
Abstract
DNA damage response (DDR) to double strand breaks is coordinated by 3 phosphatidylinositol 3-kinase-related kinase (PIKK) family members: the ataxia-telangiectasia mutated kinase (ATM), the ATM and Rad3-related (ATR) kinase and the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs). ATM and ATR are central players in activating cell cycle checkpoints and function as an active barrier against genome instability and tumorigenesis in replicating cells. Loss of ATM function is frequently reported in various types of tumors, thus placing more reliance on ATR for checkpoint arrest and cell survival following DNA damage. To investigate the role of ATR in the G2/M checkpoint regulation in response to ionizing radiation (IR), particularly when ATM is deficient, cell lines deficient of ATM, ATR, or both were generated using a doxycycline-inducible lentiviral system. Our data suggests that while depletion of ATR or ATM alone in wild-type human mammary epithelial cell cultures (HME-CCs) has little effect on radiosensitivity or IR-induced G2/M checkpoint arrest, depletion of ATR in ATM-deficient cells causes synthetic lethality following IR, which correlates with severe G2/M checkpoint attenuation. ATR depletion also inhibits IR-induced autophagy, regardless of the ATM status, and enhances IR-induced apoptosis particularly when ATM is deficient. Collectively, our results clearly demonstrate that ATR function is required for the IR-induced G2/M checkpoint activation and subsequent survival of cells with ATM deficiency. The synthetic lethal interaction between ATM and ATR in response to IR supports ATR as a therapeutic target for improved anti-cancer regimens, especially in tumors with a dysfunctional ATM pathway.
Abbreviations
| DDR | = | DNA damage response |
| ATM | = | the ataxia-telangiectasia mutated kinase |
| ATR | = | the ATM and Rad3-related |
| DNA-PKcs | = | the catalytic subunit of the DNA-dependent protein kinase |
| HME-CCs | = | human mammary epithelial cell cultures |
| IR | = | ionizing radiation |
| DSBs | = | double strand breaks |
| CHK2 | = | the checkpoint kinase 2 |
| SSBs | = | single strand breaks |
| CHK1 | = | the checkpoint kinase 1 |
| WT | = | Wild-type |
| RMI | = | relative mitotic index |
| ATP | = | adenosine triphosphate |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
Introduction
DNA damage, which can be caused by both environmental exposures and endogenous sources, is a constant challenge to genome stability and cell survival. To protect from genotoxic assaults and preserve genomic integrity, replicating cells activate cell cycle checkpoints to allow DNA damage to be repaired before resumption of their normal cell cycle, or, if the damage is too severe, cells enter into apoptosis. Central to the checkpoint machinery are 2 key phosphoinositide 3-kinase-related kinases (PIKKs), the ataxia telangiectasia mutated (ATM) and ATM and Rad 3-related (ATR) kinase. It is generally accepted that ATM detects double strand breaks (DSBs) and initiates checkpoint signaling by phosphorylating downstream targets such as the checkpoint kinase 2 (CHK2), whereas ATR is activated mainly by single strand breaks (SSBs) and triggers checkpoint signaling predominately by phosphorylating the checkpoint kinase 1 (CHK1). However, in recent years there is increasing evidence suggesting complex relationships between the ATM-CHK2 and the ATR-CHK1 pathways. It is now known that both ATM and ATR are activated in response to DSBs, with ATM activation triggering subsequent ATR activation by promoting single strand DNA formation at damage sites.Citation1,2 In addition, a large-scale analysis on the consensus sites of proteins phosphorylated following DNA damage suggests that ATM and ATR share a large number of protein substrates that are implicated in the DNA damage response (DDR).Citation3 Nevertheless, the relationship between the ATM-CHK2 and the ATR-CHK1 pathways goes beyond simple redundancy. Coordinated activation of ATM and ATR is not only required for proper activation of checkpoint arrest but also important for modulating other biological outcomes such as DNA repair and cell survival.Citation5 Therefore, targeting components in checkpoint pathways such as ATM, ATR, CHK1 and CHK2 has become a valid approach for developing small molecular inhibitors in anti-neoplastic therapies.Citation6-9 In recent years, it has become increasingly evident that targeting the DDR network for triggering synthetic lethality of tumor cells while sparing normal tissues is a promising strategy for improved anti-cancer treatments.Citation10-13
In our previous studies utilizing ATM-deficient human mammary epithelial cell cultures (HME-CCs), we have documented a compensatory relationship between the ATM-CHK2 and the ATR-CHK1 axes in response to ionizing radiation (IR)-induced DSBs that results in hyperactivation of one pathway when the other is disrupted.Citation14,15 In both studies, a slightly attenuated checkpoint arrest was observed with moderately increased radiosensitivity when one of the checkpoint pathways was disrupted, suggesting potential for a synthetically lethal interaction. In addition, enhanced CHK1 phosphorylation correlated with a well-preserved IR-induced G2/M checkpoint in ATM-deficient HME-CCs, supporting an interpretation that the ATR-CHK1 signaling is compensating for the loss of ATM and contributing to the enforcement of the G2/M checkpoint. Therefore we asked whether ATR is required in this compensatory response, and whether disruption of ATR in human mammary epithelial cell cultures (HME-CCs) could further sensitize ATM-deficient cells to DSBs generated by γ-radiation. Using doxycycline-inducible, lentiviral transduced ATR depletion HME-CCs we show that disruption of ATR function in ATM-deficient cells abrogates the IR-induced G2/M checkpoint and results in synthetic lethality following γ-radiation. Since ATM silencing, due to either ATM mutation or methylation in the promoter region, is frequently observed in multiple tumor types, we propose that this interaction may be utilized for development of synthetically lethal therapeutic regimens in cancer treatment.Citation16-18
Results
Generation of doxycycline-inducible, lentiviral ATR knockdown human mammary epithelial cell lines
Due to its critical role in stabilizing the genome during DNA replication, ATR is an essential gene for cell survival and this fact complicates investigation of its function.Citation19-21 To study in detail the effects of combined suppression of ATM and ATR in a stable genetic system, we generated lentiviral inducible cell lines. Western blotting using whole cell extracts showed that a significant decrease in ATR protein levels occurred following 24 hr exposure to doxycycline in all cell lines expressing ATR shRNA compared to their non-silencing (NS) counterparts (Fig. S1). At 48 hr, ATR expression is barely detectable in both wild-type (WT) and ATM-deficient HME-CCs that express the shRNA construct targeting the exon 8 of the ATR gene (designated as LacZ-ATR and ATM1-ATR respectively). The cell lines carrying this shRNA were then chosen for all subsequent experiments in comparison to their non-silencing counterparts. Quantification of ATR protein indicates that 5% and 2% residual ATR protein were still present in LacZ-ATR and ATM1-ATR respectively, compared to the WT HME-CC (LacZ-NS) (). As expected, in cells that are deficient of ATM alone (ATM1-NS), the ATR protein level remained essentially unchanged.
Figure 1. Validation of ATR depletion in HME-CCs and characterization of cell proliferation. (A) Quantification of ATR expression in HME-CC cell lines with representative protein gel blots. Whole cell extracts were used and results are presented in mean Relative ATR Protein Levels ±SE (n = 3). *indicates significant difference compared to LacZ-NS (P < 0.05). (B) Cell proliferation and doubling times of wild-type HME-CC (LacZ-NS) and HME-CCs deficient of ATR (LacZ-ATR), ATM (ATM1-NS) or both (ATM1-ATR). The doubling time for each cell line is displayed as mean Doubling time±SE (n = 3). *compared to LacZ-NS (P < 0.05); **compared to ATM1-NS (P < 0.05).
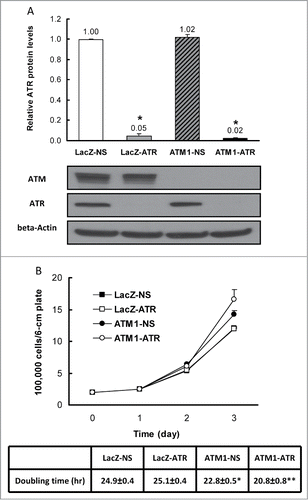
To examine the effects of ATR depletion on cell proliferation of HME-CCs, cell number increase was examined for 3 consecutive days after seeding. The average cell counts for each cell line at each time point were plotted in . It appears that ATM-deficient cells (both ATM1-NS and ATM1-ATR) replicate significantly faster than the wild-type cells. While ATR depletion alone does not affect cell proliferation significantly in the time frame examined, the absence of ATR in ATM-deficient cells further promotes cell proliferation. Calculation of doubling time confirms the above observation resulting in similar doubling times between LacZ-NS and LacZ-ATR, a shorter doubling time for ATM1-NS and a further reduced doubling time for ATM1-ATR ().
ATR depletion in ATM-deficient human mammary epithelial cells results in synthetic lethality following IR and abrogates the IR-induced G2/M checkpoint
To evaluate the effect of ATR depletion on radiosensitivity in the presence and absence of ATM, we first conducted a clonogenic assay following γ-radiation. However, LacZ-ATR and ATM1-ATR cells failed to form well-defined colonies regardless of IR treatment even though the culture media were not replenished with fresh doxycycline following IR treatment. Instead, CellTiter-Blue viability assay was performed to estimate cell viability following γ-radiation (cells were depleted of ATR for 48 hr prior to treatment). While deficiency in ATR or ATM alone caused little to moderate decrease in cell viability at 48 hr post IR, depletion of ATR in ATM-deficient cells resulted in significantly decreased cell survival compared to wild-type cells (). The decrease in the cell viability of ATM1-ATR following IR is greater than the sum of the individual decrease observed in the IR treated LacZ-ATR and ATM1-NS, suggesting a synthetic lethal effect between ATM and ATR depletion in response to γ-radiation. Statistical tests suggest this synthetic lethal interaction caused by ATR depletion in ATM-deficient HME-CCs is significant (P < 0.05).
Figure 2. Simultaneous depletion of ATR and ATM in HME-CC cells (ATM1-ATR) results in (A) Synthetic lethality to γ-radiation (Viability was evaluated using CellTiter-Blue viability assay kit at 48 hr post treatment) and (B) Abrogation of IR-induced G2/M checkpoint (2 hr post treatment). Cell viability was estimated by comparing the fluorescent signal in irradiated samples to that in the time-matched control of the same cell line. Relative mitotic index was calculated by dividing the percentage of mitotic cells in the treated sample by the percentage of mitotic cells in its respective untreated sample. The data for LacZ-NS following 3 Gy of IR treatment is not showing in the graph because the measurement is 0, which means complete G2/M checkpoint arrest. Results are presented in mean ± SE (n = 3). *means significantly different from the wild-type (LacZ-NS) (P < 0.05); **means change in cell viability due to ATM/ATR double depletion is significantly greater than the sum of that due to ATM or ATR depletion alone (P < 0.05).
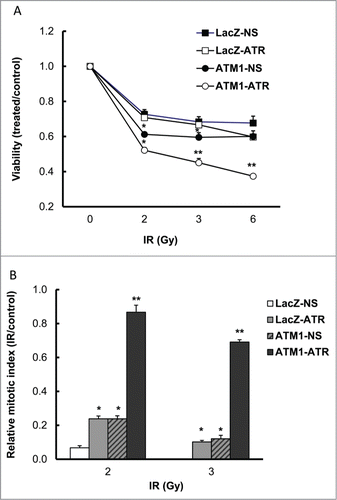
In spite of their sensitization to radiation, ATM-deficient HME-CC cells still displayed a fairly well-preserved G2/M checkpoint following IR.Citation15 To investigate the effects of ATR depletion in the IR-induced G2/M checkpoint regulation, all 4 cell lines were treated with γ-radiation and cells in mitosis were determined 2 hr post IR in both treated and untreated cells. The relative mitotic index (RMI) was then calculated for each cell line by dividing the percentage of mitotic cells in the treated sample by that in its untreated counterpart. Thus, a low RMI corresponds to a strong checkpoint, while a higher RMI indicates an attenuated checkpoint. Following 2 Gy treatment, the RMIs for LacZ-ATR and ATM1-NS were both 24%, as compared with a WT HME-CC RMI of 7%, suggesting mild checkpoint attenuation due to the individual deficiency in either ATR or ATM (). Interestingly, depletion of ATR in ATM-deficient cells led to a much significant G2/M checkpoint attenuation, resulting in a RMI of 87% in the ATM1-ATR double deficient HME-CCs. Similar results were also observed following treatment with 3 Gy. The amount of increase in RMI in ATM1-ATR cells following DNA damage is much greater than the cumulative amount of RMI increase in cells deficient of ATM or ATR alone, suggesting significant synergistic effects between ATM and ATR depletion in IR-induced G2/M checkpoint regulation. Taken together, these results clearly demonstrate that ATR function is critical for IR-induced G2/M DNA damage checkpoint activation and cell survival when ATM is depleted.
IR-induced checkpoint signaling in wild-type HME-CC, and HME-CCs deficient of ATR or ATM, and both
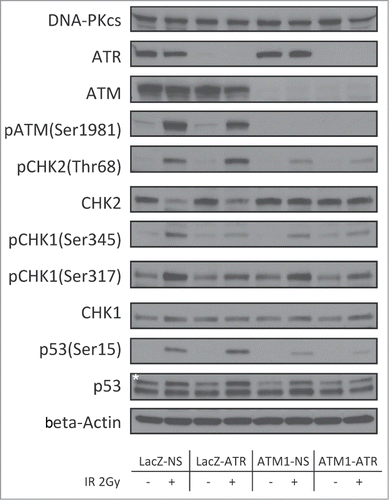
Deficiencies in the checkpoint signaling may render cells at increased risk of tumorigenesis by allowing unrepaired lesions in critical genomic regions to be carried over. To understand the differences in the checkpoint response in LacZ-NS, LacZ-ATR, ATM1-NS and ATM1-ATR, post-translational effects on DDR signaling molecules following γ-radiation at 2 Gy were examined. Depletion of ATM, ATR, or both did not affect the expression of other PIKKs, as shown in . At 2 hr post γ-radiation, LacZ-NS HME-CC displayed a typical DDR response as evidenced by IR-dependent phosphorylation of ATM (Ser1981), CHK2 (Thr68), CHK1 (Ser345 and Ser317) and p53 (Ser15) (). Depletion of ATR alone did not affect the IR-induced phosphorylation of ATM or CHK2. However, the IR-dependent CHK1 phosphorylation on Ser345 and Ser317 was diminished in LacZ-ATR cells, confirming the disruption of the ATR-CHK1 pathway. Similarly, in ATM1-NS cells, IR-dependent induction of CHK1 phosphorylation (both on Ser345 and Ser317) was largely unaffected but induction of CHK2 phosphorylation was significantly decreased compared to that in wild-type cells, indicating an impairment of the ATM-CHK2 pathway. In ATM1-ATR cells, although residual amounts of CHK1, CHK2 and p53 phosphorylation could be observed following treatment, the magnitude of these responses was the lowest among all 4 examined cell lines.
Figure 3. Regulation of checkpoint signaling at 2 hr following 2Gy of IR in wild-type HME-CCs and HME-CCs deficient in ATM, ATR or both. Whole cell extract was used for protein gel blot. "*" corresponds to the actual p53 band.
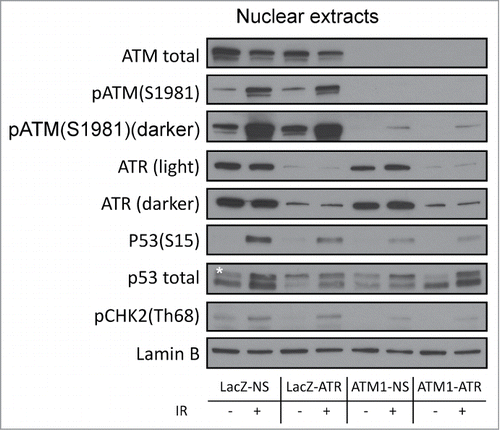
As mentioned above, the checkpoint responses observed in ATM1-ATR cells corresponded to a severe but incomplete attenuation of the G2/M checkpoint (RMI 87% following 2 Gy γ-radiation). This could potentially be due to residual ATM and ATR in the deficient cells. Western blotting analyses of nuclear extracts (see Fig. S2 for fractionation controls) clearly showed low but detectable phospho-ATM (Ser1981) and ATR as well as IR-induced phosphorylation of downstream targets in the nuclei of ATM1-ATR cells ().
Figure 4. Regulation of checkpoint signaling at 2 hr following 2 Gy of IR in wild-type HME-CCs and HME-CCs deficient in ATM, ATR or both. Nuclear extract was used for western blot. "*" corresponds to the actual p53 band.
Collectively, it appears that although a strong and intact G2/M checkpoint response to DSBs requires both ATM-CHK2 and ATR-CHK1 pathways, HME-CCs deficient of either ATR or ATM can rely on the other axis to mount a functional checkpoint response. A complete loss of checkpoint responses is possible when both the ATM-CHK2 and the ATR-CHK1 axes are simultaneously impaired.
ATR depletion inhibits DNA damage-induced autophagy and promotes apoptosis
Autophagy is a highly conserved cellular mechanism by which dysfunctional cellular components are degraded in autophagolysosomes in response to stress.Citation22 In nutrient-deprived cells, breakdown of damaged proteins or organelles can enhance cell survival by increasing adenosine triphosphate (ATP) production. In the DNA damage response, several studies have reported that autophagy can delay the onset of the DNA damage-induced apoptotic cell death and contribute to the resistance to cancer therapeutic drugs or IR treatment.Citation23-29 To investigate whether the synthetic lethal effect of ATM and ATR double deficiency in response to γ-radiation involves modulation of autophagy and apoptosis, IR-induced apoptotic and autophagic activity were measured in the wild-type HME-CC and HME-CCs deficient of ATM or ATR alone, as well as cells depleted of both kinases. At 24 hr following 2, 3 or 6 Gy of γ-radiation, LacZ-NS and ATM1-NS HME-CCs showed a dose-dependent induction in autophagic activity compared to their untreated counterparts (). Autophagy was also induced in LacZ-ATR and ATM1-ATR at 2 Gy, but to a lesser degree, and failed to increase further at higher doses. At 3 Gy, IR-induced autophagy in ATM1-ATR is significantly lower than that in LacZ-NS, or ATM1-NS cells. At 6 Gy, both ATR-deficient cell lines (LacZ-ATR and ATM1-ATR) displayed significantly lower autophagy than ATR-proficient cell lines LacZ-NS and ATM1-NS with the same treatment. In conclusion, depletion of ATR correlated with a reduced autophagic response to IR, indicating a positive role of ATR in IR-induced autophagy. Interestingly, despite the lower autophagic background when untreated, ATM1-NS HME-CC showed a very robust autophagic response following IR, similar in magnitude to the wild-type response, which could be blunted by ATR depletion. Because autophagy is considered a contributor to radioresistance following DNA damage, the lower autophagic activity secondary to the ATR depletion could be causative at least in part of the heightened radiosensitivity observed in ATM1-ATR cells.
Figure 5. IR-induced autophagy and apoptosis in HME-CCs. (A) IR-induced autophagy was inhibited in ATR-deficient HME-CCs 24 hr following IR treatment. (B) IR-induced apoptosis was enhanced in ATM and ATR double deficient cells (ATM1-ATR) 48 hr post treatment. Autophagy was evaluated at 24 hr post IR using Cyto-ID autophagy detection kit. Apoptosis Caspase3/7 activity was measured at 48 hr psot treatment using ApoTox-Glo Triplex Assay (Promega) and normalized to the number of viable cells. All of the presented values are relative to that of the untreated LacZ-NS and show in mean ± SE (n = 3). *means significantly different from wild-type (LacZ-NS) (P < 0.05); **means change in apoptosis due to ATM/ATR double depletion is significantly greater than the sum of that due to ATM or ATR depletion alone (P < 0.05).
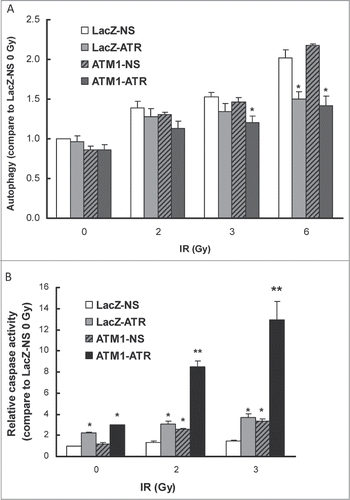
To examine the apoptotic response to IR in these cell lines, apoptosis in LacZ-NS, LacZ-ATR, ATM1-NS and ATM1-ATR cells was measured by using the ApoTox-Glo™ triplex assay 48 hr post IR. Baseline measurements indicated that ATR depletion results in significantly (P < 0.05) increased apoptotic activity in the untreated LacZ-ATR cells (2.2 fold) and ATM1-ATR (3.0 fold) compared to LacZ-NS, while depletion of ATM alone had no effect on baseline apoptosis (). Despite the difference in their basal apoptotic activity, LacZ-NS and LacZ-ATR both showed very mild induction following γ-radiation compared to their own untreated counterparts and ATM1-NS displayed a slightly higher IR-induced apoptosis (2.1 fold following 2 Gy and 2.7 fold following 3 Gy). However, ATM1-ATR HMECC showed the most robust induction in apoptosis in response to 2 Gy (2.9 fold) and 3 Gy (4.4 fold) γ-radiation. At 2 Gy, the IR-induced apoptosis in ATM1-ATR was 6.4, 2.8 and 3.3 times higher than that in LacZ-NS, LacZ-ATR and ATM1-NS, respectively. Taken together, these data suggests that ATR depletion inhibits the DNA damage-induced autophagy and promotes the IR-induced apoptosis in ATM-deficient HME-CCs, effects that could be responsible, at least partially, for the synthetic radiosensitivity conferred by concomitant depletion of ATM and ATR.
ATR depletion sensitizes HME-CCs to cancer therapeutic drugs
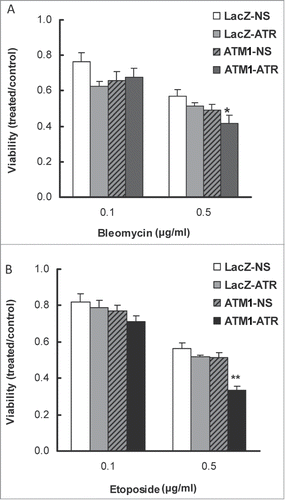
Targeting the major regulatory molecules of the DDR, such as ATM, ATR, CHK1 and CHK2 for improving the efficacy of radiation and genotoxic drugs has been widely explored in recent years.Citation30 To test whether ATR depletion can sensitize ATM-deficient HME-CCs to DNA damaging cancer therapeutic drugs, WT HME-CC (LacZ-NS) and HME-CCs deficient of ATM (ATM1-NS), ATR (LacZ-ATR), or both (ATM1-ATR) were treated with bleomycin or etoposide for 48 hr and cell viability was then evaluated using the CellTiter-Blue viability assay. As shown in , although no significant differences in cell viability were observed between the 3 deficient cell lines when treated with 0.1 μg/ml of bleomycin for 48 hr, following exposure to 0.5 μg/ml of bleomycin, the double-deficient cells (ATM1-ATR) displayed the lowest viability, suggesting that ATR depletion in ATM-deficient HME-CCs sensitizes cells to bleomycin treatment. Similarly, when cells were treated with 0.5 μg/ml of etoposide, a synthetic lethal interaction was observed between ATM and ATR depletion (). It is noteworthy that CellTiter-Blue measures cellular metabolic capacity as an indicator of cell viability and as such is not a particularly sensitive measurement of cell viability. Due to the incapability of ATR deficient cells to form colonies, we were not able to perform clonogenic assays. However we believe that the single deficient HME-CCs (LacZ-ATR, ATM1-NS) are likely more sensitive to bleomycin and etoposide than the parental cells even at the lower doses. Furthermore, depletion of ATR clearly sensitizes the ATM deficient cells to both drug treatments.
Figure 6. Depletion of ATR in ATM-deficient HME-CC cells sensitizes cells to DNA-damaging drugs (A) Bleomycin and (B) Etoposide. Cells were induced for ATR shRNA expression by doxycycline for 2 d prior to drug treatment and cell viability was evaluated by CellTiter-Blue assay at 48 h post drug treatment. All presented values were calculated by dividing the average fluorescent signal in treated cells with that in its own time-matched untreated counterpart. *means cell viability is significantly different from wild-type (LacZ-NS) (P < 0.05); **means change in cell viability due to ATM/ATR double depletion is significantly greater than the sum of that due to ATM or ATR depletion alone (P < 0.05).
Discussion
We generated lentivirally transduced cell lines that can be induced for ATR depletion in both ATM-proficient and deficient backgrounds in which more than 95% of ATR can be depleted following 48 hr exposure to 0.5 μg/ml doxycycline. While the ATR knockdown mouse is embryonically lethal, precluding many studies of ATR function, and ATR inhibitors generally lack specificity, these inducible knockdown cells allow us to study the function of 2 critical DDR proteins, ATM and ATR, in isogenic human mammary epithelial cell populations and thus investigate their functional redundancy in the DDR response. Utilizing these cell lines we showed that ATR is required for maintaining the IR-induced G2/M checkpoint in HME-CCs when ATM is deficient. While causing little change to the radiosensitivity of the wild-type cells, depletion of ATR in ATM-deficient cells results in synthetic lethal interaction in response to IR. We further showed that the synthetic lethal effect in ATM and ATR double deficient cells correlates with an imbalance between the IR-induced autophagy and apoptosis: the IR-induced autophagy is inhibited in ATM1-ATR while the IR-induced apoptosis is greatly enhanced, which leads to significant cell death.
In the current study, our data clearly shows a critical role of ATR in enforcing the IR-induced G2/M checkpoint when ATM is depleted. Indeed, depletion of either ATM or ATR alone displayed comparable small degrees of attenuation of the G2/M checkpoint, suggesting that the ATM-CHK2 pathway and ATR-CHK1 pathway are functionally redundant in this regard and both are able to independently maintain the IR-induced checkpoint arrest when the other is impaired. This is supported by evidence from protein gel blot analyses of 3 critical checkpoint signaling molecules, CHK2, CHK1, and p53, which are downstream of both ATM and ATR. When both ATM and ATR are depleted, the IR-induced phosphorylation of all 3 proteins were diminished, which correlates with a near abrogation of the G2/M checkpoint in ATM1-ATR HME-CC (RMI 87% following 2 Gy treatment). Previous work from our lab demonstrated a role of DNA-PKcs in the IR-induced G2/M checkpoint and the effect seemed to be mediated through CHK1 phosphorylation.Citation15 To test whether DNA-PKcs is contributing to the residual G2/M checkpoint in ATM1-ATR cells in addition to the incomplete shRNA-mediated depletion of ATR/ATM, DNA-PKcs inhibition in combination with ATR/ATM depletion was performed when cells were treated with 2 Gy γ-radiation. Unfortunately, the experimental variability among the biological triplicates in that experiment prevented us from drawing any firm conclusions concerning the significance of the effect of treatment with the DNA-PKcs inhibitor on the G2/M checkpoint in the cells depleted of both ATM and ATR. We speculate that the compensatory effect of DNA-PKcs in the IR-induced G2/M checkpoint may be mediated through ATR, ATM, or both, but further investigation is needed to dissect the underlying mechanism.
ATR regulates mitotic entry in a time-dependent manner. Upon double strand breaks, ATR activation is subsequent to ATM activation and is triggered by single strand DNA formation at damage sites, which is promoted by ATM activation.2 ATR contributes to the early delay of mitotic entry by phosphorylating CHK1 (Ser345) and p53 (Ser15). ATR also plays a critical role in the late delay of mitotic entry. This has been reported by Brown and Baltimore in mouse embryonic fibroblasts using a Cre/lox-conditional system.Citation5 Our data of G2/M checkpoint function in human mammary epithelial cell culture agrees well with the above report. At 6 hr post 2 Gy or 3 Gy IR, delay in mitotic entry was observed only in ATR-proficient cells (LacZ-NS and ATM1-NS) but not in cells depleted of ATR (LacZ-ATR and ATM1-ATR), suggesting ATR is regulating the late delay of mitotic entry following IR (Fig. S3).
Synthetic lethality arises from the combined effects of disruption of 2 or more genes or pathways regulating survival while change in either one will not cause cell death.Citation31,32 Activation of checkpoint arrest is critical for cell survival following DNA damage in proliferating cells.Citation9 Our results on radiosensitivity of HME-CCs correlated very well with the G2/M checkpoint regulation following γ-radiation. Depletion of ATM alone rendered HME-CC cells moderately more sensitive to irradiation compared to wild-type cells and depletion of ATR alone did not change the radiosensitivity of the cells. However, in ATM1-ATR HME-CC cells, while ATM and ATR depletion conferred a growth advantage, the double deficiency also caused a significant reduction in viability following IR that proved beyond the additive effect of ATM or ATR depletion alone. This clearly shows that simultaneous depletion of ATM and ATR abrogates the G2/M checkpoint response and leads to synthetic lethal effects to double strand breaks. We postulate that tumor cells deficient in ATM function have adapted through compensatory signaling through ATR and/or DNA-PKcs, and hence have become essentially addicted to this compensatory signaling for proliferation. Or the other way around, addiction to the ATR pathways leads to inactivation of the redundant ATM pathway and such cells become target of synthetic lethality.Citation31 Thus by targeting ATR in such ATM-deficient tumors, one could enhance therapeutic efficacy of killing cancer cells while sparing normal cells through a synthetic lethality. This is particularly important for the development of novel anti-cancer regimens, as a more efficacious tumor killing could be achieved at lower doses of chemotherapeutic drugs by targeting both arms of the DDR pathway.
Both ATM and ATR have been explored as therapeutic targets for development of small molecular inhibitors in anti-neoplastic therapies. It has been reported that ATR overexpression in cancer cells can induce prolonged G2/M arrest and contribute to resistance to ionizing radiation.Citation33 On the other hand, ATR mutation seems to be associated with a trend toward improved disease-free survival in colon cancers and ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy, which highlight the potential of using ATR inhibitors in anti-cancer regimens.Citation34,35 Loss of ATM function, either due to low levels of ATM expression or defects in upstream or downstream signaling, has been reported in many types of tumors.Citation16-18 This renders cells more dependent on ATR for checkpoint arrest and cell survival following DNA damage. Targeting ATR in these ATM-deficient tumors could be potentially beneficial for improved therapeutic responses to radiation and chemotherapy. The concept of synthetic lethality between the ATM and ATR pathways is also supported by a few studies from other researchers using cancer cell lines. Sangster-Guity and colleagues reported that genetic inhibition of ATR expression selectively enhanced cisplatin sensitivity in human colorectal cancer cells with inactivated p53, which is a major substrate of ATM.Citation12 Knock-in of functional p53 in ATR-deficient cells restored checkpoint function. In another report, ATR inhibition through VE-821, a selective small-molecule inhibitor, showed profound synthetic lethal interaction in cells deficient in ATM-p53 pathway when treated with DNA-damaging agents.Citation36 Therefore, deficiency in other components in the ATM-CHK2-p53 pathway may also sensitize cells to ATR disruption. In tumors harboring ATR mutations, ATM may be targeted to enhance the response to radiation of chemotherapy.
The functional relationship between autophagy and apoptosis in response to cellular stress is complex because depending on the circumstances autophagy can either suppress apoptosis to promote cell survival or lead to autophagic cell death.Citation37 The DNA damage-induced autophagy has been reported to contribute to the resistance of tumor cell to IR or drug treatment.Citation25,29,38 Autophagy is currently a therapeutic target for anti-cancer drug development as its inhibition can sensitize tumor cells to DNA damage.Citation24,27,39 In this paper, the IR-induced autophagy is clearly inhibited in ATM/ATR double deficient HME-CCs. The inhibition seems to be a consequence of the ATR depletion, as both LacZ-ATR and ATM1-ATR cells had reduced autophagic responses compared to LacZ-NS and ATM1-NS cell lines at 24 hr post IR treatments. In addition to autophagy inhibition, IR-induced apoptosis dramatically increased in ATM/ATR double deficient cells compared to cells deficient of ATM alone. This supports that in the absence of ATM cell survival relies heavily on ATR following IR and ATR suppression is an effective approach to sensitize tumor cells that are deficient in ATM while being less toxic to normal cells. Collectively, our data shows that depletion of ATR in ATM-deficient HME-CCs results in reduced autophagy and increased apoptotic response to IR. The imbalance between autophagy and apoptosis in ATM/ATR double deficient cells leads to dramatically increased cell death following IR.
In the last decade or so, inhibitors targeting the DNA damage response network have been of great interest in the development of cancer therapeutic drugs.Citation30,40,41 Many of these inhibitors target major signaling molecules of the G2/M checkpoint, such as ATM, ATR, CHK1 and CHK2.Citation6-8,Citation40,42,43 Some of these inhibitors are already in clinical trials and various therapeutic results have been reported. In addition, radiation therapy remains a valid strategy for cancer treatments as an inducer of double stand breaks. Understanding the basis of the cellular choices in response to double strand breaks is essential for designing personalized treatment regimens that would direct tumor cells into apoptosis while sparing the healthy tissues. Overall, this study provides additional evidence for developing personalized therapeutic strategies in anti-neoplastic treatments. The exploration of a potentially synthetically lethal interaction between the ATR and the ATM pathways provides an attractive opportunity to deliver anticancer drugs that increase the efficacy of established DNA-damaging agents.
Materials and Methods
Chemicals and reagents
Etoposide (Catalog #: E1383) was purchased from Sigma Aldrich and dissolved in DMSO at a concentration of 20 mg/ml. Bleomycin sulfate was from Santa Cruz (Catalog #: sc-200134) and dissolved in DMSO at a concentration of 10 mg/ml. Blasticidin S HCl (10 mg/ml) was obtained from Invitrogen (Cat#: A11139-03). All stock solutions were stored at −20°C. TRIPZ inducible lentiviral ATR shRNA (Catalog #: RHS4696) and TRIPZ inducible lentiviral non-silencing shRNA control (Catalog #: RHS4743) were purchased from Thermo Scientific.
Cell culture and generation of ATR knockdown cell lines
The wild-type and ATM-deficient (>95% depletion) human mammary epithelial cell lines, HME-CC-LacZ and HME-CC-ATM1, were described previously.Citation15 Cells were maintained in HuMEC ready medium (Invitrogen, 12752-010) with 4 μg/ml blasticidin at 37°C in a 5% CO2 incubator. To conditionally knock down ATR in HME-CC cell lines, HME-CC-LacZ and HME-CC-ATM1 cells were transduced with either TRIPZ inducible lentiviral non-silencing shRNA control or an individual TRIPZ inducible lentiviral ATR shRNA construct. Two ATR shRNA constructs were used to target different regions of the ATR gene, with one targeting the exon 8 (mature sense sequence CCGCTAATCTTCTAACATT), and the other targeting the exon 9/10 junction (mature sense sequence CCAAGATTCTTATAGATAA). Transduction was performed in multiple wells on a 6-well plate and successful transduction of the constructs was confirmed by visualization of TurboRFP reporter expression after adding doxycycline (0.5 μg/ml) to one of the transduced wells. Puromycin was then added to the rest of the transduced wells (without doxycycline) at a concentration of 0.5 μg/ml for further stable cell line selection. Cells were grown in puromycin for 9 d and propagated as needed before freezing down for long term storage. For later experiments the transduced cell lines containing TRIPZ inducible lentiviral shRNA constructs were first recovered from the frozen stock in HuMEC ready medium with 4 μg/ml blasticidin and 0.5 μg/ml puromycin (referred to as growth medium) for at least one passage. The subcultures were then induced for shRNA expression in growth medium for 2 d to suppress ATR expression before being used for treatment.
Proliferation assay
Cell proliferation was performed in 6-cm tissue culture plates. Exponentially growing HME-CC cells were seeded at a density of 2 × 105 per dish with doxycycline added to the growth medium to suppress ATR expression. Cells were collected by trypsinization 24 hr, 48 hr and 72 hr after plating and cell number was determined using a Cellometer Auto T4 cell counter (Nexcelom Bioscience). Results of the growth curve shown were from 3 independent experiments and reported as Mean ± SE (n = 3). Cell doubling time was calculated using an online calculator (http://www.doubling-time.com/compute.php) by inputting 4 data points (0, 24, 48 and 72 hr) from each experiment. The doubling time for each cell line was estimated by taking the average of 3 experiments and displayed as mean doubling time ± SE (n = 3).
Cell viability assay
Cell viability assays were performed in a 96-well plate format using the CellTiter-Blue viability assay kit following the manufacturer's instruction (Promega G8081). Briefly, exponentially growing cells were plated in growth medium at a density of 1.7 × 103 per well and doxycycline was added at a final concentration of 0.5 μg/ml to induce ATR shRNA expression. Cells were grown for 2 d to allow ATR suppression before being exposed to either γ-radiation using a 137Cs source or one of the cancer therapeutic drugs. Cell viability at indicated time points was then determined by adding 20 μl of CellTiter-Blue and further incubated for 2 hours. Fluorescent signal at 590 nm was recorded using a FLUOstar Omega plate reader with an excitation filter of 544 nm (BMG Labtech). For each treatment, 6–9 duplicate wells were plated in a single experiment and the cell viability value for each cell line at a given time point was calculated by dividing the average fluorescent signal in treated cells with that in its own time-matched untreated counterpart. Three independent experiments were performed and results were presented as mean ± SE (n = 3).
G2/M checkpoint and flow cytometry
G2/M checkpoint analysis following γ-radiation was performed as described previously.Citation14 Briefly, treated and control cells were harvested by trypsinization 2 hr after IR and fixed in 4% paraformaldehyde (BioLegend 420801). 4′,6-diamidino-2-phenylindole (DAPI) stain was used for determination of DNA content and labeling of phosphorylated histone H3 (Cell Signaling 9706) was used to evaluate the proportion of cells in mitosis. Readings were taken using an LSRII flow cytometer (Becton Dickinson) and data were analyzed using FACSDiva analysis software. A relative mitotic index (RMI) was calculated by comparing the percentage of mitotic cells in the treated sample to that of untreated sample of the same cell line and presented as a percentage of the untreated counterpart. While a small RMI close to 0% in treated samples indicates a fully functional G2/M checkpoint, a higher percentage RMI is interpreted as an impaired or attenuated G2/M checkpoint. A RMI close to 100% indicates abolishment of the G2/M checkpoint.
Autophagy following IR
Autophagy following IR was determined by flow cytometry using the Cyto-ID autophagy detection kit according to the manufacturer's instructions (Enzo Life Science, ENZ-51031-K200). Cells were plated on 10-cm plates in growth medium with 0.5 μg/ml doxycycline and allowed to grow for 2 d before treatment with γ-radiation. At the indicated time points post IR, cells were collected by trypsinization, resuspended in diluted Cyto-ID Green stain solution, and incubated for 30 minutes at 37°C. The fluorescent signal was recorded using an LSRII flow cytometer and data were analyzed in FlowJo. Three independent experiments were performed with the results presented as mean ± SE (n = 3).
Apoptosis following IR
Cellular apoptosis following IR was determined using the ApoTox-Glo™ triplex assay from Promega in a 96-well plate format following manufacturer's instructions (Catalog #: G6320). Briefly, cells were plated in growth medium at a density of 1.7 × 103 per well and allowed to grow for 2 d with 0.5 μg/ml doxycycline before being treated with γ-radiation. Apoptosis following IR at indicated time points was determined by the luminescence measurement of the caspase3/7 activity on the luminogenic substrate using a FLUOstar Omega plate reader. The fluorescence measurement of the live-cell protease activity in the same well was also recorded and used as a build-in control to normalize cellular apoptotic activity. For each treatment, 6–9 duplicate wells were plated in a single experiment and 3 independent experiments were performed. Results are presented as mean ± SE (n = 3).
Western blotting
Whole cell lysates were prepared using RIPA buffer (1X PBS, 1% Nonidet P-40, 0.5% Na Deoxycholate, 0.1% SDS and 10% Glycerol) supplemented with phosphatase and protease inhibitors from Pierce (Catalog #: 78442). Cells were washed twice with ice-cold PBS then scrapped off plates into ice-cold RIPA buffer. Cell lysates were then sonicated for 15 seconds. The insoluble portion of the lysate was removed by centrifugation for 15 minutes at 14,000 rpm and 4°C. Soluble protein in the supernatant was quantitated using the Pierce BCA protein assay kit (Catalog # 23225) to ensure equal amounts of protein were loaded for SDS-PAGE gel separation and protein gel blot analysis. beta-Actin was probed to confirm equal loading and satisfactory protein transfer of whole cell extracts.
Nuclear protein was prepared using the Pierce NE-PER nuclear and cytoplasmic extraction reagents following the manufacturer's instruction (Catalog # 78833). Equal numbers of cells from each sample were used for cell fractionation. The separation of nuclear and cytoplasmic protein was confirmed by protein gel blot analysis of known nuclear or cytoplasmic proteins (Fig. S2). Lamin B was used as a control for equal loading and protein transfer of nuclear extracts.
Antibodies to ATR, CHK1 and Lamin B were purchased from Santa Cruz Biotechnology (Catalog # sc-1887, sc-8408 and sc-6216). Antibodies to ATM, phospho-ATM (Ser1981), and p53 were obtained from Epitomics (Catalog # 1549-1, 2152-1, and 1026-1). Antibodies to phospho-CHK1 (Ser345, Ser317), phospho-CHK2 (Th68), and phospho-p53 (Ser15) were from Cell Signaling Technology (Catalog numbers 2348 S, 8191 S, 2661 S and 9284 S). Antibodies to CHK2 and DNA-PKcs were from Stressgen (Catalog # KAM-CC112 and KAP-P1001E). Antibody to beta-Actin was from Sigma (Catalog #: A5316). Peroxidase-conjugated secondary antibodies and enhanced chemiluminescence were used in detection of primary antibodies (Pierce 34080). ImageQuant TL v2005 software was used for protein gel blot quantitation (GE Healthcare).
Statistical analysis
One-way (or 2-way if there was a combination of cell lines and different doses of treatments) ANOVA was used for significance tests. Least significant difference procedure was used for multiple comparisons. The interaction term (ATM X ATR) in a 2-way ANOVA (ATM deficiency and ATR deficiency) was evaluated to test the hypothesis that ATM and ATR double depletion can generate synthetic lethal effects.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
960729_Supplementary_Materials.zip
Download Zip (1.6 MB)Acknowledgments
We would like to thank the Viral Core at NIEHS for their assistance in generating the lentiviral particles, the NIEHS Flow Cytometry Center for training and support, and Drs. Carl Anderson and Daniel Menendez for critical reading of the paper.
Funding
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01ES021157).
Supplemental Materials
Supplemental data for this article can be found on the publisher's website.
References
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GCM, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8:37-U13; PMID:16327781; http://dx.doi.org/10.1038/ncb1337
- Zou L, Shiotani B. Single-Stranded DNA Orchestrates an ATM-to-ATR Switch at DNA Breaks. Mol Cell 2009; 33:547-58; PMID:19285939; http://dx.doi.org/10.1016/j.molcel.2009.01.024
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao ZM, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160-6; PMID:17525332; http://dx.doi.org/10.1126/science.1140321
- Smith J, Tho LM, Xu NH, Gillespie DA. The ATM-Chk2 and ATR-Chk1 Pathways in DNA Damage Signaling and Cancer. Advances in Cancer Research, Vol 108. San Diego: Elsevier Academic Press Inc, 2010:73-112.
- Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 2003; 17:615-28; PMID:12629044; http://dx.doi.org/10.1101/gad.1067403
- Morgan MA, Parsels LA, Zhao LL, Parsels JD, Davis MA, Hassan MC, Arumugarajah S, Hylander-Gans L, Morosini D, Simeone DM, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G(2) checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res 2010; 70:4972-81; PMID:20501833; http://dx.doi.org/10.1158/0008-5472.CAN-09-3573
- Fokas E, Prevo R, Hammond EM, Brunner TB, McKenna WG, Muschel RJ. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat Rev 2013; Rev 2014; 40(1):109-17; PMID:23583268; http://dx.doi.org/10.1016/j.ctrv.2013.03.002
- Fokas E, Prevo R, Pollard JR, Reaper PM, Charlton PA, Cornelissen B, Vallis KA, Hammond EM, Olcina MM, McKenna WG, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis 2012; 3:e441; PMID:23222511; http://dx.doi.org/10.1038/cddis.2012.181
- Chen T, Stephens PA, Middleton FK, Curtin NJ. Targeting the S and G2 checkpoint to treat cancer. Drug Discov Today 2012; 17:194-202; PMID:22192883; http://dx.doi.org/10.1016/j.drudis.2011.12.009
- Sultana R, Abdel-Fatah T, Perry C, Moseley P, Albarakti N, Mohan V, Seedhouse C, Chan S, Madhusudan S. Ataxia telangiectasia mutated and rad3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. Plos One 2013; 8(2):e57098; PMID:23451157; 10.1371/journal.pone.0057098
- Schoppy DW, Brown EJ. Chk'ing p53-deficient breast cancers. J Clin Invest 2012; 122:1202-5; PMID:22446183; http://dx.doi.org/10.1172/JCI63205
- Sangster-Guity N, Conrad BH, Papadopoulos N, Bunz F. ATR mediates cisplatin resistance in a p53 genotype-specific manner. Oncogene 2011; 30:2526-33; PMID:21258400; http://dx.doi.org/10.1038/onc.2010.624
- Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB, Hemann MT. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev 2009; 23:1895-909; PMID:19608766; http://dx.doi.org/10.1101/gad.1815309
- Palii SS, Cui YX, Innes CL, Paules RS. Dissecting cellular responses to irradiation via targeted disruptions of the ATM-CHK1-PP2A circuit. Cell Cycle 2013; 12:1105-18; PMID:23462183; http://dx.doi.org/10.4161/cc.24127
- Arlander SJH, Greene BT, Innes CL, Paules RS. DNA protein kinase-dependent G(2) checkpoint revealed following knockdown of ataxia-telangiectasia mutated in human mammary epithelial cells. Cancer Res 2008; 68:89-97; PMID:18172300; http://dx.doi.org/10.1158/0008-5472.CAN-07-0675
- Bolt J, Vo QN, Kim WJ, McWhorter AJ, Thomson J, Hagensee ME, Friedlander P, Brown KD, Gilbert J. The ATM/p53 pathway is commonly targeted for inactivation in squamous cell carcinoma of the head and neck (SCCHN) by multiple molecular mechanisms. Oral Oncol 2005; 41:1013-20; PMID:16139561; http://dx.doi.org/10.1016/j.oraloncology.2005.06.003
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455:1069-75; PMID:18948947; http://dx.doi.org/10.1038/nature07423
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Edkins S, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007; 446:153-8; PMID:17344846; http://dx.doi.org/10.1038/nature05610
- Baltimore D, Brown EJ. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 2000; 14:397-402; PMID:10691732
- de Klein A, Muijtjens M, van OsR, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JHJ. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 2000; 10:479-82; PMID:10801416; http://dx.doi.org/10.1016/S0960-9822(00)00447-4
- Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem J 2011; 436:527-36; PMID:21615334; http://dx.doi.org/10.1042/BJ20102162
- Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8:528-39; PMID:21587219; http://dx.doi.org/10.1038/nrclinonc.2011.71
- Amaravadi RK, Yu DN, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326-36; PMID:17235397; http://dx.doi.org/10.1172/JCI28833
- Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res 2008; 68:1485-94; PMID:18316613; http://dx.doi.org/10.1158/0008-5472.CAN-07-0562
- Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol 2011; 99:287-92; PMID:21722986; http://dx.doi.org/10.1016/j.radonc.2011.06.002
- Chen LH, Loong CC, Su TL, Lee YJ, Chu PM, Tsai ML, Tsai PH, Tu PH, Chi CW, Lee HC, et al. Autophagy inhibition enhances apoptosis triggered by BO-1051, an N-mustard derivative, and involves the ATM signaling pathway. Biochem Pharmacol 2011; 81:594-605; PMID:21184746; http://dx.doi.org/10.1016/j.bcp.2010.12.011
- Chen SN, Rehman SK, Zhang W, Wen AD, Yao LB, Zhang JA. Autophagy is a therapeutic target in anticancer drug resistance. Biochim Biophys Acta-Rev Cancer 2010; 1806:220-9; http://dx.doi.org/10.1016/j.bbcan.2010.07.003
- Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J Cell Mol Med 2009; 13:3644-54; PMID:20196784; http://dx.doi.org/10.1111/j.1582-4934.2009.00663.x
- Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Diff 2007; 14:548-58; PMID:16946731; http://dx.doi.org/10.1038/sj.cdd.4402030
- Furgason JM, Bahassi E. Targeting DNA repair mechanisms in cancer. Pharmacol Ther 2013; 137:298-308; PMID:23107892; http://dx.doi.org/10.1016/j.pharmthera.2012.10.009
- Blagosklonny MV. NCI's provocative questions on cancer: some answers to ignite discussion. Oncotarget 2011; 2:1352-67; PMID:22267462
- Fang B. Development of synthetic lethality anticancer therapeutics. J Med Chem 2014; PMID:24893124; 10.1021/jm500415t
- Kim YM, Lee YM, Park SY, Pyo H. Ataxia telangiectasia and Rad3-Related overexpressing cancer cells induce prolonged G(2) arrest and develop resistance to ionizing radiation. DNA and Cell Biol 2011; 30:219-27; PMID:21294646; http://dx.doi.org/10.1089/dna.2010.1141
- Lewis KA, Bakkum-Gamez J, Loewen R, French AJ, Thibodeau SN, Cliby WA. Mutations in the ataxia telangiectasia and rad3-related-checkpoint kinase I DNA damage response axis in colon cancers. Genes Chromosomes Cancer 2007; 46:1061-8; PMID:17879369; http://dx.doi.org/10.1002/gcc.20486
- Huntoon CJ, Flatten KS, Hendrickson AEW, Huehls AM, Sutor SL, Kaufmann SH, Karnitz LM. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res 2013; 73:3683-91; PMID:23548269; http://dx.doi.org/10.1158/0008-5472.CAN-13-0110
- Reaper PM, Griffiths MR, Long JM, Charrier JD, MacCormick S, Charlton PA, Golec JMC, Pollard JR. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011; 7:428-30; PMID:21490603; http://dx.doi.org/10.1038/nchembio.573
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741-52; PMID:17717517; http://dx.doi.org/10.1038/nrm2239
- Lomonaco SL, Finniss S, Xiang CL, DeCarvalho A, Umansky F, Kalkanis SN, Mikkelsen T, Brodie C. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer 2009; 125:717-22; PMID:19431142; http://dx.doi.org/10.1002/ijc.24402
- Zois CE, Koukourakis MI. Radiation-induced autophagy in normal and cancer cells Towards novel cytoprotection and radio-sensitization policies? Autophagy 2009; 5:442-50; PMID:19164950; http://dx.doi.org/10.4161/auto.5.4.7667
- Toledo LI, Murga M, Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: A new model for new (and old) drugs. Molecular Oncology 2011; 5:368-73; PMID:21820372; http://dx.doi.org/10.1016/j.molonc.2011.07.002
- Finlay MRV, Griffin RJ. Modulation of DNA repair by pharmacological inhibitors of the PIKK protein kinase family. Bioorg Med Chem Lett 2012; 22:5352-9; PMID:22835870; http://dx.doi.org/10.1016/j.bmcl.2012.06.053
- Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, Muschel RJ, Brunner TB. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther 2012; 13:1072-81; PMID:22825331; http://dx.doi.org/10.4161/cbt.21093
- Montano R, Chung I, Garner KM, Parry D, Eastman A. Preclinical Development of the Novel Chk1 Inhibitor SCH900776 in Combination with DNA-Damaging Agents and Antimetabolites. Mol Cancer Ther 2012; 11:427-38; PMID:22203733; http://dx.doi.org/10.1158/1535-7163.MCT-11-0406