptional levels. FEBS Lett 1999; 463:350-4; PMID:10606752; http://dx.doi.org/10.1016/S0014-5793(99)01640-3
- Bessa M, Saville MK, Watson RJ. Inhibition of cyclin A/Cdk2 phosphorylation impairs B-Myb transactivation function without affecting interactions with DNA or the CBP coactivator. Oncogene 2001; 20:3376-86; PMID:11423988; http://dx.doi.org/10.1038/sj.onc.1204439
- Saville MK, Watson RJ. The cell-cycle regulated transcription factor B-Myb is phosphorylated by cyclin A/Cdk2 at sites that enhance its transactivation properties. Oncogene 1998; 17:2679-89; PMID:9840932; http://dx.doi.org/10.1038/sj.onc.1202503
- Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev 2012; 26:474-89; PMID:22391450; http://dx.doi.org/10.1101/gad.181933.111
- Zhan M, Riordon DR, Yan B, Tarasova YS, Bruweleit S, Tarasov KV, Li RA, Wersto RP, Boheler KR. The B-MYB transcriptional network guides cell cycle progression and fate decisions to sustain self-renewal and the identity of pluripotent stem cells. PLoS One 2012; 7:e42350; PMID:22936984; http://dx.doi.org/10.1371/journal.pone.0042350
- Mamely I, van Vugt MA, Smits VA, Semple JI, Lemmens B, Perrakis A, Medema RH, Freire R. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr Biol 2006; 16:1950-5; PMID:16934469; http://dx.doi.org/10.1016/j.cub.2006.08.026
- Listovsky T, Oren YS, Yudkovsky Y, Mahbubani HM, Weiss AM, Lebendiker M, Brandeis M. Mammalian Cdh1/Fzr mediates its own degradation. Embo J 2004; 23:1619-26; PMID:15029244; http://dx.doi.org/10.1038/sj.emboj.7600149
- Gao D, Inuzuka H, Korenjak M, Tseng A, Wu T, Wan L, Kirschner M, Dyson N, Wei W. Cdh1 regulates cell cycle through modulating the claspin/Chk1 and the Rb/E2F1 pathways. Mol Biol Cell 2009; 20:3305-16; PMID:19477924; http://dx.doi.org/10.1091/mbc.E09-01-0092
- Bernis C, Vigneron S, Burgess A, Labbe JC, Fesquet D, Castro A, Lorca T. Pin1 stabilizes Emi1 during G2 phase by preventing its association with SCF(betatrcp). EMBO Rep 2007; 8:91-8; PMID:17159919; http://dx.doi.org/10.1038/sj.embor.7400853
- Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 2005; 7:126-36; PMID:15654331
- Laoukili J, Alvarez M, Meijer LA, Stahl M, Mohammed S, Kleij L, Heck AJ, Medema RH. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol Cell Biol 2008; 28:3076-87; PMID:18285455; http://dx.doi.org/10.1128/MCB.01710-07
- Astuti P, Boutros R, Ducommun B, Gabrielli B. Mitotic phosphorylation of Cdc25B Ser321 disrupts 14-3-3 binding to the high affinity Ser323 site. J Biol Chem 2010; 285:34364-70; PMID:20801879; http://dx.doi.org/10.1074/jbc.M110.138412
- Pevarello P, Brasca MG, Orsini P, Traquandi G, Longo A, Nesi M, Orzi F, Piutti C, Sansonna P, Varasi M, et al. 3-Aminopyrazole inhibitors of CDK2/cyclin A as antitumor agents. 2. Lead optimization. J Med Chem 2005; 48:2944-56; PMID:15828833; http://dx.doi.org/10.1021/jm0408870
- Caldon CE, Sergio CM, Sutherland RL, Musgrove EA. Differences in degradation lead to asynchronous expression of cyclin E1 and cyclin E2 in cancer cells. Cell Cycle 2013; 12:596-605; PMID:23324394; http://dx.doi.org/10.4161/cc.23409
- Gabrielli BG, De Souza CP, Tonks ID, Clark JM, Hayward NK, Ellem KA. Cytoplasmic accumulation of cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J Cell Sci 1996; 109:1081-93; PMID:8743955