
Abstract
Selective targeting of cancer stem cells (CSCs), implicated in tumor relapse, holds great promise in the treatment of colorectal cancer. Overexpression of C-terminal binding protein (CtBP), an NADH dependent transcriptional regulator, is often observed in colon cancer. Of note, TCF-4 signaling is also up-regulated in colonic CSCs. We hypothesized that CtBP, whose dehydrogenase activity is amenable to pharmacological inhibition by 4-methylthio-2-oxobutyric acid (MTOB), positively regulates TCF-4 signaling, leading to CSC growth and self-renewal. CSCs demonstrated significant upregulation of CtBP1 and CtBP2 levels (mRNA and protein) and activity partly due to increased NADH/NAD ratio, as well as increased TCF/LEF transcriptional activity, compared to respective controls. Depletion of CtBP2 inhibited, while its overexpression enhanced, CSC growth (1° spheroids) and self-renewal (2°/3° spheroids). Similarly, MTOB caused a robust inhibition of spheroid growth and self-renewal in a dose dependent manner. MTOB displayed significantly greater selectivity for growth inhibition in the spheroids, at least in part through induction of apoptosis, compared to monolayer controls. Moreover, MTOB inhibited basal as well as induced (by GSK-3β inhibitor) TCF/LEF activity while suppressing mRNA and protein levels of several β-catenin target genes (CD44, Snail, C-MYC and LGR5). Lastly, CtBP physically interacted with TCF-4, and this interaction was significantly inhibited in the presence of MTOB. The above findings point to a novel role of CtBPs in the promotion of CSC growth and self-renewal through direct regulation of TCF/LEF transcription. Moreover, small molecular inhibition of its function can selectively target CSCs, presenting a novel approach for treatment of colorectal cancer focused on targeting of CSCs.
Abbreviations
| 1° | = | primary |
| 2° | = | secondary |
| 3° | = | tertiary |
| CtBP | = | c-terminal binding protein |
| CSC | = | cancer stem cell |
| TCF-4 | = | transcription factor 4 |
Introduction
In recent years, the cancer stem cell (CSC) hypothesis has generated significant interest in the scientific community as a unifying hypothesis explaining many of the shortcomings of current anti-cancer therapeutics.Citation1,2 The outcomes for advanced colorectal cancer remain poor, mainly due to disease relapse.Citation3 The survival of a smaller population of CSCs with the ability to self-renew, resulting in reconstitution of the entire tumor, is at the heart of such relapses.Citation1 Hence, a therapeutic paradigm aimed at selectively targeting CSCs is gaining momentum as a singular approach to achieve long-term disease control or cure.Citation4 In addition, mechanistic similarities at the CSC level among different tumor types suggest that targeting CSCs represents a promising approach to treat cancer.
Despite this promise, the discovery of selective anti-CSC agents is sluggish. Several challenges, including the relative paucity of CSCs in “bulk” tumor cell populations, and a lack of clearly identified therapeutic targets, has contributed to slow progress.Citation5,6 Although the precise mechanisms governing stem cell self-renewal are poorly understood, it is widely accepted that epigenetic modulation of gene expression programs regulates stem cell fate.Citation7 Hypoxia, which promotes a reduced redox state (decreased oxidized nicotinamide adenosine dinucleotide [NAD] i.e. NAD+/NADH ratio),Citation8 is known to promote CSC initiation and growth.Citation9,10 Moreover, reduced redox state has been shown to tightly regulate stem cell self-renewal.Citation11 Hence, by logical deduction we hypothesized that NAD+/NADH sensing epigenetic regulators might govern CSCs growth and self-renewal.
Of various proteins that use NAD+/NADH as ligand, cofactor or substrate, the C-terminal binding protein (CtBP) class of proteins are true chemical sensors of changes in cellular NADH levels.Citation12 Upon binding to NADH, CtBP causes target gene repression/activation by interacting with DNA binding transcription factors and modulating the epigenetic machinery.Citation13 Hence, CtBP appears to be an important molecular link between cellular redox potential and gene expression regulation. Given the importance of cellular redox potential in governing CSCs growth and self-renewal, it is highly probable that CtBP plays a role in regulation of CSC phenotype.Citation8,14 In fact, in an in vitro model of estrogen receptor responsive and triple negative breast cancer, CtBP knockdown resulted in attenuation of CD44/CD24 ratio suggestive of inhibition of CSC phenotype.Citation15
Increasing evidence points to a tumor promoter role for CtBP in various cancers including colorectal cancer.Citation15 In fact, Straza et al. demonstrated that almost 2 thirds of colorectal cancers overexpress CtBP compared to adjacent normal tissue.Citation16 Traditionally, CtBP was regarded as a repressor of gene expression.Citation17 However, more recent evidence points to a more diverse role in which CtBP can be both an activator as well as repressor of gene expression depending on the partnering transcription factor, as well as cell context.Citation15,18 In fact, in a drosophila model, CtBP was proposed as an activator of TCF-4 signaling in gene and context specific manner.Citation19 We and others have shown that TCF-4 signaling plays a key role in colonic CSC growth and self-renewal.Citation20,21 In fact, calcein-effluxing subpopulation (Clop) of human colon cancer cells was shown to possess in vivo self-renewal capacity indicating enrichment of CSCs in Clop.Citation22 These CloP cells also demonstrated increased presence of nuclear β-catenin and β-catenin siRNA significantly depleted CloP in colon cancer cell lines.Citation22 Based on the above observations, we hypothesized that CtBP might positively regulate colon CSC growth and self-renewal via activation of β-catenin/TCF-4 signaling.
Recently, Straza et al. demonstrated that the penultimate compound in the methionine salvage pathway, 2-keto-4-methyl-triobutryate (MTOB) promotes cytotoxicity in colon cancer cells through apoptosis induction by inhibiting CtBP repressor activity on the Bik promoter.Citation16 Molecular mechanism of MTOB mediated suppression of CtBP's recruitment to target promoter was not studied in detail;Citation16 however, it is proposed that MTOB binding might cause conformational changes in CtBP substrate-binding domain leading to reduced CtBP interaction with promoter bound transcription factors.Citation23 Although, MTOB produces meaningful antitumor activity in xenograft models, it induces apoptosis in vitro only at very high concentrations; raising the possibility that MTOB might selectively target relatively rare cells, such as CSCs, in the tumor. However, MTOB's effect on colonic CSCs has not been studied in detail. Herein, we demonstrate that CtBP expression and activity is significantly enhanced in CSCs more so than in non-CSCs. Moreover, CtBP depletion or MTOB treatment caused inhibition of CSC growth and self-renewal, in part, through inhibition of TCF-4 signaling.
Results
Determination of CtBP expression and activity in CSCs
Among diverse cellular phenomena regulated by CtBP, many are reminiscent of CSC phenotypes such as epithelial-mesenchymal transition, invasion, and metastasis.Citation14,24 Moreover, CtBP was shown to regulate expression of breast CSC markers.Citation15 Hence, we hypothesized that CtBP, a redox regulated transcriptional modulator, which is frequently overexpressed in colorectal cancerCitation16 will regulate colonic CSC growth and self-renewal.
We and others have shown that colonic spheroids are several-fold enriched in CSCs than their monolayer counterparts.Citation20,25 In fact, we confirmed a several fold increase in CD133(hi)/CXCR4(hi) [Dual (hi)] cells in HCT-116 spheroids compared to monolayer controls (Fig. S1). Next, we sought to determine the expression and activity of CtBP in colonic CSCs. We observed a modest increase in CtBP2 and a mild increase in CtBP1 levels in HCT-116 spheroids compared to monolayer controls (Fig. S2). Although spheroid cultures are enriched in CSCs, they represent a mixture of stem and progenitor cells. In order to further enrich for CSCs, we performed fluorescent activated cell-sorting (FACS) for 2 colonic CSCs markers CD133 and CXCR4 in HT-29 spheroid cells. Indeed, Dual (hi), showed approximately fold3- increase in spheroid formation compared to CD133(low)/CXCR4(low) [Dual (lo)], controls suggesting that Dual (hi) cells represent CSCs (Fig. S3). Upon further analyses, we observed a robust 2- and fold5- increase in CtBP1 and CtBP2 expression respectively in Dual (hi) compared to controls (). This increased expression of CtBP in Dual (hi) CSCs is due in part to increased gene transcription as evident by an approximately fold2- increase in mRNA expression of both CtBP homologues in Dual (hi) cells compared to controls (). Consistent with increased levels, CtBP activity was higher in Dual (hi) cells as evident by 2 to fold4- changes in gene expression of its bona fide repression and activation targets, namely BIK and DKK1, respectively (). Hence, Dual (hi) colonic CSCs show increased expression and activity of both paralogs of CtBP with a preference for CtBP2.
Figure 1. CtBP expression and activity is upregulated in colon CSCs. (A) An immunoblot analysis revealing higher expression of CtBPs in Dual (hi) [CD133(hi)/CXCR4(hi), CSCs] compared to Dual (lo) [CD133(low)/CXCR4 (low), non-CSCs] HT-29 spheroid cells. GAPDH is used as a loading control. (B) Real-time quantitative reverse transcriptase polymerase chain reaction (QPCR) showing increase in CTBP(s) levels in Dual (hi) CSCs, in part, due to increased gene transcription. Data was normalized to GAPDH. (C) QPCR analysis of differential expression of CtBP target proteins such as BIK (repression), and DKK1 (activation) in Dual (hi) compared to Dual (lo) cells suggesting higher CtBP activity in CSCs. (D) Colonospheres, enriched in Dual (hi) CSCs (Fig. S1), demonstrating decreased NAD+/NADH ratio compared to monolayer controls in both HCT-116 and HT-29 colon cancer cells, suggesting a reduced redox state in the spheroids. (E and F) Immunoblot and QPCR analyses of BIK, a CtBP repression target, suggesting higher CtBP activity in HCT-116 spheroid cells compared to monolayer controls. Actin and GAPDH were used as loading control respectively. Data is represented as percent of vehicle-treated cells. Error bars represent ±1 SEM. *P < 0 .01.
![Figure 1. CtBP expression and activity is upregulated in colon CSCs. (A) An immunoblot analysis revealing higher expression of CtBPs in Dual (hi) [CD133(hi)/CXCR4(hi), CSCs] compared to Dual (lo) [CD133(low)/CXCR4 (low), non-CSCs] HT-29 spheroid cells. GAPDH is used as a loading control. (B) Real-time quantitative reverse transcriptase polymerase chain reaction (QPCR) showing increase in CTBP(s) levels in Dual (hi) CSCs, in part, due to increased gene transcription. Data was normalized to GAPDH. (C) QPCR analysis of differential expression of CtBP target proteins such as BIK (repression), and DKK1 (activation) in Dual (hi) compared to Dual (lo) cells suggesting higher CtBP activity in CSCs. (D) Colonospheres, enriched in Dual (hi) CSCs (Fig. S1), demonstrating decreased NAD+/NADH ratio compared to monolayer controls in both HCT-116 and HT-29 colon cancer cells, suggesting a reduced redox state in the spheroids. (E and F) Immunoblot and QPCR analyses of BIK, a CtBP repression target, suggesting higher CtBP activity in HCT-116 spheroid cells compared to monolayer controls. Actin and GAPDH were used as loading control respectively. Data is represented as percent of vehicle-treated cells. Error bars represent ±1 SEM. *P < 0 .01.](/cms/asset/b336d676-976b-466d-8641-fe3a3a2d97b7/kccy_a_958407_f0001_c.gif)
Furthermore, we observed a relatively reduced redox state, as evident by increased NADH/NAD+ ratio, in CSC-enriched spheroids compared to their monolayer counterparts in 2 colonic cell lines with different genetic backgrounds with respect to K-RAS and p53 mutations, 2 most common mutations in colon cancers (). CtBP is a highly sensitive redox sensor which upon binding to NADH forms homo- or hetero-dimers, leading to its increased binding to target gene promoters.Citation26 In support, we observed a significant decrease in BIK mRNA and protein levels in spheroids compared to monolayer controls ( ), consistent with CtBP-mediated repression of BIK promoter, as reported previously.Citation16 In summary, CtBP2 expression and activity is significantly increased in CSCs, indicating its role as a therapeutic target to selectively inhibit colonic CSCs.
Modulation of CtBP2 levels regulates CSC phenotype
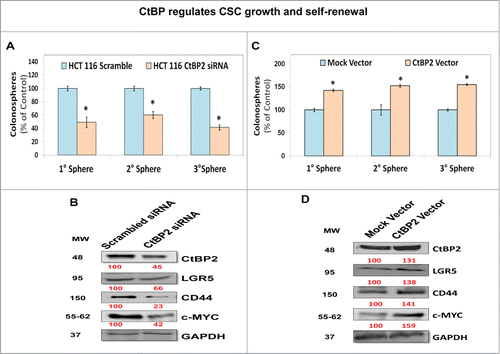
In order to determine consequences of increased CtBP2 levels/activity, we modulated CtBP2 levels by siRNA (depletion) or CtBP2 expression vector (overexpression) and examined CSC growth by determining primary spheroid formation as well as CSC self-renewal by enumerating secondary/tertiary spheroids derived from primary spheroids. CtBP2 siRNA transfected HCT-116 cells demonstrated an approximately 50% reduction in basal CtBP2 expression, resulting in significant inhibition of CSC growth and self-renewal (). In contrast, a modest increase in CtBP2 level following transfection with CtBP2 expression vector resulted in a moderate increase in CSC growth and self-renewal (). In support of the morphological changes above, CtBP2 siRNA treated spheroids showed significant reduction in levels of the CSC markers CD44 and LGR5 as well as c-MYC, a CSC self-renewal factor ().Citation27 In contrast, CtBP2 overexpression caused an increase in the levels of CD44, LGR5 and c-MYC (). Above findings suggest that CtBP2 levels positively regulate CSC growth and self-renewal.
Figure 2. CtBP regulates CSC growth and self-renewal. (A) CtBP2 depletion using siRNA resulted in inhibition of 1°, 2°, and 3° colonosphere formation suggesting attenuation of CSC growth and self-renewal. (B) Immunoblot demonstrating reduced expression of CSC markers CD44 and LGR5 as well as self-renewal factor c-MYC in CtBP2 knockdown colonospheres compared to scrambled siRNA transfected controls. GAPDH was used as a loading control. (C) Overexpression of CtBP2 in HCT-116 cells promoted CSC growth (1° spheroids) and self-renewal (2° and 3° spheroids). (D) Immunoblot analysis demonstrating increased expression of CSC markers CD44 and LGR5 as well as self-renewal factor c-MYC in CtBP2 vector transfected colonosphere compared to mock transfected controls. The latter findings (D) are in stark contrast to those observed with CtBP2 depletion (B). Data is represented as percent of vehicle-treated cells. Error bars represent ±1 SEM. *P < 0 .05. Numbers under the blot represent relative densitometry values.
Pharmacological inhibition of CtBP selectively attenuates CSC growth
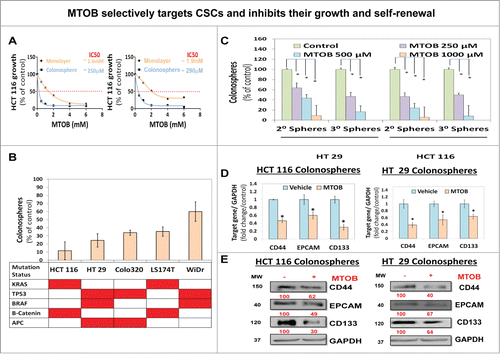
We previously demonstrated that the CtBP dehydrogenase substrate 4-methylthio-2-oxobutyric acid (MTOB) can act as a CtBP inhibitor, albeit at high concentrations in colon cancer cells grown in monolayer condition. Based on our observation that CtBP activity is significantly enhanced in CSCs, we hypothesized that MTOB might selectively target CSCs for inhibition. In fact, we observed that MTOB is at least fold5- more potent in inhibiting CSC growth at day 5 in spheroid cultures (IC50: 250–300 μM) than in monolayer condition (IC50: 1.5–2 mM) in both HCT-116 and HT-29, cells, suggesting that MTOB selectively targets CSCs (). Moreover, MTOB consistently inhibited CSC growth with IC50s ranging from 250 μM to >1 mM and a robust (40–90%) inhibition in spheroid formation at 1 mM concentration () across a spectrum of colon cancer cells harboring various combination of commonly found genetic mutations in human colorectal cancer.Citation28 More importantly, MTOB robustly inhibited CSC self-renewal (at ≥250 μM) in both HCT-116 and HT-29 cells (). Moreover, MTOB treatment resulted in significant inhibition of both mRNA and protein levels of CSC markers, including CD44, Epithelial adhesion molecule (EPCAM) and CD133 () in HCT-116 and HT-29 cells.Citation27 Thus, MTOB-induced molecular changes are consistent with the cellular growth changes observed above. Moreover, MTOB treatment (250–500 μM) following siRNA mediated knockdown of CtBP2 resulted in a significantly greater inhibition in HCT-116 spheroid formation compared to either CtBP2 depletion or MTOB treatment alone (Fig. S4). In summary, pharmacological inhibition of CtBP with MTOB selectively inhibited growth of colonic CSCs and simultaneous depletion of CtBP2 levels along with its functional inhibition (with MTOB) resulted in greater inhibition of CSCs growth compared to either strategy alone.
Figure 3. MTOB selectively targets CSCs and inhibits their growth and self-renewal. (A) Effect of MTOB on growth in spheroid (1° spheroid formation) and monolayer (MTT assay, values derived as OD @ 490 nM wavelength) conditions at 120 h suggesting selectivity toward inhibiting growth in CSC enriched spheroid condition by MTOB. (B) Robust effect of 1 mM MTOB treatment on inhibition of 1° spheroid formation was observed in multiple colon cancer cells and across varied genetic backgrounds. (C) Inhibitory effect of MTOB (250 μM → 1 mM) on CSC self-renewal (2°/3° spheroids) in HCT-116 and HT-29 cells. (D and E) Attenuation of CSCs markers such as CD44, EPCAM (epithelial adhesion molecule) and CD133 at both mRNA (QPCR) (D) and protein (immunoblot) (E) levels following treatment with MTOB (1 mM) at 24 h in HCT-116 and HT-29 cells. GAPDH was used as housekeeping control. Data is represented as percent of vehicle-treated cells. Error bars represent ±1 SEM. *P<0 .05. Numbers under the blot represent relative densitometry values.
MTOB treatment selectively inhibits CSCs growth, in part, via induction of apoptosis
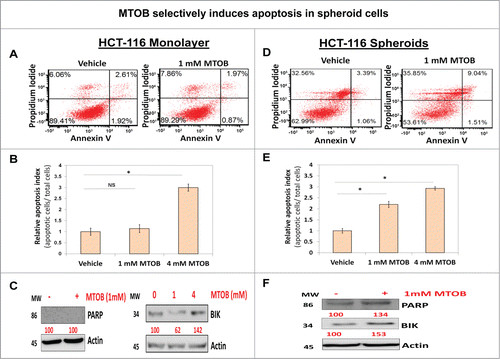
To understand the mechanism of selective CSC growth inhibition by MTOB, we examined apoptosis induction, an established effect of MTOB on colon cancer cells,Citation16 in spheroid and monolayer conditions. Treatment with 1 mM MTOB caused a robust 2–fold3- induction of apoptosis in HCT-116 spheroids () but not in monolayer cells () at this lower dose of MTOB. Similarly, levels of cleaved PARP were only elevated in spheroid cells treated with 1 mM MTOB (). However, consistent with our earlier observation,Citation16 a significantly higher dose of MTOB (4 mM) was able to induce apoptosis in both spheroids and monolayer HCT-116 cells to a similar extent (). Next, to determine a molecular mechanism associated with apoptosis induction, we examined expression of BIK, a BH3 domain proapoptotic protein which is known to be repressed by CtBP, following treatment with MTOB.Citation16 Consistent with above observations on apoptosis induction, treatment with low dose (1 mM) MTOB significantly increased BIK levels in HCT-116 spheroid cells (). In contrast, there was a modest inhibition in BIk expression in HCT-116 monolayer at low dose MTOB treatment, the reasons for which are not entirely clear but the findings are entirely consistent with lack of PARP cleavage and lack of apoptosis in monolayer cells at this dose (). However, there was an appropriate increase in BIK expression following treatment with higher dose (4 mM) MTOB () confirming our previous findings.Citation16 Hence, MTOB-induced selective CSC growth inhibition is mediated in part via selective induction of apoptosis.
Figure 4. MTOB selectively induces apoptosis in spheroid cells (A and D). Flow cytometric analyses of Annexin V expression demonstrating strong induction of apoptosis in HCT116 spheroids (D) but not in monolayer cells (A) at 24 h following treatment with a lower dose MTOB (1 mM). Additionally, (B and E) confirm that using a different method for quantitation of apoptosis, acridine orange/ ethidium bromide staining, in HCT-116 cells shows selective induction of apoptosis in cells grown as spheroids (E) but not monolayer (B) following low dose MTOB (1 mM). However, at significantly higher concentration of MTOB (4 mM), the selectivity fades away (B and E). Moreover, (C and F) show upregulation of cleaved PARP and pro-apoptotic molecule BIK following low dose MTOB treatment (1 mM) in spheroid but not monolayer cells. However, BIK induction was observed in monolayer cells at higher MTOB (4 mM) dose (C). Both monolayer and spheroids were treated for 24 h prior to preparing cell lysate. Actin was used as a loading control. Numbers under the blot represent relative densitometry values. Data is represented as percent of vehicle-treated cells. Error bars represent ±1 SEM, n = 3. *P < 0 .01.
MTOB mediated CSC growth inhibition is mediated via its effect on CtBP and not through the ornithine decarboxylase (ODC) pathway
Besides serving as a substrate for the CtBP dehydrogenase, MTOB is also an inhibitor of ODC, which links the methionine biosynthesis and polyamine pathways. In monolayer conditions, ODC inhibition was thought not to be responsible for MTOB's cytotoxicity as supplementation with polyamines did not reverse MTOB induced cell death.Citation29 However, the existence of an alternative mechanism of action in CSCs has not been studied thus far. Supplementation with 2 different polyamines, spermine and spermidine, was unable to rescue MTOB-mediated inhibition of CSC growth in HCT-116 cells (), suggesting that MTOB mediates its selective effect on CSCs through a target other than ODC.
Figure 5. MTOB induced CSC growth inhibition is mediated via its effect on CtBP and not through ornithine decarboxylase (ODC) pathway. (A) No significant effect of supplementation with ODC pathway metabolites (spermine and sperimidine, [0.5 mM]) on 1° spheroid formation following MTOB (1 mM) treatment in HCT-116 cells. (B) CtBP2 overexpression reverses the effect of MTOB (1 mM) on 1°, 2°, and 3° spheroid formation in HCT-116 cells suggesting its ability to partly rescue MTOB mediated effect on CSCs. Data is represented as percent of mock-transfected cells treated with vehicle. Moreover, (C) shows similar partial reversal by CtBP2 overexpression on MTOB mediated decrease in levels of LGR5 and c-MYC (CSC markers) as well as increase BIK (CtBP2 target) expression (C). Numbers under the blot represent relative densitometry values. Error bars represent ±1 SEM. *P < 0.01
![Figure 5. MTOB induced CSC growth inhibition is mediated via its effect on CtBP and not through ornithine decarboxylase (ODC) pathway. (A) No significant effect of supplementation with ODC pathway metabolites (spermine and sperimidine, [0.5 mM]) on 1° spheroid formation following MTOB (1 mM) treatment in HCT-116 cells. (B) CtBP2 overexpression reverses the effect of MTOB (1 mM) on 1°, 2°, and 3° spheroid formation in HCT-116 cells suggesting its ability to partly rescue MTOB mediated effect on CSCs. Data is represented as percent of mock-transfected cells treated with vehicle. Moreover, (C) shows similar partial reversal by CtBP2 overexpression on MTOB mediated decrease in levels of LGR5 and c-MYC (CSC markers) as well as increase BIK (CtBP2 target) expression (C). Numbers under the blot represent relative densitometry values. Error bars represent ±1 SEM. *P < 0.01](/cms/asset/8a9b442e-63b2-4073-a9f1-9b56b8e7b721/kccy_a_958407_f0005_c.gif)
To further substantiate the notion that MTOB mediates its selective inhibition of CSCs through inhibition of CtBP activity, we transfected HCT-116 cells with CtBP2 expression vector or a mock control and examined the effect of MTOB treatment on CSCs growth and self-renewal. CtBP2 overexpression significantly reversed the MTOB-mediated inhibition of 1°/2°/3° spheroid formation in mock transfected control (). The degree of rescue of CSC growth by CtBP overexpression is entirely consistent with a modest increase in the levels of CtBP2 following transfection with CtBP2 vector compared to mock controls (). More importantly, CtBP2 overexpression partly reverted inhibition of LGR5 and c-MYC induced by MTOB treatment in mock transfected controls (). On the other hand, MTOB failed to induce Bik levels in CtBP2 overexpressed cells, while it did so in mock transfected controls (). Overall, MTOB inhibits CSCs growth and self-renewal through inhibition of CtBP activity and not through ODC inhibition.
MTOB inhibits TCF-4/LEF signaling to attenuate CSCs growth and self-renewal
We have previously demonstrated the importance of β-catenin signaling in regulation of colonic CSC growth.Citation20 To further study the regulation of β-catenin signaling in CSCs, we generated colon cancer cells stably expressing TCF/LEF reporter plasmids in which the firefly luciferase gene is under the control of TCF/LEF responsive elements (Fig. S5A). Two genetically distinct colon cancer cells [HCT-116 (p53 wt, KRAS mut) and HT-29 (p53 mut, KRAS wt)] demonstrated increased TCF/LEF reporter activity in spheroids compared to monolayer cells (Fig. S5A), suggesting a basal increase in TCF/LEF transcriptional activity in CSCs. Moreover, we found that mRNA as well as protein levels of select direct targets of β-catenin-TCF/LEF signaling that are of relevance to CSC growth and self-renewal such as c-MYC and LGR5 were significantly upregulated in spheroids compared to monolayer control (Fig. S5B).
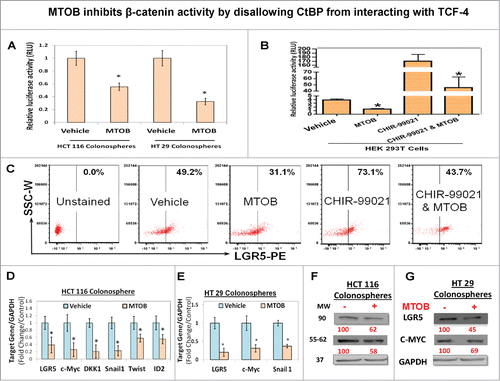
Since CtBP activity is significantly upregulated in CSCs, we hypothesized that the effect of CtBP on CSCs is mediated through regulation of TCF/LEF signaling. In fact, MTOB treatment significantly inhibited TCF/LEF reporter activity in both HCT-116 and HT-29 spheroids (). In order to determine the effect of MTOB on TCF/LEF transcriptional activity following induction of canonical β-catenin signaling, we examined TCF/LEF luciferase reporter in HEK-293 cells following treatment with or without CHIR-99021, a GSK3β inhibitor (that would be expected to induce b-catenin activity by inhibiting its phosphorylation and degradation). MTOB treatment modestly inhibited basal TCF/LEF activity. Furthermore, as expected, CHIR-99021 significantly induced TCF/LEF transcriptional activity which was robustly inhibited by pretreatment with MTOB (). In addition, FACS analyses revealed a 36% decrease in the levels of LGR5, a TCF-4 target gene and a CSC marker,Citation30 in unstimulated HT-29 spheroids following MTOB treatment. Moreover, while GSK3β inhibitor increased LGR5 expression by 44% above baseline in vehicle treated control, MTOB treatment resulted in an equally significant 40% inhibition in LGR5 expression (). In addition, MTOB treatment resulted in reduction of mRNA and proteins levels of several TCF/LEF target genes including LGR5, c-MYC ()Citation30,31 as well as ID2, SNAIL, TWIST, and DKK1Citation32–35 in HCT-116 spheroids (). Moreover, MTOB treatment resulted in reduction of LGR5 and c-MYC mRNA and protein level in HT-29 spheroids (). Overall, these results suggest that MTOB, an inhibitor of CtBP activity, potently inhibits basal as well as induced TCF/LEF signaling.
Figure 6. MTOB inhibits β-catenin activity by disallowing CtBP from interacting with TCF-4. (A) Significant inhibition of de novo TCF/LEF reporter activity following treatment with MTOB (1 mM) in HCT116 and HT 29 colonosphere, stably transduced with TCF/LEF luciferase reporter construct. Additionally, (B) shows significant inhibition of TCF/LEF reporter activity in basal condition as well as following activation of canonical wnt signaling by inhibition of GSK3β (CHIR-99021, 100 nM) in HEK 293T cells transient transfected with TCF/LEF luciferase reporter construct. (C) Flow cytometric analyses (FACS) of LGR5, a TCF-4 target gene and a CSC marker, expression at 24 h following MTOB (1 mM) treatment in HT 29 colonosphere in both unstimulated state as well as following CHIR-99021 pretreatment (12 h). (C). (D and E) show mRNA expression (QPCR) and (F)and G) show protein levels (immunoblot) of select TCF-4 target genes in HCT-116 and HT-29 colonospheres. GAPDH was used as housekeeping control for both QPCR and immunoblot analyses. Numbers under the blot represent relative densitometry values. Error bars represent ±1 SEM. *P < 0.05
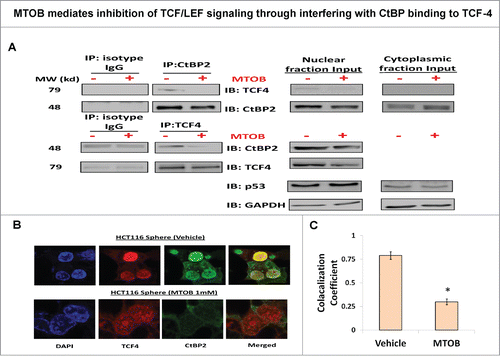
MTOB mediates inhibition of TCF/LEF signaling through interfering with CtBP binding to TCF-4
Studies in lower vertebrates suggest that CtBP might interact with components of the β-catenin transcriptional machinery.Citation19 Moreover, TCF4 was found to colocalize with CtBPCitation36 but direct interaction between CtBP, especially CtBP2, and TCF4 as well as the role of MTOB in this interaction remains to be elucidated. To determine if CtBP2 directly binds to TCF4 to regulate its transcription, we carried out reciprocal co-immunoprecipitation with anti-CtBP2 and anti-TCF4 antibodies in the nuclear extract of HCT-116 spheroid cells. A fraction of nuclear CtBP2 was found to bind to TCF4 and vice versa (). More importantly, MTOB treatment resulted in a robust abrogation of such interaction (). Additionally, we performed immunofluorescence microscopy to examine colocalization of CtBP2 and TCF4. We observed that spheroids show strong TCF4 expression in the nucleus (). On the other hand, while CtBP2 is present in the cytoplasm as well as nucleus, it was predominantly localized in the nucleus in the HCT-116 spheroids, and MTOB treatment significantly inhibited nuclear presence of CtBP2 (). In addition, TCF4 and CtBP2 colocalized in the nucleus and MTOB treatment resulted in a robust inhibition of this interaction (). In summary, the above results suggest that CtBP, a transcriptional co-regulator, binds to TCF4 and thus might directly regulate its transcriptional activity. Overall, we report a novel role of CtBP2 in promotion of CSC growth and self-renewal through direct regulation of TCF/LEF transcription which can be inhibited by MTOB, a small molecular inhibitor of CtBP function, resulting in selective targeting of CSCs.
Figure 7. MTOB mediates inhibition of TCF/LEF signaling through interfering with CtBP binding to TCF-4. (A) Physical interaction between TCF4 and CtBP2 proteins using reciprocal co-immunoprecipitation with anti-CtBP2 and anti-TCF4 antibodies in nuclear extract derived from spheroids, which is readily abrogated by MTOB treatment. p53 and GAPDH served as loading controls for nuclear and cytoplasmic fractions respectively. (B) Immunofluorescence confocal (IFC) microscopy analyses of spheroids cells incubated with fluorophore conjugated secondary antibodies against anti-CtBP2 (FITC-green) and anti-TCF4 (PE-red) showing nuclear colocalization between the 2 molcules which is decreased upon MTOB (1 mM) treatment. (C) Quantitative analysis of IFC microscopy images demonstrates decreased colocalization in MTOB treated samples. All the experiments were carried out in HCT-116 cells. Error bars represent ±1 SEM. *P < 0.05
Discussion
Primary and acquired resistance to cytotoxic therapies plays a major role in cancer recurrence, which arises from the survival of a small population of tumor cells with the ability to self-renew and reconstitute the entire tumor.Citation1,2,4,6 Growing evidence indicates that these so-called cancer stem-like cells (CSCs) should be the focus of anti-cancer drug discovery effects to cure the disease.Citation1,4 Yet, efforts to develop CSC-selective agents have achieved limited success, partly due to a dearth of druggable targets.Citation37 The only success reported so far relies on systemic or random chemical biology approaches without a defined target.Citation37-41 Our data clearly suggest that CtBP can be one such target that offers up a possibility of developing selective anti-CSC agents. Moreover, given CtBP's role in other aggressive cancer phenotypes including epithelial-mesenchymal transition, these drugs might have broader anti-cancer effects.Citation15,42
The confidence in the selective targeting of CSCs by inhibition of CtBP lies in consistent and complementary observations at both cellular and molecular levels. Additionally, both genetic and pharmacological modulations result in a similar phenotype, supporting the notion that the effect of MTOB is likely mediated through inhibition of CtBP activity. The latter contention is further reinforced by a series of observations in which MTOB mediated inhibition of the CSC phenotype was partially reversed by overexpression of CtBP2, but not by addition of polyamines, suggesting that inhibition of CtBP, but not ornithine decarboxylase, another target of MTOB, is important for its anti-CSC effects.
CtBP has a diverse role in epigenetic modification in response to changes in cellular energetics.Citation12-14,17,42 In earlier investigations, its role in transcriptional repression was highlighted.Citation17 However, accumulating evidence suggest that its role in transcriptional activation is just as important in mediating its oncogenic activities.Citation15,18 Specifically, our group demonstrated that CtBP mediated activation of TIAM-1 plays a critical role in activating the migration of colon cancer cells.Citation18 Our current findings support its role in activation of TCF-4 signaling. Earlier reports (including Valenta et al.Citation43) demonstrated physical interaction between TCF-4 and CtBP1 which resulted in repression of TCF target gene expression.Citation43,44 However, these experiments were carried out with supraphysiological levels of CtBP1 in non-transformed cells. A subsequent study clearly demonstrated that the outcome of interaction between CtBP and TCF family members is gene and context dependent.Citation19 Our data is in line with the latter contention and is not contradictory to the earlier report by Valenta et al. as several differences exist between studies. We have studied the role of CtBP2 mediated TCF-4 transcription in the context of physiological levels of CTBPs using MTOB as a pharmacologic inhibitor in the neoplastic cells; all of which can account for differences between ours and Valenta et al. studies. Nonetheless, our observation that CtBP2 promotes activation of TCF-4 signaling in colon CSCs is supported not only by physical interaction between TCF-4 and CtBP2, but also by robust and consistent inhibition of basal and activated TCF-4 luciferase reporter activity and expression of its target genes by MTOB. Even though the exact mechanism of CtBP mediated activation of TCF-4 signaling remains to be explored in colon cancer cells, it was suggested that CtBP might compete with Groucho, an inhibitor of TCF-4 signaling, in drosophila cells.Citation19 This will be an important focus of future investigations into the role of CtBP in regulating TCF-4 signaling in epithelial CSCs, particularly colorectal CSCs. In fact, targeting Wnt/β-catenin/TCF-4 signaling pathway remains one of the most attractive targets for development of anti-CSC therapies,Citation45 as it is significantly upregulated in many epithelial CSCs including colorectal CSCs.Citation20,45 Current tactics to inhibit Wnt signaling includes a multipronged approach with agents being developed to target ligand-receptor interaction, β-catenin stability as well as β-catenin/TCF-4 transactivation machinery.Citation45 As most colorectal cancers have constitutive activation of β-catenin signaling downstream of APC complex,Citation46 disruption of β-catenin/TCF-4 mediated transactivation of target gene remains the most viable option to inhibit CSCs. Our data provide proof-of-principle evidence that small molecular inhibition of TCF-4 mediated gene transactivation by targeting CtBP dehydrogenase domain is a promising therapeutic approach for selectively targeting colorectal CSCs.
This is the first comprehensive investigation of CtBP's role in the CSC phenotype. In breast cancer MCF-7 and MDA-MB-231 cells, MTOB treatment caused inhibition of CSC markers expression, similar to our observation in colorectal cancer cells.Citation15 However, the detailed effect on CSC phenotype was not examined in that work.Citation15 Another report suggested that inhibition, and not upregulation, of CtBP by tumor-infiltrating myeloid-derived suppressor cells promoted the CSCs phenotype.Citation47 This is in contrast to the observations in colon and breast cancer cells here and as reported.Citation15 These differences could be explained by context-dependent modulation of gene transcription by CtBP. For example, in ovarian cancer cells there was promotion of core CSC self-renewal factors such as NANOG, whereas we observed a significant reduction in NANOG (Fig. S6) as well as c-MYC, also a core transcription factor and a downstream target of TCF-4 signaling.Citation48 As TCF-4 signaling play a critical role in both colon and breast CSCs, it is likely that inhibition of this pathway by MTOB could lead to inhibition of CSCs in cells which exhibit activated TCF-4 signaling.Citation5
Overall, we have established a novel role of CtBP in promoting colorectal CSCs growth through activation of TCF-4 signaling, a key oncogenic pathway in colorectal and other epithelial malignancies. Based on our previous finding of suitability of CtBP for pharmacological inhibition, our data should bolster efforts to develop selective anti-CSC agents to target TCF-4 signaling. This would be a paradigm-shifting approach in the treatment of colorectal cancer, in particular, and for TCF-4 driven epithelial malignancies in general.
Materials and Methods
Cell culture and transfection
HCT-116, HT-29, WiDR, LS174T, Colo-320 human colon cancer cells were obtained from ATCC. These cells were maintained in a tissue cultured treated plates as monolayer in DMEM / F-12 supplemented with 10% fetal bovine serum (FBS) and 1% streptomycin/ penicillin. The cells were passaged using trypsin -EDTA before they reached 70% confluence. All reagents were obtained from Gibco (Cat#: 11320–033, 10438–026, 15240–0062, and 25300–054). Mammalian expression plasmids (4 μg/well) were transfected using lipofectamine® in a 6-well plate. Lipofectamine (4 μl/per well) was incubated with 24 μl of OptiMEM media for 10 minutes at room temperature (RT). Subsequently, 3 μl of either control (scramble) siRNA or CtBP2 specific siRNA, and 97 μl of OptiMEM media was added to the lipofectamine reaction mix and incubated for 20 minutes. Reaction mix was then added drop wise to 2 ml of the appropriate medium in each of the wells to be transfected, while gently swirling the media in the wells.
Cell proliferation assay
MTT cell proliferation assay was performed as per manufacturer protocol (Sigma # M 2128). Briefly, 2500 cells were plated in 96-well tissue culture treated plate. After overnight incubation at 37ºC, Vehicle (control) or MTOB was added at the desired concentration. Following 60 h of incubation, 10 μl of 5 mg/ml MTT solution (Sigma) made in PBS buffer was added to each well. After 3 h of incubation, crystal was dissolved after adding 150 micro liters of 4 mM HCl in isopropanol solution. The plates were read at 590 nm using a microplate reader and inhibition was calculated as compared to percent control.
Colonosphere formation
Primary spheroid culture was performed after plating 100 cells in non-treated 96 well in stem cell media (SCM)- DMEM/F12 (Gibco # 11320–023, Gibco # 15240–0062), supplemented with 1× B27 supplement (Gibco # 17504–044), 20 ng/ml Epidermal growth factors (Sigma # E9644), and 10 ng/ml fibroblast growth factor (Sigma # 354060). as described previously (DOI: 10.1021/cb500402f). After 4 hours of incubation, vehicle (control) or MTOB were added at the desired concentrations. On day 5, numbers of spheres ranging from 50–150 Âμm in diameter were counted using a phase contrast microscope. For self-renewal experiments, primary sphere were trypsinized and dissociated with mechanical disruption and then re-plated at 100 cells per well in SCM media in a low adhesion plate without any further treatment. For CtBP si-RNA and CtBP overexpression vector or appropriate control vector transfected monolayer cells were plated in SCM approximately 72 hours after the transfection and MTOB or vehicle treated were added as above.
Western blotting and immunoprecipitation analyses
Western blot analysis was performed as described previously (DOI: 10.1021/cb500402f). Briefly, colonosphere grown in stem cell media were treated with MTOB for 24 h and cell lysate was prepared in lysis buffer (20 mM Na3PO4, 100 mM NaCl, 2 mM EDTA, 1% Nonidet P-40, 2.5 mM Na3VO4) supplemented with a protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail 2 and 3 (Sigma). For Immunoprecipitation analysis, 300 μg nuclear fraction using NE-PER® nuclear and cytoplasmic Extraction Reagents kit, Fisher Scientific), diluted in the lysis buffer, were pre-cleared with protein G agarose resin (GenScript) for 60 minute at 4°C. The lysate was incubated overnight with either 3 μg anti- mouse CtBP2, TCF-4 antibody (BD transduction laboratories) or mouse IgG antibody. Immunocomplexes were recovered by incubation with Protein G-agarose beads for 2 h at 4°C with constant rotation. Beads were washed 3 times with lysis buffer and boiled with 2 X SDS buffer and pelleted by centrifugation. Equal amount of protein was separated by SDS-PAGE and was transferred to PVDF membranes (Bio-Rad). Blocking was performed with 5% low fat milk powder for 1 h followed by overnight incubation with primary antibody. Protein expression of anti-CD44 (Cell signaling #3570S), and anti-Epcam (Cell signaling #2929S); anti-CXCR4 (Abcam#Ab2074),CtBP2 (BD Biosciences) and TCF4 (Cell signaling); following incubation with appropriate secondary antibodies, protein bands were visualized using the ECL detection system and imaged with LAS-3000 Imaging system (FUJIFILM). Densitometry was analyzed by AIDA image analyzer software (Raytest) and the results were calculated as relative intensity compared to control. All experiments were performed at least 3 times.
Real-time polymerase chain reaction (QPCR) analysis
Total RNA was isolated using the mirVana™ miRNA Isolation Kit (Life technologies). 1 μg total RNA was reverse transcribed using First-Strand cDNA synthesis Kit using hexamer reverse primer (Affymetrix). Real time QPCR was performed using RT2 SYBR® Green qPCR Mastermix (Qiagen) in a 7500 fast real time machine (Applied Biosystem). Relative expressions of mRNA were calculated using ΔΔCT methods using GAPDH as a loading control. List of primers and their sequence has been described in Table S1.
FACS analysis
Human colon cancer HCT-116 cells, grown in spheroid or adherent condition and treated with vehicle or MTOB for 24 hours and single cells were re-suspended at 1 × 106 cells/ml in PBS. Cells were incubated with conjugated antibody for 30 minutes at 40°C and washed with PBS buffer prior to analysis. Following antibody and dilution were used: CD133/1 (AC133)-APC (1:33 dilution) (Miltenyi Biotec), CXCR4-PE conjugated clone 2B11 (Dilution 1:50) (ebioscience), LGR5-PE (Dilution 1:50) (Origene), Cell sorting was performed using FACSAria™ II High-Speed Cell Sorter (BD Biosciences) and data were analyzed using FCS express 4 flow research edition (De-Novo Software).
Dual Luciferase Assay
Cignal Lenti TRE reporter lentivirus particles, which express a luciferase gene driven by multiple TCF/LEF (AGATCAAAGGGGGTA) repeats and Cignal Lenti Renilla control lentivirus particles, were purchased from SA Biosciences. HCT116 and HT29 cell were transduced with TCF/LEF promoter along with Renilla luciferase (selected with 2 μg puromycin) as described previously.Citation49 Stable pooled clones over expressing reporter construct were grown as a spheroid for 3 d and then treated with 1 mM MTOB for 24 hours. For transient transduction experiment, HEK293T cells were infected with 1:500 diluted TCF/LEF and Renilla lentivirus (diluted in 8 μg/ml polybrene containing media) for 16 h and media was replaced. After 48 h of transduction, Cells were pre-treated with CHIR99021 (GSK inhibitor) followed by MTOB treatment. Luciferase activity was measured on Titertek Berthold luminometer by using the Dual Luciferase reporter assay (Promega, Madison, WI) according to the manufacturer's protocol.
Immunofluorescence staining and confocal microscopy
HCT116 colonosphere, grown in L-ornithine coated glass coverslip, were treated with 1 mM MTOB for 24 h. Cells were fixed with 4% paraformaldehyde for 15 minutes at room temperature and permeabilized for 5 min in 0.5% Triton X-100. Colonosphere were then blocked in PBS containing 1% BSA for one hour and incubated with primary antibodies for 3 hours. Colonosphere were then washed, incubated for 60 min with Alexa Fluor conjugate secondary antibodies, rinsed with blocking buffer and mounted on slides with DAPI containing Prolong antifade Reagent (Invitrogen). Fluorescently-labeled colonosphere were examined using a Zeiss LSM700 laser scanning confocal microscope (Carl-Zeiss Inc.). The Alexa Fluor 488 and Alexa Flour 555 signals were imaged sequentially in frame-interlaced mode to eliminate cross talk between channels. The images were processed and colocalization coefficient was calculated using Zen 2011 software.
Statistical analysis
All data are expressed as means ± SEM unless otherwise indicated. The results were analyzed using the unpaired, 2-tailed Student's t-test. P < 0.05 was designated as the level of significance unless specified otherwise.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
JP and SB performed CSC cultures and mechanistic studies, analyzed the results and assisted in writing the manuscript; IL performed mechanistic studies; NP performed mechanistic studies and analyzed the results; SRG analyzed the results and wrote the manuscript; and BBP directed the project, analyzed the results and wrote the manuscript.
958407_Supplementary_Materials.zip
Download Zip (896.7 KB)Funding
This work was supported by a VA Merit award to BBP and Research Scholar Grant from the American Cancer Society to SRG. The use of VCU microscopy core laboratory was supported, in part, by the NIH-NINDS center core grant 5P30NS047463. The use of Flow Cytometry Core laboratory was supported, in part, by funding from the NIH-NCI Cancer Center Support Grant (P30 CA016059).
References
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006; 66:9339-44; PMID:16990346; http://dx.doi.org/10.1158/0008-5472.CAN-06-3126
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414:105-11; PMID:11689955; http://dx.doi.org/10.1038/35102167
- Edwards MS, Chadda SD, Zhao, Z, Barber BL, Sykes DP. A systematic review of treatment guidelines for metastatic colorectal cancer. Colorectal Dis 2012; 14:e31-47; PMID:21848897; http://dx.doi.org/10.1111/j.1463-1318.2011.02765.x
- Gangemi R Paleari L, Orengo AM, Cesario A, Chessa L, Ferrini S, Russo P. Cancer stem cells: a new paradigm for understanding tumor growth and progression and drug resistance. Curr Med Chem 2009; 16:1688-703; PMID:19442140; http://dx.doi.org/10.2174/092986709788186147
- Korkaya H, Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. BioDrugsy 2007; 21:299-310; PMID:17896836; http://dx.doi.org/10.2165/00063030-200721050-00002
- Yi SY, Hao YB, Nan KJ, Fan TL. Cancer stem cells niche: a target for novel cancer therapeutics. Cancer Treat Rev 2013; 39:290-6; PMID:23219150; http://dx.doi.org/10.1016/j.ctrv.2012.10.004
- Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science 2013; 339:1567-70; PMID:23539597; http://dx.doi.org/10.1126/science.1230184
- Khan S, O'Brien PJ. Modulating hypoxia-induced hepatocyte injury by affecting intracellular redox state. Biochim Biophys Acta 1995; 1269:153-61; PMID:7488648
- Salnikov AV, Liu L, Platen M, Gladkich J, Salnikova O, Ryschich E, Mattern J, Moldenhauer G, Werner J, Schemmer P, et al. Hypoxia induces EMT in low and highly aggressive pancreatic tumor cells but only cells with cancer stem cell characteristics acquire pronounced migratory potential. PloS One 2012; 7, e46391; PMID:23050024; http://dx.doi.org/10.1371/journal.pone.0046391
- Yeung TM, Gandhi SC, Bodmer WF. Hypoxia and lineage specification of cell line-derived colorectal cancer stem cells. Proc Natl Acad Sci U S A 2011; 108:4382-87; PMID:21368208; http://dx.doi.org/10.1073/pnas.1014519107
- Diehn, M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009; 458:780-3; PMID:19194462; http://dx.doi.org/10.1038/nature07733
- Zhang, Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science 2002; 295:1895-7; PMID:11847309; http://dx.doi.org/10.1126/science.1069300
- Deng, Y, Liu J, Han G, Lu SL, Wang SY, Malkoski S, Tan AC, Deng C, Wang XJ, Zhang Q. Redox-dependent Brca1 transcriptional regulation by an NADH-sensor CtBP1. Oncogene 2010; 29:6603-8; PMID:20818429; http://dx.doi.org/10.1038/onc.2010.406
- Byun JS, Gardner K. C-Terminal Binding Protein: A Molecular Link between Metabolic Imbalance and Epigenetic Regulation in Breast Cancer. Int J Cell Biol 2013; 2013:647975; PMID:23762064; http://dx.doi.org/10.1155/2013/647975
- Di LJ, Byun JS, Wong MM, Wakano C, Taylor T, Bilke S, Baek S, Hunter K, Yang H, Lee M, et al. Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat Commun 2013; 4:1449; PMID:23385593; http://dx.doi.org/10.1038/ncomms2438
- Straza MW, Paliwal S, Kovi RC, Rajeshkumar B, Trenh P, Parker D, Whalen GF, Lyle S, Schiffer CA, Grossman SR. Therapeutic targeting of C-terminal binding protein in human cancer. Cell Cycle 2010; 9:3740-50; PMID:20930544; http://dx.doi.org/10.4161/cc.9.18.12936
- Sewalt RG, Gunster MJ, van der Vlag, J, Satijn DP, Otte AP. C-Terminal binding protein is a transcriptional repressor that interacts with a specific class of vertebrate Polycomb proteins. Mol Cell Biol 1999; 19:777-87; PMID:9858600
- Paliwal, S, Ho, N, Parker D, Grossman SR. CtBP2 Promotes Human Cancer Cell Migration by Transcriptional Activation of Tiam1. Genes Cancer 2012; 3:481-90; PMID:23264848; http://dx.doi.org/10.1177/1947601912463695
- Fang, M, Li J, Blauwkamp T, Bhambhani C, Campbell N, Cadigan KM. C-terminal-binding protein directly activates and represses Wnt transcriptional targets in Drosophila. EMBO J 2006; 25:2735-45; PMID:16710294; http://dx.doi.org/10.1038/sj.emboj.7601153
- Kanwa SS, Yu, Y, Nautiyal, J, Patel BB, Majumdar AP. The Wnt/beta-catenin pathway regulates growth and maintenance of colonospheres. Mol Cancer 2010; 9:212; PMID:20691072; http://dx.doi.org/10.1186/1476-4598-9-212
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005; 434:843-50; PMID:15829953; http://dx.doi.org/10.1038/nature03319
- Allen JE, El-Deiry WS. Calcein-effluxing human colon cancer cells are enriched for self-renewal capacity and depend on beta-catenin. Oncotarget 2013; 4:184-91; PMID:23468473
- Zhao LZ, Chinnadurai G. Incapacitating CtBP to kill cancer. Cell Cycle 2010; 9:3645-6; PMID:20930508; http://dx.doi.org/10.4161/cc.9.18.13221
- Chinnadurai G. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol Cell 2002; 9:213-24; PMID:11864595; http://dx.doi.org/10.1016/S1097-2765(02)00443-4
- Dontu, G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev 2003; 17:1253-70; PMID:12756227; http://dx.doi.org/10.1101/gad.1061803
- Balasubramanian, P, Zhao LJ, Chinnadurai G. Nicotinamide adenine dinucleotide stimulates oligomerization, interaction with adenovirus E1A and an intrinsic dehydrogenase activity of CtBP. FEBS Lett 2003; 537:157-60; PMID:12606049; http://dx.doi.org/10.1016/S0014-5793(03)00119-4
- Vaiopoulos AG, Kostakis ID, Koutsilieris M, Papavassiliou AG. Colorectal cancer stem cells. Stem Cells 2012; 30:363-71; PMID:22232074; http://dx.doi.org/10.1002/stem.1031
- Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318:1108-13; PMID:17932254; http://dx.doi.org/10.1126/science.1145720
- Tang, B, Kadariya, Y, Murphy ME, Kruger WD. The methionine salvage pathway compound 4-methylthio-2-oxobutanate causes apoptosis independent of down-regulation of ornithine decarboxylase. Biochem Pharmacol 2006; 72:806-15; PMID:16870157; http://dx.doi.org/10.1016/j.bcp.2006.06.018
- Barker, N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007; 449:1003-7; PMID:17934449; http://dx.doi.org/10.1038/nature06196
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science 1998; 281:1509-12; PMID:9727977; http://dx.doi.org/10.1126/science.281.5382.1509
- Howe LR, Watanabe, O, Leonard J, Brown AM. Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res 2003; 63:1906-13; PMID:12702582
- ten Berge, D, Koole W, Fuerer C, Fish M, Eroglu E, Nusse R. Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell 2008; 3:508-18; PMID:18983966; http://dx.doi.org/10.1016/j.stem.2008.09.013
- Rockman SP, Currie SA, Ciavarella M, Vincan E, Dow C, Thomas RJ, Phillips WA. Id2 is a target of the beta-catenin/T cell factor pathway in colon carcinoma. J Biol Chem 2001; 276:45113-9; PMID:11572874; http://dx.doi.org/10.1074/jbc.M107742200
- Gujral TS, MacBeath G. A system-wide investigation of the dynamics of Wnt signaling reveals novel phases of transcriptional regulation. PloS One 2010; 5, e10024; PMID:20383323; http://dx.doi.org/10.1371/journal.pone.0010024
- Cuilliere-Dartigues, P, El-Bchiri J, Krimi A, Buhard O, Fontanges P, Fléjou JF, Hamelin R, Duval A. TCF-4 isoforms absent in TCF-4 mutated MSI-H colorectal cancer cells colocalize with nuclear CtBP and repress TCF-4-mediated transcription. Oncogene 2006; 25:4441-8; PMID:16547505; http://dx.doi.org/10.1038/sj.onc.1209471
- Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009; 138:645-59; PMID:19682730; http://dx.doi.org/10.1016/j.cell.2009.06.034
- Mohammed, A, Janakiram NB, Brewer M, Ritchie RL, Marya A, Lightfoot S, Steele VE, Rao CV. Antidiabetic Drug Metformin Prevents Progression of Pancreatic Cancer by Targeting in Part Cancer Stem Cells and mTOR Signaling. Transl Oncol 2013; 6:649-59; PMID:24466367; http://dx.doi.org/10.1593/tlo.13556
- Hirsch HA, Iliopoulos, D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res 2009; 69:7507-11; PMID:19752085; http://dx.doi.org/10.1158/0008-5472.CAN-09-2994
- Kanwar SS, Yu Y, Nautiyal J, Patel BB, Padhye S, Sarkar FH, Majumdar AP. Difluorinated-curcumin (CDF): a novel curcumin analog is a potent inhibitor of colon cancer stem-like cells. Pharm Res 2011; 28:827-38; PMID:21161336; http://dx.doi.org/10.1007/s11095-010-0336-y
- Carmody, LA, Germain, B, Morgan, L, VerPlank, C, Fernandez, E, Forbeck, A, Ting, Y, Feng, J, Perez, S, Dandapani, B et al. Identification of a selective small-molecule inhibitor of breast cancer stem cells - Probe 1. Probe Reports from the NIH Molecular Libraries Program 2010.
- Grooteclaes, M, Deveraux Q, Hildebrand J, Zhang Q, Goodman RH, Frisch SM. C-terminal-binding protein corepresses epithelial and proapoptotic gene expression programs. Proc Natl Acad Sci U S A 2003; 100:4568-73; PMID:12676992; http://dx.doi.org/10.1073/pnas.0830998100
- Valenta, T, Lukas J, Korinek V. HMG box transcription factor TCF-4's interaction with CtBP1 controls the expression of the Wnt target Axin2/Conductin in human embryonic kidney cells. Nucleic Acids Res 2003; 31:2369-80; PMID:12711682; http://dx.doi.org/10.1093/nar/gkg346
- Brannon, M, Brown JD, Bates, R, Kimelman D, Moon RT. XCtBP is a XTcf-3 co-repressor with roles throughout Xenopus development. Development 1999; 126, 3159-70; PMID:10375506
- Curtin JC, Lorenzi MV. Drug discovery approaches to target Wnt signaling in cancer stem cells. Oncotarget 2010; 1:552-66; PMID:21317452
- Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487:330-7; PMID:22810696; http://dx.doi.org/10.1038/nature11252
- Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, Vatan L, Szeliga W, Mao Y, Thomas DG, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013; 39:611-21; PMID:24012420; http://dx.doi.org/10.1016/j.immuni.2013.08.025
- van de Wetering, M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002; 111:241-50; PMID:12408868
- Kutner RH, Zhang XY, Reiser J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 2009; 4:495-505; PMID:19300443; http://dx.doi.org/10.1038/nprot.2009.22