
Abstract
Regulation of chromatin structure is an essential component of the DNA damage response (DDR), which effectively preserves the integrity of DNA by a network of multiple DNA repair and associated signaling pathways. Within the DDR, chromatin is modified and remodeled to facilitate efficient DNA access, to control the activity of repair proteins and to mediate signaling. The mammalian ISWI family has recently emerged as one of the major ATP-dependent chromatin remodeling complex families that function in the DDR, as it is implicated in at least 3 major DNA repair pathways: homologous recombination, non-homologous end-joining and nucleotide excision repair. In this review, we discuss the various manners through which different ISWI complexes regulate DNA repair and how they are targeted to chromatin containing damaged DNA.
Abbreviations
| ACF1 | = | ATP-utilizing Chromatin assembly and remodeling Factor 1 |
| BER | = | Base Excision Repair |
| DDR | = | DNA Damage Response |
| DSB | = | Double Strand Break |
| GG-NER | = | Global Genome Nucleotide Excision Repair |
| HR | = | Homologous Recombination |
| ISWI | = | Imitation SWItch |
| MRN | = | MRE11/Rad50/NBS1 |
| NER | = | Nucleotide Excision Repair |
| NHEJ | = | Non-Homologous End Joining |
| PAR | = | Poly(ADP-Ribose) |
| RNApolII | = | RNA Polymerase II |
| RSF1 | = | Remodeling and Spacing Factor 1 |
| SMARCA | = | SWI-SNF-related Matrix-associated Actin-dependent Regulator of Chromatin A |
| TC-NER | = | Transcription-Coupled Nucleotide Excision Repair |
| WSTF | = | Williams Syndrome Transcription Factor |
DNA damage has a major impact on health and is believed to be the underlying cause of both cancer and aging.Citation1 To protect against the adverse effects of DNA damage, organisms are equipped with diverse mechanisms of DNA repair and associated DNA damage signaling pathways, collectively called the DNA damage response (DDR).Citation2 Although the repair mechanisms of most DDR pathways are known in detail, how they function within chromatin and how chromatin configuration and reconfiguration facilitates the DDR has only recently obtained wider attention. In this review, we discuss novel insights that demonstrate an essential function for the ISWI family of ATP-dependent chromatin remodelers in mammalian DDR regulation. As changes and defects in both chromatin remodeling and in the DDR are linked to human malignancies and aging,Citation1,3,4 it is crucial to understand the connection between both processes.
Chromatin remodeling
The basal packaging units of the eukaryotic genome are nucleosomes, which each consist of 147 bp DNA wrapped around a histone octamer containing 2 copies of each histone (H2A, H2B, H3 and H4).Citation5 This way of packaging of DNA into chromatin not only allows the large DNA molecule to fit in the nucleus but also serves as an efficient and important mechanism to actively regulate DNA transacting processes such as transcription, replication and DNA repair.Citation6 Modification of chromatin regulates access of proteins to DNA and also provides cues to attract proteins or initiate signaling cascades.
Chromatin is modified by the specific activity of histone modifiers, histone chaperones and ATP-dependent chromatin remodelers. Histone modifying enzymes predominantly covalently modify the N-terminal tails of histone proteins. This in turn may lead to the modulation of nucleosome dynamics by altering histone-DNA contacts or to the chromatin association of proteins that act on DNA or mediate signaling responses.Citation7 Many different types of histone modifications play essential roles in orchestrating DNA repair and DNA damage signaling, including acetylation, methylation, phosphorylation, ubiquitylation and ADP-ribosylation.Citation8
ATP-dependent chromatin remodeling complexes catalyze the disruption of DNA-histone contacts and can slide or evict nucleosomes by using the energy from ATP hydrolysis.Citation9 In addition, they control nucleosome assembly and composition, in cooperation with histone chaperones, by exchanging histones and histone variants. Four structurally related, evolutionary conserved families have been described among various chromatin remodeling complexes: SWI/SNF, INO80, CHD and ISWI. Central to these complexes is a catalytic subunit harboring a SWI2/SNF2-family ATPase domain.Citation10 The remodeling families are functionally different because of the unique domains that reside within or adjacent to this domain. In addition, tissue context and specific subunits that are associated with most ATPases determine the particular function of each complex. Some ATP-dependent chromatin remodeling complexes, such as SWI/SNF, form large protein assemblies composed of up to 14 subunits, while others consist of only one or a few additional subunits, such as ISWI and some CHD complexes. In spite of their differences, all 4 families are involved in transcription, DNA replicationCitation9 and DDRCitation11 by regulating access to DNA, efficient recruitment of repair factors and DNA damage signaling. In this review, we specifically focus on recent findings that implicate ISWI chromatin remodeling complexes in the DDR.
ISWI family of ATP-dependent chromatin remodelers
ISWI (Imitation SWItch) family remodeling complexes were first discovered in Drosophila melanogasterCitation12-15 and later shown to be conserved in many other organisms. So far, 7 different mammalian ISWI complexes have been described: WICH, NoRC, RSF, ACF, CHRAC, NURF and CERF ().Citation16,17 Each of the complexes contain one of 2 conserved ATPase subunits: SMARCA5 (SWI-SNF-related Matrix-associated Actin-dependent Regulator of Chromatin A5, also known as SNF2H) or SMARCA1 (also known as SNF2L) besides one or more accessory subunits. SMARCA5 is found in all ISWI complexes except CERFCitation18 and NURF,Citation19 which contain SMARCA1 (), although this ATPase selectivity may in part be cell type specificCitation20. The ATPase domain of both ATPases, which is comprised of a DExx and HELICc region, belongs to the superfamily of DEAD⁄H-helicasesCitation10,21 and is located in the N-terminal half of the proteins (). In addition, both proteins contain a HAND, SANT and SLIDE domain in the C terminus,Citation22 which regulate the activity and specificity of the ATPase domain ().Citation23 In line with this, the remodeling activity of Drosophila and yeast ISWI complexes is reduced when either the SANT domain or the SLIDE domain is deleted.Citation22,24 Especially the SLIDE domain, which interacts with extranucleosomal DNA, stimulates ATPase activity and is required to move DNA along the nucleosome.Citation24,25
Figure 1. Mammalian ISWI family chromatin remodeling complexes. (A) Depicted are the 7 currently known mammalian ISWI family ATP-dependent chromatin remodeling complexes. WICH, NoRC, RSF, ACF1 and CHRAC all share the SMARCA5 ATPase subunit. NURF and CERF share the SMARCA1 ATPase subunit. (B) Schematic representation of the domains within SMARCA1/SMARCA5. The catalytic activity of both SMARCA1 and SMARCA5 is defined by its ATPase domain that is split in 2 parts: DExx and HELICc. This domain, which is located in the N-terminal half, is part of the superfamily of DEAD⁄H-helicases and is shared by SWI2/SNF2-like ATP dependent chromatin remodelers. In addition, both proteins contain HAND, SANT and SLIDE domains that are characteristic for the ISWI family in their C-terminus. These domains regulate the activity of the ATPase domain and are involved in DNA binding.
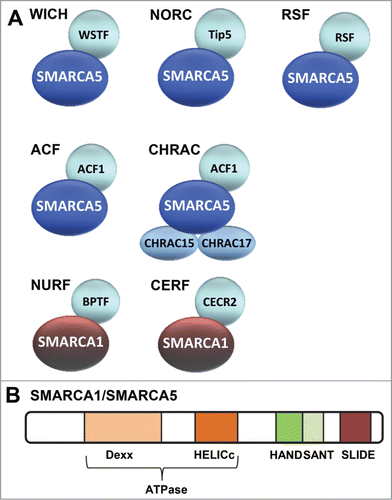
ISWI complexes exhibit various activities, e.g. ACF and CHRAC and yeast Isw regularly space nucleosomes, whereas others, such as NURF, show the opposite activity.Citation13,14,26,27 By controlling nucleosome spacing, these complexes regulate transcription, either by repression or activation, stimulate replication through heterochromatin and generate and maintain higher order chromatin and chromosome organization.Citation28-30 The accessory subunits in each complex most probably modulate the ATPase's function and activity and are important for target recognition and specificity.Citation18,31,32
ISWI function in base excision repair
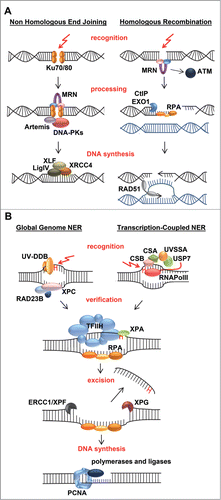
The continuous induction of a wide diversity of DNA lesions necessitates the action of distinct DNA repair mechanisms, which remove DNA damage depending on the type of lesion, location in the genome and cell cycle phase in which lesions are encountered.Citation33 The major DNA repair mechanisms that have thus far been associated with ISWI chromatin remodeling are Base Excision Repair (BER) and Nucleotide Excision Repair (NER), which both repair lesions that only affect one DNA strand, and Double Strand Break (DSB) repair ().
Figure 2. Mechanisms of double strand break and nucleotide excision repair. (A) Most double strand breaks (DSBs) are repaired either via Non-Homologous End-Joining (NHEJ) or Homologous Recombination (HR) and their detection initiates a DNA damage signaling cascade mediated by the PI3 kinase ATM. In NHEJ, detection of the break by Ku70/80 leads to recruitment of downstream factors including MRN, Artemis, and DNA-PK that process the DNA ends that are finally joined by XLF/XRCC4/LigIV. In HR, detection of the break by MRN leads to 5'-3' end resection, assisted by CtIP and EXO1, which creates ssDNA coated by RPA. RPA is replaced by RAD51, which mediates strand invasion and pairing of a homologous DNA strand that serves as template for error-free repair. The invading DNA end is extended by novel DNA synthesis and either dissociates and re-anneals with its original template, or a second DNA end invades the homologous template after which the resultant joint structures are resolved by specific endonucleases. Finally ligation takes place to complete repair. (B) UV-induced photolesions, monoadducts and other bulky lesions that distort the double helix are repaired by Nucleotide Excision Repair (NER). Lesions are either detected by the global genome NER machinery, via the concerted action of the UV-DDB and XPC/RAD23B complexes, or by the transcription-coupled NER machinery, involving CSA, CSB and UVSSA/USP7 that are recruited to DNA-damage-stalled RNA Polymerase II (RNApolII). Lesion detection leads to recruitment of the transcription factor H (TFIIH) complex, which unwinds DNA and verifies the presence of DNA damage. XPA binds to the lesion in the unwound DNA and RPA covers the non-damaged strand. The endonucleases ERCC1/XPF and XPG bind at both sites of the lesion and excise the damaged strand. The resulting gap is filled in by DNA synthesis and sealed by ligases.
BER mainly deals with non-bulky small nucleobase lesions, such as oxidative and alkylated bases, by excising and replacing incorrect or damaged bases.Citation34,35 Lesion-specific DNA-glycosylase enzymes recognize specific types of DNA damage, flip the damaged base out of the double helix and cleave the N-glycosidic bond between the substrate base and the 2′-deoxyribose. Next, AP-endonuclease cleaves the abasic site in the sugar-phosphate backbone. The resulting single strand break is then filled in by BER-specific DNA polymerase β and ligated by the XRCC1/ligase III complex (short patch BER). Alternatively, PCNA-dependent DNA polymerase δ polymerizes several nucleotides while displacing the nicked DNA strand, which is removed by the flap-structure endonuclease FEN1, after which DNA ligase I seals the ends (long-patch BER).
How BER functions within chromatin and the role of chromatin remodeling in BER is only poorly understood. In vitro, BER efficiency is inhibited by the presence of nucleosomes and compact chromatin, which can be overcome by ATP-dependent chromatin remodeling.Citation36-39 Evidence for a potential involvement of ISWI in BER comes from an in vitro study showing that gap-filling by DNA Polymerase β in oligonucleosome arrays is stimulated by yeast ISWI complexes ISW1 and ISW2.Citation40 This may indicate that ATP-dependent nucleosome displacement facilitates access of DNA polymerase β to DNA. Although it is not understood whether ISWI complexes are similarly required in vivo, SMARCA5, SMARCA1 and ACF1 are shown to be recruited to 405 nm laser induced DNA damage in human cells.Citation41 This type of irradiation mainly induces oxidative damage and single strand breaks,Citation42,43 which are lesions that are commonly repaired by BER and a variant of BER called single-strand break repair. These findings suggest that human ISWI complexes may assist in vivo BER.
Double strand break repair
DSBs can be induced by specific chemicals or reactive oxygen compounds produced by e.g. ionizing radiation or the cell's metabolism. In addition, processing of stalled replication on different types of lesions may eventually lead to the formation of DSBs. As both DNA strands are damaged, DBSs are very hazardous to the cell and can cause chromosome instability and genome rearrangements if not repaired properly. Two main mechanisms are employed to repair DSBs: Homologous Recombination (HR)Citation44 and Non-Homologous End Joining (NHEJ).Citation45
HR only takes place in S and G2 phase of the cell cycle when the sister chromatid is present, which is used as template to repair DSBs (), thereby providing a principally error-free repair of these hazardous lesions. HR is initiated when the MRE11/Rad50/NBS1 (MRN) complex binds to a DSB and together with CtIP and EXO1 resects the broken DNA ends. The created 3′overhangs are bound by the single strand binding protein RPA, which is subsequently exchanged for RAD51 by BRCA2. The resulting RAD51 nucleoprotein filament mediates strand invasion into the homologous sister leading to strand exchange such that the homologous sister chromatid serves as template for novel DNA synthesis. Hereafter, the invading DNA strand dissociates or the other DNA end migrates as well and the resultant joined structures are resolved. Finally, ligation takes place to complete repair.Citation44 In the NHEJ pathway, which can take place in any cell cycle, broken DNA ends are detected by the Ku70/80 heterodimer, which recruits subsequent repair factors ().Citation45 Following detection, the free DNA ends are processed by Artemis, DNA-PK and the MRN complex and finally joined together by the XLF/XRCC4/LigaseIV complex. NHEJ can be more error-prone than HR, because during end-processing nucleotide loss may occur prior to ligation.
DSBs further lead to an extensive signaling cascade in which chromatin serves as an important signal amplifier. This cascade is initiated by the binding of the MRN complex to DSBs which recruits and activates the PI3-kinase ATM. Activated ATM phosphorylates a range of downstream target proteins, including histone variant H2AX at residue S139 and the E3 ubiquitin ligase BRCA1.Citation46 H2AX phosphorylation is a key signaling event that spreads into adjacent chromatin, thereby amplifying the signal, and is readily visible under the microscope when stained with specific antibodies - as discernable sub-nuclear foci referred to as ionizing radiation induced foci.Citation47 The signaling protein MDC1 binds to phosphorylated H2AX and recruits the E3 ubiquitin ligases RNF8 and RNF168 that ubiquitylate H2A/H2AX, after which a range of signaling proteins are recruited, among which are 53BP1 and BRCA1. BRCA1 interacts with several DNA repair proteins including CtIP and, together with 53BP1, regulates whether DSBs are repaired by either NHEJ or HR.Citation48,49 BRCA1 furthermore regulates cell cycle arrest in concert with cell cycle checkpoint proteins that are also phosphorylated by ATM in response to DNA damage.Citation50 DSB repair and signaling also involve poly(ADP-ribose) (PAR) polymerases PARP1, PARP2 and PARP3, which are thought to PARylate both histone and non-histone proteins to regulate the recruitment of specific PAR-binding repair and chromatin remodeling proteins to stimulate HR or NHEJ.Citation51
ISWI chromatin remodelers in double strand break repair
Most evidence for a role of ATP-dependent chromatin remodeling complexes in DDR comes from studies of DSB repair.Citation8,11 Research on DSB repair is attractive because of its significance for cancer biology. Moreover, cellular DSB repair studies are particularly boosted by the developed technology to induce a single break in a cell which allows the analysis of DSB repair factor binding in intact cells.Citation52,53 Several studies have shown that different chromatin remodelers, including SMARCA5 and its binding partners, function in DSB repair. Intriguingly, however, many different activities have been ascribed to these complexes, pointing to multiple parallel functions ().Citation54-57 Their interplay and the exact molecular mechanism, in which they actually remodel chromatin, is currently not well understood.
Figure 3. ISWI chromatin remodeling complexes in double strand break repair. Depicted is a model that includes the various functions of different ISWI complexes at sites of double strand break (DSB) repair. DSBs activate the PI3 kinase ATM, which phosphorylates histone variant H2AX at S139 (indicated with S-P), leading to the recruitment of MDC1, RNF8 and RNF168, which mediate a ubiquitylation signaling cascade. RNF168 is PARylated by PARP1 and interacts with SMARCA5. SMARCA5, whose recruitment is also regulated by NuMA and PARP3, stimulates RNF168-mediated histone ubiquitylation (indicated with Ub). At DSBs, SIRT6 deacetylates H3 at K56 (indicated with Ac), after which RNF20 is recruited to ubiquitylate H2B at K120 (indicated with Ub), coinciding with the methylation of H3 at K4 (indicated with M) and recruitment of SMARCA5, which interacts with SIRT6. SMARCA5 is furthermore recruited together with ACF1, CHRAC15 and CHRAC17 as part of the CHRAC complex and with WSTF as part of the WICH complex. WSTF interacts with and phosphorylates H2AX at Y142 (indicated with Y-P) to maintain S139 phosphorylation (indicated with S-P). Finally, RSF promotes the loading of histone-fold proteins CENP-S and CENP-X at or near DSB sites independently of SMARCA5. The recruitment of SMARCA5, ACF1, WSTF and RSF likely leads to the remodeling of chromatin that is necessary for efficient recruitment of repair factors, including BRCA1, RAD51, Ku70/80 and XRCC4, to facilitate repair by homologous recombination and non-homologous end-joining.
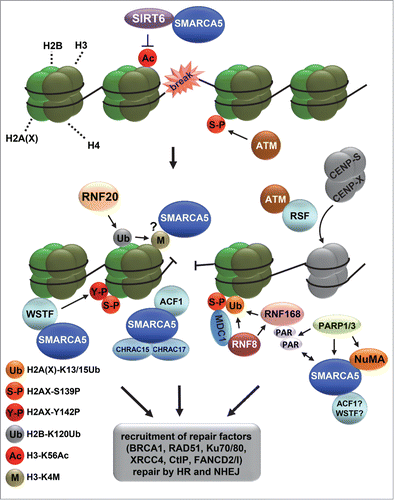
Both human ISWI ATPases SMARCA5 and SMARCA1 are rapidly recruited to DSBs and their knockdown renders cells hypersensitive to DNA damage.Citation54-59 SMARCA5 is recruited to DSBs together with RNF168, in a PARP1-dependent manner, to regulate HR and NHEJ.Citation56 SMARCA5 binds to PARylated RNF168 and stimulates RNF168-mediated histone ubiquitylation that leads to the recruitment of BRCA1. Another study showed that SMARCA5 recruitment is regulated by PARP3 and the structural nuclear protein NuMA, which interacts with SMARCA5 and is needed for efficient HR.Citation59 Single and dual silencing of NuMA and SMARCA5 led to similar defects in the DSB-induced appearance of ubiquitin foci and CtIP, BRCA1 and RAD51 recruitment, suggesting that both proteins act in a common pathway. Furthermore, SMARCA5 was found to be recruited to DSBs depending on the E3 ubiquitin ligase RNF20.Citation54 RNF20 is recruited to DNA damage and ubiquitylates residue K120 of H2B at DSBs to promote the methylation of H3K4. As yeast Isw1 is recruited to chromatin by methylated H3K4,Citation60 this suggests that methylated H3K4 also facilitates the binding of mammalian SMARCA5 at sites of DNA damage. Depletion of RNF20 and SMARCA5 or expression of a ubiquitylation-defective H2B mutant leads to impaired DNA end resection and RAD51 and BRCA1 foci formation and thus defective HR repair. This RNF20-associated defective HR repair may relate to the frequently observed mutations in RNF20 which are seen in colorectal cancers.Citation61 Finally, SMARCA5 recruitment to DSBs was shown to depend on an interaction with the deacetylase SIRT6, which localizes early to DSBs to deacetylate H3K56 and is together with SMARCA5 required for efficient DSB signaling and repair.Citation58 As RNF20 recruitment and H2B K120 ubiquitylation were impaired in SIRT6 knockout cells, it was suggested that SIRT6 acts upstream of RNF20 in DSB repair. Intriguingly, SIRT6-deficient mice showed reduced chromatin enrichment of SMARCA5, though only in brain and pancreas but not in liver and heart tissue. These data point to tissue-specific differences in genomic organization involving SMARCA5, which may not only influence tissue-specific transcription, but also DDR.
The importance of ISWI remodeling complexes in genome maintenance is further confirmed by the implication of additional ISWI subunits in chromatin reorganization during DSB repair. ACF1 (ATP-utilizing Chromatin assembly and remodeling Factor 1, also known as BAZ1A) is a noncatalytic subunit of the human ISWI complexes ACF and CHRAC (). It is thought to enhance the efficiency of nucleosome sliding and to regulate the template specificity of SMARCA5, depending on the DNA flanking the nucleosomes.Citation62-64 The ACF complex assists DNA replication through heterochromatinCitation28 and regulates transcription in concert with other histone modifying enzymes and transcriptional regulators.Citation65 The CHRAC complex additionally contains the histone-fold proteins CHRAC15 and CHRAC17 (), which facilitate ATP-dependent nucleosome sliding by SMARCA5 and ACF1.Citation66 ACF1 is implicated in G2/M checkpoint control in response to DSBs and replication stressCitation57 and functions together with CHRAC15, CHRAC17 and SMARCA5 in both HR and NHEJ.Citation55 ACF1 is also rapidly recruited to DSBs, where it interacts with and stimulates the binding of NHEJ-joining KU70/80 proteins to DSBs. However, the interaction of ACF1 with SMARCA5 is only partially required for ACF1 recruitmentCitation57 and SMARCA5 is dispensable for KU70/80 recruitment.Citation55 This may indicate that SMARCA5 recruitment follows that of ACF1. Since SMARCA5 recruitment is regulated by SIRT6, RNF20, PARP and NuMA, it would be interesting to test whether ACF1 binding to DSBs also depends on these proteins and to determine whether the involvement of ACF1 and SMARCA5 in DSB repair is part of the same process or whether both proteins can act (partially) independent.
Another ISWI complex, WICH, consists of SMARCA5 and the noncatalytic subunit WSTF (Williams syndrome transcription factor, also known as BAZ1B), which is a paralog of ACF1 ().Citation67 WSTF interacts with the sliding clamp PCNA during replication to promote an open chromatin structure together with SMARCA5.Citation29 WSTF is also recruited to DSBsCitation57 and implicated in DDR signaling by regulating phosphorylation of H2AX.Citation68 In unchallenged conditions, WSTF interacts with H2AX and harbors an unexpected intrinsic kinase activity that constitutively phosphorylates Tyr142 of H2AX, which upon DNA damage induction is needed for the maintenance and amplification of the canonical Ser139 phosphorylation and the sustained recruitment of ATM and MDC1 at damaged sites.Citation68 Remarkably, however, in response to DNA damage, Tyr142 of H2AX is dephosphorylated by the tyrosine phosphatases EYA1 and EYA3 which is a prerequisite for the initial efficient MDC1 binding to phosphorylated Ser139 of H2AX.Citation69,70 Because the pro-apoptotic kinase JNK1 associates with H2AX when it is phosphorylated on both Ser139 and Tyr142, it was therefore suggested that Tyr142 phosphorylation acts as a molecular switch for a cell to decide between repair and apoptosis.Citation71 Thus, besides their molecular activity in providing access for repair proteins, on a larger scale ISWI complexes may function in concert with other proteins, such as signaling kinases and phosphatases, to determine cell fate in response to DNA damage.
RSF is another ISWI complex implicated in regulating cell fate. It consists of SMARCA5 and RSF1 (Remodeling and spacing factor 1, also known as HBXAP), a protein that is frequently found to be overexpressed in many types of cancer. Also RSF seems to be involved in genome maintenance, because RSF1 overexpression induces DNA strand breaks via an unknown mechanism.Citation72 If p53 is defective, as is the case for most ovarian serous carcinomas in which RSF1 is overexpressed, DNA damage checkpoint signaling is absent and tumor cells proliferate in the presence of DNA strand breaks, resulting in chromosomal aberrations. Thus, this suggests that the expression of chromatin remodelers must be tightly regulated to prevent genomic instability and inadequate repair. RSF1 was also shown to actively participate in DDR itself, although, surprisingly, independently of SMARCA5. Upon DSB induction, RSF interacts with and is phosphorylated by ATM.Citation73 Furthermore, RSF1 accumulates at DSB sites without SMARCA5 and promotes NHEJ and HR by facilitating the loading of centromeric histone-fold proteins CENP-S and CENP-X at damaged chromatin, which assist the recruitment of the NHEJ factor XRCC4/LigIVCitation74 and the interstrand crosslink repair proteins FANCD2 and FANCI with which RSF also interacts.Citation73 Furthermore, RSF1 was found to promote HR through a distinct, but unresolved mechanism not involving the loading of CENP-S or CENP-X.Citation74
The current evidence suggests that distinct ISWI complexes have multiple functions in DSB repair (). The significance of these complexes for the DDR is emphasized by the notion that HR and NHEJ are impaired when SMARCA5 or associated subunits are depleted. It remains however to be determined whether the association of ISWI complexes with different components of the repair machinery, such as with H2AX, RNF20, RNF168 and Ku70/80, are all part of the same chromatin remodeling event or whether these represent distinct or subsequent steps of the repair reaction.
ISWI chromatin remodelers in nucleotide excision repair
NER removes a large variety of single-stranded helix-distorting DNA lesions, including UV induced DNA damage.Citation75,76 It consists of 2 damage recognition subpathways (): Global genome repair (GG-NER), which detects damage throughout the whole genome, and transcription-coupled repair (TC-NER), which detects and repairs damage specifically in the transcribed strand of active genes. GG-NER is initiated by the damage detecting protein complex XPC/HR23B, which is - for certain less bulky lesions - assisted by the UV-DDB complex. The UV-DDB complex is particularly important for regulating chromatin organization during GG-NER initiation, as it recruits several ATP-dependent chromatin remodeling proteinsCitation77-79 and induces chromatin decondensation.Citation80 TC-NER is initiated upon stalling of RNA Polymerase II (RNApolII) at a DNA lesion, which serves as a signal to attract the TC-NER factors CSA, CSB and UVSSA/USP7. Subsequent steps are similar for TC-NER and GG-NER. First, the transcription factor IIH unwinds the DNA helix and verifies the presence of DNA damage, after which XPA and RPA stabilize the repair complex and properly orient the structure specific endonucleases XPF/ERCC1 and XPG to excise the damaged strand (). The resulting ∼30 nt single stranded DNA gap is filled by DNA synthesis and ligated.
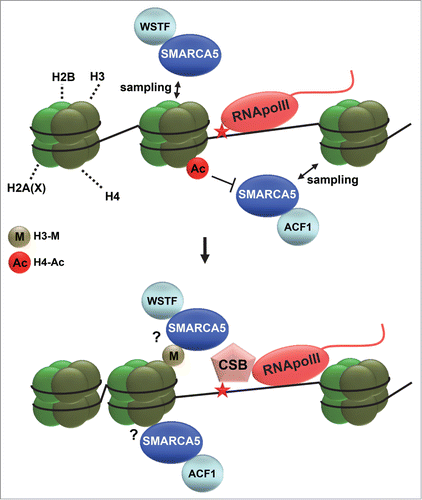
Several studies suggest a role for ATP dependent chromatin remodelers in the initiation of mammalian NER, including SWI/SNF, INO80 and the TC-NER protein CSB itself, which harbors a functional SWI2/SNF2 domain necessary for its function.Citation11,78,79,81,82 Because NER intermediates induce a similar DNA damage signaling response as is observed during DSB repair, involving H2AX phosphorylation leading to MDC1 recruitment and an RNF8/RNF168 and ubiquitin-mediated signaling cascade,Citation83-85 it is to be expected that ISWI complexes function similarly in this response when induced by NER. Strangely, however, although SMARCA5 and RNF168 interact after ionizing radiation, they do not interact after UV.Citation56 Intriguingly, recent evidence from our lab shows that ISWI complexes have yet another additional function in regulating UV damage repair, which differs from their identified roles in DSB repair. Within a genetic screen, isw-1, the C. elegans ortholog of SMARCA1/SMARCA5, was identified as a novel chromatin-associated protein involved in the UV-induced DDR.Citation86 Follow up experiments in mammals showed that knockdown of SMARCA5, but also of ACF1 and WSTF, renders cells sensitive to UV irradiation and defective in the TC-NER dependent re-initiation of transcription after UV.Citation87 SMARCA5, ACF1 and WSTF are recruited to photolesions induced by 266 nm UV-C laser irradiation and like CSB, SMARCA5 recruitment is transcription-dependent. Moreover, SMARCA5, ACF1 and WSTF are needed for efficient recruitment of CSB to UV damage, suggesting that chromatin remodeling by at least 2 distinct ISWI complexes, ACF1/CHRAC and WICH, facilitates TC-NER ().
Figure 4. ISWI chromatin remodeling during NER. Depicted is a hypothetical model of how ISWI complexes function in transcription-coupled nucleotide excision repair. ISWI complexes, including ACF (SMARCA5 and ACF1) and WICH (SMARCA5 and WSTF), continuously sample DNA and only associate with chromatin when encountering specific signals. Such signals could be dependent on RNA polymerase II (RNApolII) arrest at DNA damage and could involve histone deacetylation (acetylation is indicated with Ac) and methylation (indicated with M). DNA damage arrested RNApolII binds with more affinity to the essential repair protein CSB and is itself reverse translocated, likely to make the lesion accessible for repair. SMARCA5 is necessary for efficient loading of CSB at sites of UV-induced transcriptional arrest, suggesting that chromatin remodeling facilitates access to DNA. Reverse translocation of RNApolII probably also requires chromatin remodeling.
Targeting ISWI complexes to DNA damage
Despite the clear evidence that ISWI complexes function in DDR, several questions remain, such as: 1) How do ISWI complexes find sites of DNA damage and identify their target nucleosomes?; 2) What are the exact functional differences between the different ISWI complexes when it comes to actual chromatin remodeling in specific cases?; 3) What determines the choice of chromatin remodeling complex to be used?; 4) What are the interactions of the different complexes in vivo? It appears that ISWI complexes localize in a different manner to DSBs, repaired by HR and NHEJ, than to UV-C induced photolesions, repaired by NER. The rapid recruitment to DSBs involves an interaction with RNF168 and depends on PARP activity but not on transcription.Citation54,56 In contrast, SMARCA5 does not interact with RNF168 after UV and its recruitment to 266 nm induced UV-C damage is independent of PARP but dependent on active transcription.Citation56,87 Furthermore, the SLIDE but not the ATPase domain of SMARCA5 is sufficient for DSB recruitment,Citation55 whereas in contrast both domains of SMARCA5 are required for recruitment to UV-C damage.Citation87 Finally, however, recruitment to DSBs and UV-C damage may both depend on H3 methylation.Citation54,87
ISWI chromatin remodelers were proposed to continuously sample nucleosomes by transient binding to chromatin until they encounter a signal that permits them to bind longer and with higher affinity to allow remodeling.Citation88 Particularly, specific chromatin modifications or targeting molecules may represent such a cue to target and activate ISWI complexes. Indeed, SMARCA5 activity is stimulated by unacetylated histone H4 tails and specifically inhibited by H4K16 acetylation.Citation89-92 Furthermore, H3K4 methylation was shown in yeast to recruit Isw1 during transcription.Citation60 In addition, binding and orientation of yeast SMARCA5 orthologs depends on the presence of a specific length of extranucleosomal DNA, involving an interaction with the SLIDE domain ().Citation25,Citation93-96 Thus, multiple cues involving different histone modifications and DNA configurations may act synergistically in determining binding site selectivity and initiation of remodeling activity.
The relatively high concentration of SMARCA5 and its dynamic sampling of chromatin, as observed in live cell imaging experiments, was suggested to ensure efficient and fast loading of SMARCA5 onto its targets, including DNA damaged chromatin.Citation41,88 This sampling model also provides a kinetic framework for the swift localization of ISWI complexes to UV-C damaged DNA, which represent sites where RNApolII is stalled (). SMARCA5 recruitment to UV-C damage is attenuated when specific cues cannot be detected, i.e. following treatment with chemical inhibitors of histone deacetylation or methylation and when the SLIDE domain that interacts with extranucleosomal DNA is deleted.Citation87 Because recruitment to UV-C damage is also dependent on transcription, these specific histone mark cues that recruit SMARCA5 may be induced as a consequence of lesion-stalled RNApolII. A similar selection mechanism involving histone modifications could also be envisioned for DSB repair, as H3K4 methylation and H3K56 deacetylation coincide with SMARCA5 enrichment at DSBs.Citation54,58 Additionally, these recruitment cues could be formed by specific protein interactions, such as with SIRT6, PARylated RNF168 and NuMA. It is also likely that accessory subunits such as ACF1 participate in site selection, as it was shown that a small ACF1 interaction motif in SMARCA5 is necessary for its localization to DSBs.Citation55
The recruitment of SMARCA5 to UV-C damage, but not to DSBs, depends on ATP hydrolysis.Citation55,87 SMARCA5 was proposed to use the energy of ATP to associate to substrate nucleosomes, as part of a kinetic proofreading mechanism.Citation97,98 In this model, ISWI complexes act as dimers, containing 2 ATPasesCitation99 in which the first ATP-hydrolysis is used to become committed to a nucleosomeal target, after sampling for cues, which is followed by a second ATP-hydrolysis event that is used for DNA-nucleosome translocation (remodeling). This model is supported by the observation that ATP hydrolysis promotes a more stable and extensive binding of yeast ISW1a and ISW2 to template nucleosomes, before actual translocation takes place.Citation100 Thus, ATPase dependent recruitment to UV-C damage may imply that SMARCA5 employs both a sampling as well as a proofreading mechanism to identify and subsequently associate with substrate nucleosomes near or containing UV-damaged DNA.
Chromatin remodeling activity of ISWI complexes at sites of DNA damage
Following target identification, several observations suggest that SMARCA5, as expected, indeed remodels nucleosomes to promote an open chromatin environment to stimulate repair. The requirement of RNF20, SIRT6 and SMARCA5 in HR can be bypassed by forced chemical chromatin relaxation and DNA near a DSB is less accessible to nuclease digestion if SIRT6 is knocked down.Citation54,58 Furthermore, ATPase activity of SMARCA5 is necessary for both BRCA1 accumulationCitation56 and its stimulation of NHEJ.Citation55 Both at DSBs and at UV-C lesions, SMARCA5 has been observed to re-localize to the periphery of a DNA damaged area upon initial recruitment which may represent the remodeling of chromatin. In case of DSBs, this re-localization depends on PARP1 activity and involves chromatin expansion and spreading of SMARCA5 and downstream DSB factors throughout damaged chromatin.Citation56 Although PARP1 also stimulates chromatin remodeling and the re-localization of histones during GG-NER initiation,Citation77,80 a similar chromatin expansion is not observed at sites of laser-induced UV-C damageCitation101 and SMARCA5 spreading to the periphery of these sites also does not depend on PARP activity.Citation87 This suggests that chromatin remodeling activities of ISWI complexes at sites of DSBs and UV lesions are different, but it is not known which substrates define these differences. Dedicated research is thus required to disclose the molecular mechanisms of nucleosome and/or histone displacement by ISWI complexes at different DNA lesions. Furthermore, understanding how ISWI complexes localize to DNA damage may prove to be very helpful to uncover how exactly chromatin remodeling complexes identify their target nucleosomes in vivo.
Concluding remarks
In summary, it appears that distinct ISWI complexes carry out a variety of different functions in the mammalian DDR and have multiple different ways of accommodating chromatin structure to facilitate efficient DNA repair and signaling. Importantly, chromatin organization involving SMARCA5 complexes may be an important determinant for cancer development and treatment. SMARCA5 expression is regulated by the miR-99 family of microRNAs, consisting of miR-99a and miR-100, which is induced upon DNA damage inductionCitation102 but shows reduced expression in more advanced cancers.Citation103 Differences in radiation sensitivity between breast tumor cell lines were attributed to the ability of cells to express the miR-99 family in response to DNA damage.Citation103 Upregulated miR-99a and miR-100 expression leads to SMARCA5 downregulation, which in turn reduces DNA repair, making cells more sensitive to DNA damage and especially to multiple rounds of DNA damage induction such as used in cancer therapy. Therefore, decreased expression of this miRNA family in cancers may be a mechanism for tumor cells to upregulate DNA repair and acquire resistance to DNA damaging agents. Thus, SMARCA5 may be a component in cancer resistance to DNA damaging agents and therefore a potential therapeutic target. Interestingly, inhibition of the other ISWI ATPase, SMARCA1, selectively activates the DDR and leads to growth inhibition and apoptosis of highly malignant tumor cells,Citation104 implying that ISWI complexes in general may be promising targets for cancer treatment.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med 2009; 361:1475-85; PMID:19812404; http://dx.doi.org/10.1056/NEJMra0804615
- Giglia-Mari G, Zotter A, Vermeulen W. DNA damage response. Cold Spring Harb Perspect Biol 2011; 3:a000745; PMID:20980439; http://dx.doi.org/10.1101/cshperspect.a000745
- Garraway LA, Lander ES. Lessons from the cancer genome. Cell 2013; 153:17-37; PMID:23540688; http://dx.doi.org/10.1016/j.cell.2013.03.002
- O’Sullivan RJ, Karlseder J. The great unravelling: chromatin as a modulator of the aging process. Trends Biochem Sci 2012; 37:466-76; http://dx.doi.org/10.1016/j.tibs.2012.08.001
- Luger K, Dechassa ML, Tremethick DJ. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 2012; 13:436-47; PMID:22722606; http://dx.doi.org/10.1038/nrm3382
- Soria G, Polo SE, Almouzni G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 2012; 46:722-34; PMID:22749398; http://dx.doi.org/10.1016/j.molcel.2012.06.002
- Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol 2013; 20:259-66; PMID:23463310; http://dx.doi.org/10.1038/nsmb.2470
- Luijsterburg MS, van Attikum H. Chromatin and the DNA damage response: the cancer connection. Mol Oncol 2011; 5:349-67; PMID:21782533; http://dx.doi.org/10.1016/j.molonc.2011.06.001
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem 2009; 78:273-304; PMID:19355820; http://dx.doi.org/10.1146/annurev.biochem.77.062706.153223
- Eisen JA, Sweder KS, Hanawalt PC. Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res 1995; 23:2715-23; PMID:7651832; http://dx.doi.org/10.1093/nar/23.14.2715
- Lans H, Marteijn JA, Vermeulen W. ATP-dependent chromatin remodeling in the DNA-damage response. Epigenetics Chromatin 2012; 5:4; PMID:22289628; http://dx.doi.org/10.1186/1756-8935-5-4
- Tsukiyama T, Daniel C, Tamkun J, Wu C. ISWI, a member of the SWI2/SNF2 ATPase family, encodes the 140 kDa subunit of the nucleosome remodeling factor. Cell 1995; 83:1021-6; PMID:8521502; http://dx.doi.org/10.1016/0092-8674(95)90217-1
- Tsukiyama T, Wu C. Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell 1995; 83:1011-20; PMID:8521501; http://dx.doi.org/10.1016/0092-8674(95)90216-3
- Varga-Weisz PD, Wilm M, Bonte E, Dumas K, Mann M, Becker PB. Chromatin-remodelling factor CHRAC contains the ATPases ISWI and topoisomerase II. Nature 1997; 388:598-602; PMID:9252192; http://dx.doi.org/10.1038/41587
- Ito T, Bulger M, Pazin MJ, Kobayashi R, Kadonaga JT. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell 1997; 90:145-55; PMID:9230310; http://dx.doi.org/10.1016/S0092-8674(00)80321-9
- Erdel F, Rippe K. Chromatin remodelling in mammalian cells by ISWI-type complexes–where, when and why? FEBS J 2011; 278:3608-18; PMID:21810179; http://dx.doi.org/10.1111/j.1742-4658.2011.08282.x
- Toto M, D’Angelo G, Corona DF. Regulation of ISWI chromatin remodelling activity. Chromosoma 2014; 123(1-2):91-102; PMID:24414837; http://dx.doi.org/10.1007/s00412-013-0447-4
- Banting GS, Barak O, Ames TM, Burnham AC, Kardel MD, Cooch NS, Davidson CE, Godbout R, McDermid HE, Shiekhattar R. CECR2, a protein involved in neurulation, forms a novel chromatin remodeling complex with SNF2L. Hum Mol Genet 2005; 14:513-24; PMID:15640247; http://dx.doi.org/10.1093/hmg/ddi048
- Barak O, Lazzaro MA, Lane WS, Speicher DW, Picketts DJ, Shiekhattar R. Isolation of human NURF: a regulator of Engrailed gene expression. EMBO J 2003; 22:6089-100; PMID:14609955; http://dx.doi.org/10.1093/emboj/cdg582
- Thompson PJ, Norton KA, Niri FH, Dawe CE, McDermid HE. CECR2 is involved in spermatogenesis and forms a complex with SNF2H in the testis. J Mol Biol 2012; 415:793-806; PMID:22154806; http://dx.doi.org/10.1016/j.jmb.2011.11.041
- Bork P, Koonin EV. An expanding family of helicases within the ‘DEAD/H’ superfamily. Nucleic Acids Res 1993; 21:751-2; PMID:8382805; http://dx.doi.org/10.1093/nar/21.3.751
- Grune T, Brzeski J, Eberharter A, Clapier CR, Corona DF, Becker PB, Muller CW. Crystal structure and functional analysis of a nucleosome recognition module of the remodeling factor ISWI. Mol Cell 2003; 12:449-60; PMID:14536084; http://dx.doi.org/10.1016/S1097-2765(03)00273-9
- Mueller-Planitz F, Klinker H, Ludwigsen J, Becker PB. The ATPase domain of ISWI is an autonomous nucleosome remodeling machine. Nat Struct Mol Biol 2013; 20:82-9; PMID:23202585; http://dx.doi.org/10.1038/nsmb.2457
- Hota SK, Bhardwaj SK, Deindl S, Lin YC, Zhuang X, Bartholomew B. Nucleosome mobilization by ISW2 requires the concerted action of the ATPase and SLIDE domains. Nat Struct Mol Biol 2013; 20:222-9; PMID:23334290; http://dx.doi.org/10.1038/nsmb.2486
- Dang W, Bartholomew B. Domain architecture of the catalytic subunit in the ISW2-nucleosome complex. Mol Cell Biol 2007; 27:8306-17; PMID:17908792; http://dx.doi.org/10.1128/MCB.01351-07
- Gkikopoulos T, Schofield P, Singh V, Pinskaya M, Mellor J, Smolle M, Workman JL, Barton GJ, Owen-Hughes T. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science 2011; 333:1758-60; PMID:21940898; http://dx.doi.org/10.1126/science.1206097
- Yang JG, Madrid TS, Sevastopoulos E, Narlikar GJ. The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat Struct Mol Biol 2006; 13:1078-83; PMID:17099699; http://dx.doi.org/10.1038/nsmb1170
- Collins N, Poot RA, Kukimoto I, Garcia-Jimenez C, Dellaire G, Varga-Weisz PD. An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat Genet 2002; 32:627-32; PMID:12434153; http://dx.doi.org/10.1038/ng1046
- Poot RA, Bozhenok L, van den Berg DL, Steffensen S, Ferreira F, Grimaldi M, Gilbert N, Ferreira J, Varga-Weisz PD. The Williams syndrome transcription factor interacts with PCNA to target chromatin remodelling by ISWI to replication foci. Nat Cell Biol 2004; 6:1236-44; PMID:15543136; http://dx.doi.org/10.1038/ncb1196
- Corona DF, Tamkun JW. Multiple roles for ISWI in transcription, chromosome organization and DNA replication. Biochim Biophys Acta 2004; 1677:113-9; PMID:15020052; http://dx.doi.org/10.1016/j.bbaexp.2003.09.018
- Langst G, Becker PB. ISWI induces nucleosome sliding on nicked DNA. Mol Cell 2001; 8:1085-92; PMID:11741543; http://dx.doi.org/10.1016/S1097-2765(01)00397-5
- Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell 2002; 108:475-87; PMID:11909519; http://dx.doi.org/10.1016/S0092-8674(02)00654-2
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/10.1038/nature08467
- Kim YJ, Wilson DM, 3rd. Overview of base excision repair biochemistry. Curr Mol Pharmacol 2012; 5:3-13; PMID:22122461; http://dx.doi.org/10.2174/1874467211205010003
- Dianov GL, Hubscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res 2013; 41:3483-90; PMID:23408852; http://dx.doi.org/10.1093/nar/gkt076
- Beard BC, Wilson SH, Smerdon MJ. Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc Natl Acad Sci U S A 2003; 100:7465-70; PMID:12799467; http://dx.doi.org/10.1073/pnas.1330328100
- Menoni H, Gasparutto D, Hamiche A, Cadet J, Dimitrov S, Bouvet P, Angelov D. ATP-dependent chromatin remodeling is required for base excision repair in conventional but not in variant H2A.Bbd nucleosomes. Mol Cell Biol 2007; 27:5949-56; PMID:17591702; http://dx.doi.org/10.1128/MCB.00376-07
- Menoni H, Shukla MS, Gerson V, Dimitrov S, Angelov D. Base excision repair of 8-oxoG in dinucleosomes. Nucleic Acids Res 2012; 40:692-700; PMID:21930508; http://dx.doi.org/10.1093/nar/gkr761
- Nilsen H, Lindahl T, Verreault A. DNA base excision repair of uracil residues in reconstituted nucleosome core particles. EMBO J 2002; 21:5943-52; PMID:12411511; http://dx.doi.org/10.1093/emboj/cdf581
- Nakanishi S, Prasad R, Wilson SH, Smerdon M. Different structural states in oligonucleosomes are required for early versus late steps of base excision repair. Nucleic Acids Res 2007; 35:4313-21; PMID:17576692; http://dx.doi.org/10.1093/nar/gkm436
- Erdel F, Rippe K. Binding kinetics of human ISWI chromatin-remodelers to DNA repair sites elucidate their target location mechanism. Nucleus 2011; 2:105-12; PMID:21738833; http://dx.doi.org/10.4161/nucl.2.2.15209
- Lan L, Nakajima S, Komatsu K, Nussenzweig A, Shimamoto A, Oshima J, Yasui A. Accumulation of Werner protein at DNA double-strand breaks in human cells. J Cell Sci 2005; 118:4153-62; PMID:16141234; http://dx.doi.org/10.1242/jcs.02544
- Kielbassa C, Roza L, Epe B. Wavelength dependence of oxidative DNA damage induced by UV and visible light. Carcinogenesis 1997; 18:811-6; PMID:9111219; http://dx.doi.org/10.1093/carcin/18.4.811
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 2008; 77:229-57; PMID:18275380; http://dx.doi.org/10.1146/annurev.biochem.77.061306.125255
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 2010; 79:181-211; PMID:20192759; http://dx.doi.org/10.1146/annurev.biochem.052308.093131
- Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol 2011; 13:1161-9; PMID:21968989; http://dx.doi.org/10.1038/ncb2344
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146:905-16; PMID:10477747; http://dx.doi.org/10.1083/jcb.146.5.905
- Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009; 459:460-3; PMID:19357644; http://dx.doi.org/10.1038/nature07955
- Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014; 15:7-18; PMID:24326623; http://dx.doi.org/10.1038/nrm3719
- Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet 2002; 30:285-9; PMID:11836499; http://dx.doi.org/10.1038/ng837
- Pines A, Mullenders LH, van Attikum H, Luijsterburg MS. Touching base with PARPs: moonlighting in the repair of UV lesions and double-strand breaks. Trends Biochem Sci 2013; 38:321-30; PMID:23562323; http://dx.doi.org/10.1016/j.tibs.2013.03.002
- Lisby M, Mortensen UH, Rothstein R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol 2003; 5:572-7; PMID:12766777; http://dx.doi.org/10.1038/ncb997
- Soutoglou E, Dorn JF, Sengupta K, Jasin M, Nussenzweig A, Ried T, Danuser G, Misteli T. Positional stability of single double-strand breaks in mammalian cells. Nat Cell Biol 2007; 9:675-82; PMID:17486118; http://dx.doi.org/10.1038/ncb1591
- Nakamura K, Kato A, Kobayashi J, Yanagihara H, Sakamoto S, Oliveira DV, Shimada M, Tauchi H, Suzuki H, Tashiro S, et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol Cell 2011; 41:515-28; PMID:21362548; http://dx.doi.org/10.1016/j.molcel.2011.02.002
- Lan L, Ui A, Nakajima S, Hatakeyama K, Hoshi M, Watanabe R, Janicki SM, Ogiwara H, Kohno T, Kanno S, et al. The ACF1 complex is required for DNA double-strand break repair in human cells. Mol Cell 2010; 40:976-87; PMID:21172662; http://dx.doi.org/10.1016/j.molcel.2010.12.003
- Smeenk G, Wiegant WW, Marteijn JA, Luijsterburg MS, Sroczynski N, Costelloe T, Romeijn RJ, Pastink A, Mailand N, Vermeulen W, et al. Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J Cell Sci 2013; 126:889-903; PMID:23264744; http://dx.doi.org/10.1242/jcs.109413
- Sanchez-Molina S, Mortusewicz O, Bieber B, Auer S, Eckey M, Leonhardt H, Friedl AA, Becker PB. Role for hACF1 in the G2/M damage checkpoint. Nucleic Acids Res 2011; 39:8445-56; PMID:21745822; http://dx.doi.org/10.1093/nar/gkr435
- Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, Sebastian C, Cosentino C, Martinez-Pastor B, Giacosa S, et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell 2013; 51:454-68; PMID:23911928; http://dx.doi.org/10.1016/j.molcel.2013.06.018
- Vidi PA, Liu J, Salles D, Jayaraman S, Dorfman G, Gray M, Abad P, Moghe PV, Irudayaraj JM, Wiesmuller L, et al. NuMA promotes homologous recombination repair by regulating the accumulation of the ISWI ATPase SNF2h at DNA breaks. Nucleic Acids Res 2014; PMID:24753406
- Santos-Rosa H, Schneider R, Bernstein BE, Karabetsou N, Morillon A, Weise C, Schreiber SL, Mellor J, Kouzarides T. Methylation of histone H3 K4 mediates association of the Isw1p ATPase with chromatin. Mol Cell 2003; 12:1325-32; PMID:14636589; http://dx.doi.org/10.1016/S1097-2765(03)00438-6
- Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S, et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci U S A 2008; 105:3443-8; PMID:18299561; http://dx.doi.org/10.1073/pnas.0712384105
- Eberharter A, Ferrari S, Langst G, Straub T, Imhof A, Varga-Weisz P, Wilm M, Becker PB. Acf1, the largest subunit of CHRAC, regulates ISWI-induced nucleosome remodelling. EMBO J 2001; 20:3781-8; PMID:11447119; http://dx.doi.org/10.1093/emboj/20.14.3781
- Ito T, Levenstein ME, Fyodorov DV, Kutach AK, Kobayashi R, Kadonaga JT. ACF consists of two subunits, Acf1 and ISWI, that function cooperatively in the ATP-dependent catalysis of chromatin assembly. Genes Dev 1999; 13:1529-39; PMID:10385622; http://dx.doi.org/10.1101/gad.13.12.1529
- He X, Fan HY, Garlick JD, Kingston RE. Diverse regulation of SNF2h chromatin remodeling by noncatalytic subunits. Biochemistry 2008; 47:7025-33; PMID:18553938; http://dx.doi.org/10.1021/bi702304p
- Yasui D, Miyano M, Cai S, Varga-Weisz P, Kohwi-Shigematsu T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 2002; 419:641-5; PMID:12374985; http://dx.doi.org/10.1038/nature01084
- Kukimoto I, Elderkin S, Grimaldi M, Oelgeschlager T, Varga-Weisz PD. The histone-fold protein complex CHRAC-15/17 enhances nucleosome sliding and assembly mediated by ACF. Mol Cell 2004; 13:265-77; PMID:14759371; http://dx.doi.org/10.1016/S1097-2765(03)00523-9
- Bozhenok L, Wade PA, Varga-Weisz P. WSTF-ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO J 2002; 21:2231-41; PMID:11980720; http://dx.doi.org/10.1093/emboj/21.9.2231
- Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, Ishibe-Murakami S, Wang B, Tempst P, Hofmann K, et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 2009; 457:57-62; PMID:19092802; http://dx.doi.org/10.1038/nature07668
- Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009; 458:591-6; PMID:19234442; http://dx.doi.org/10.1038/nature07849
- Krishnan N, Jeong DG, Jung SK, Ryu SE, Xiao A, Allis CD, Kim SJ, Tonks NK. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J Biol Chem 2009; 284:16066-70; PMID:19351884; http://dx.doi.org/10.1074/jbc.C900032200
- Stucki M. Histone H2A.X Tyr142 phosphorylation: a novel sWItCH for apoptosis? DNA Repair (Amst) 2009; 8:873-6; PMID:19446503; http://dx.doi.org/10.1016/j.dnarep.2009.04.003
- Sheu JJ, Guan B, Choi JH, Lin A, Lee CH, Hsiao YT, Wang TL, Tsai FJ, Shih Ie M. Rsf-1, a chromatin remodeling protein, induces DNA damage and promotes genomic instability. J Biol Chem 2010; 285:38260-9; PMID:20923775; http://dx.doi.org/10.1074/jbc.M110.138735
- Pessina F, Lowndes NF. The RSF1 histone-remodelling factor facilitates DNA double-strand break repair by recruiting centromeric and fanconi anaemia proteins. PLoS Biol 2014; 12:e1001856; PMID:24800743; http://dx.doi.org/10.1371/journal.pbio.1001856
- Helfricht A, Wiegant WW, Thijssen PE, Vertegaal AC, Luijsterburg MS, van Attikum H. Remodeling and spacing factor 1 (RSF1) deposits centromere proteins at DNA double-strand breaks to promote non-homologous end-joining. Cell Cycle 2013; 12:3070-82; PMID:23974106; http://dx.doi.org/10.4161/cc.26033
- Scharer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol 2013; 5:a012609; PMID:24086042; http://dx.doi.org/10.1101/cshperspect.a012609
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 2014; 15:465-81; PMID:24954209; http://dx.doi.org/10.1038/nrm3822
- Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S, et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol 2012; 199:235-49; PMID:23045548; http://dx.doi.org/10.1083/jcb.201112132
- Jiang Y, Wang X, Bao S, Guo R, Johnson DG, Shen X, Li L. INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proc Natl Acad Sci U S A 2010; 107:17274-9; PMID:20855601; http://dx.doi.org/10.1073/pnas.1008388107
- Zhang L, Zhang Q, Jones K, Patel M, Gong F. The chromatin remodeling factor BRG1 stimulates nucleotide excision repair by facilitating recruitment of XPC to sites of DNA damage. Cell Cycle 2009; 8:3953-9; PMID:19901545; http://dx.doi.org/10.4161/cc.8.23.10115
- Luijsterburg MS, Lindh M, Acs K, Vrouwe MG, Pines A, van Attikum H, Mullenders LH, Dantuma NP. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J Cell Biol 2012; 197:267-81; PMID:22492724; http://dx.doi.org/10.1083/jcb.201106074
- Citterio E, Rademakers S, van der Horst GT, van Gool AJ, Hoeijmakers JH, Vermeulen W. Biochemical and biological characterization of wild-type and ATPase-deficient Cockayne syndrome B repair protein. J Biol Chem 1998; 273:11844-51; PMID:9565609; http://dx.doi.org/10.1074/jbc.273.19.11844
- Selzer RR, Nyaga S, Tuo J, May A, Muftuoglu M, Christiansen M, Citterio E, Brosh RM, Jr., Bohr VA. Differential requirement for the ATPase domain of the Cockayne syndrome group B gene in the processing of UV-induced DNA damage and 8-oxoguanine lesions in human cells. Nucleic Acids Res 2002; 30:782-93; PMID:11809892; http://dx.doi.org/10.1093/nar/30.3.782
- Marteijn JA, Bekker-Jensen S, Mailand N, Lans H, Schwertman P, Gourdin AM, Dantuma NP, Lukas J, Vermeulen W. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J Cell Biol 2009; 186:835-47; PMID:19797077; http://dx.doi.org/10.1083/jcb.200902150
- Marini F, Nardo T, Giannattasio M, Minuzzo M, Stefanini M, Plevani P, Muzi Falconi M. DNA nucleotide excision repair-dependent signaling to checkpoint activation. Proc Natl Acad Sci U S A 2006; 103:17325-30; PMID:17088560; http://dx.doi.org/10.1073/pnas.0605446103
- Hanasoge S, Ljungman M. H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis 2007; 28:2298-304; PMID:17615256; http://dx.doi.org/10.1093/carcin/bgm157
- Lans H, Marteijn JA, Schumacher B, Hoeijmakers JH, Jansen G, Vermeulen W. Involvement of global genome repair, transcription coupled repair, and chromatin remodeling in UV DNA damage response changes during development. PLoS Genet 2010; 6:e1000941; PMID:20463888; http://dx.doi.org/10.1371/journal.pgen.1000941
- Aydin OZ, Marteijn JA, Ribeiro-Silva C, Rodriguez Lopez A, Wijgers N, Smeenk G, van Attikum H, Poot RA, Vermeulen W, Lans H. Human ISWI complexes are targeted by SMARCA5 ATPase and SLIDE domains to help resolve lesion-stalled transcription. Nucleic Acids Res 2014; 42:8473-85; PMID:24990377; http://dx.doi.org/10.1093/nar/gku565
- Erdel F, Schubert T, Marth C, Langst G, Rippe K. Human ISWI chromatin-remodeling complexes sample nucleosomes via transient binding reactions and become immobilized at active sites. Proc Natl Acad Sci U S A 2010; 107:19873-8; PMID:20974961; http://dx.doi.org/10.1073/pnas.1003438107
- Alenghat T, Yu J, Lazar MA. The N-CoR complex enables chromatin remodeler SNF2H to enhance repression by thyroid hormone receptor. EMBO J 2006; 25:3966-74; PMID:16917504; http://dx.doi.org/10.1038/sj.emboj.7601280
- Corona DF, Clapier CR, Becker PB, Tamkun JW. Modulation of ISWI function by site-specific histone acetylation. EMBO Rep 2002; 3:242-7; PMID:11882543; http://dx.doi.org/10.1093/embo-reports/kvf056
- Clapier CR, Langst G, Corona DF, Becker PB, Nightingale KP. Critical role for the histone H4 N terminus in nucleosome remodeling by ISWI. Mol Cell Biol 2001; 21:875-83; PMID:11154274; http://dx.doi.org/10.1128/MCB.21.3.875-883.2001
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006; 311:844-7; PMID:16469925; http://dx.doi.org/10.1126/science.1124000
- Dang W, Kagalwala MN, Bartholomew B. Regulation of ISW2 by concerted action of histone H4 tail and extranucleosomal DNA. Mol Cell Biol 2006; 26:7388-96; PMID:17015471; http://dx.doi.org/10.1128/MCB.01159-06
- Zofall M, Persinger J, Bartholomew B. Functional role of extranucleosomal DNA and the entry site of the nucleosome in chromatin remodeling by ISW2. Mol Cell Biol 2004; 24:10047-57; PMID:15509805; http://dx.doi.org/10.1128/MCB.24.22.10047-10057.2004
- Kagalwala MN, Glaus BJ, Dang W, Zofall M, Bartholomew B. Topography of the ISW2-nucleosome complex: insights into nucleosome spacing and chromatin remodeling. EMBO J 2004; 23:2092-104; PMID:15131696; http://dx.doi.org/10.1038/sj.emboj.7600220
- Stockdale C, Flaus A, Ferreira H, Owen-Hughes T. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J Biol Chem 2006; 281:16279-88; PMID:16606615; http://dx.doi.org/10.1074/jbc.M600682200
- Narlikar GJ. A proposal for kinetic proof reading by ISWI family chromatin remodeling motors. Curr Opin Chem Biol 2010; 14:660-5; PMID:20833099; http://dx.doi.org/10.1016/j.cbpa.2010.08.001
- Blossey R, Schiessel H. Kinetic proofreading in chromatin remodeling: the case of ISWI/ACF. Biophys J 2011; 101:L30-2; PMID:21843463; http://dx.doi.org/10.1016/j.bpj.2011.07.001
- Racki LR, Yang JG, Naber N, Partensky PD, Acevedo A, Purcell TJ, Cooke R, Cheng Y, Narlikar GJ. The chromatin remodeller ACF acts as a dimeric motor to space nucleosomes. Nature 2009; 462:1016-21; PMID:20033039; http://dx.doi.org/10.1038/nature08621
- Gangaraju VK, Prasad P, Srour A, Kagalwala MN, Bartholomew B. Conformational changes associated with template commitment in ATP-dependent chromatin remodeling by ISW2. Mol Cell 2009; 35:58-69; PMID:19595716; http://dx.doi.org/10.1016/j.molcel.2009.05.013
- Dinant C, Ampatziadis-Michailidis G, Lans H, Tresini M, Lagarou A, Grosbart M, Theil AF, van Cappellen WA, Kimura H, Bartek J, et al. Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol Cell 2013; 51:469-79; PMID:23973375; http://dx.doi.org/10.1016/j.molcel.2013.08.007
- Sun D, Lee YS, Malhotra A, Kim HK, Matecic M, Evans C, Jensen RV, Moskaluk CA, Dutta A. miR-99 family of MicroRNAs suppresses the expression of prostate-specific antigen and prostate cancer cell proliferation. Cancer Res 2011; 71:1313-24; PMID:21212412; http://dx.doi.org/10.1158/0008-5472.CAN-10-1031
- Mueller AC, Sun D, Dutta A. The miR-99 family regulates the DNA damage response through its target SNF2H. Oncogene 2013; 32:1164-72; PMID:22525276; http://dx.doi.org/10.1038/onc.2012.131
- Ye Y, Xiao Y, Wang W, Wang Q, Yearsley K, Wani AA, Yan Q, Gao JX, Shetuni BS, Barsky SH. Inhibition of expression of the chromatin remodeling gene, SNF2L, selectively leads to DNA damage, growth inhibition, and cancer cell death. Mol Cancer Res 2009; 7:1984-99; PMID:19996304; http://dx.doi.org/10.1158/1541-7786.MCR-09-0119