
Abstract
The low survival and differentiation rates of stem cells after either transplantation or neural injury have been a major concern of stem cell-based therapy. Thus, further understanding long-term survival and differentiation of stem cells may uncover new targets for discovery and development of novel therapeutic approaches. We have previously described the impact of mitochondrial apoptosis-related events in modulating neural stem cell (NSC) fate. In addition, the endogenous bile acid, tauroursodeoxycholic acid (TUDCA) was shown to be neuroprotective in several animal models of neurodegenerative disorders by acting as an anti-apoptotic and anti-oxidant molecule at the mitochondrial level. Here, we hypothesize that TUDCA might also play a role on NSC fate decision. We found that TUDCA prevents mitochondrial apoptotic events typical of early-stage mouse NSC differentiation, preserves mitochondrial integrity and function, while enhancing self-renewal potential and accelerating cell cycle exit of NSCs. Interestingly, TUDCA prevention of mitochondrial alterations interfered with NSC differentiation potential by favoring neuronal rather than astroglial conversion. Finally, inhibition of mitochondrial reactive oxygen species (mtROS) scavenger and adenosine triphosphate (ATP) synthase revealed that the effect of TUDCA is dependent on mtROS and ATP regulation levels. Collectively, these data underline the importance of mitochondrial stress control of NSC fate decision and support a new role for TUDCA in this process.
Abbreviations
| NSC | = | neural stem cells |
| TUDCA | = | tauroursodeoxycholic acid |
| mtROS | = | mitochondrial reactive oxygen species |
| ATP | = | adenosine triphosphate |
| ROS | = | reactive oxygen species |
| mtDNA | = | mitochondrial DNA |
| cdk | = | cyclin-dependent kinase |
| MnSOD | = | manganese superoxide dismutase |
| OGG1 | = | 8-oxoguanine DNA glycosylase |
| UDCA | = | ursodeoxycholic acid |
| CsA | = | cyclosporin A |
| OligA | = | oligomycin A |
| GAPDH | = | glyceraldehyde 3-phosphate dehydrogenase |
| VDAC | = | voltage-dependent anion channel |
| FACS | = | fluorescence-activated cell sorting analysis |
| BrdU | = | bromodeoxyuridine |
| Sox2 | = | sex determining region Y- box 2 |
| GFAP | = | glial fibrillary acidic protein |
| DiOC6(3) | = | 3, 3′-dihexyloxacarbocyanine iodide |
Introduction
Several efforts have been made to strategically manipulate neural stem cell (NSC) fate. Interestingly, recent evidence has suggested that mitochondria may influence stem cell fate by mechanisms defined as retrograde signaling.Citation1,2 Indeed, the integrity and functional stage of mitochondria are crucial for cell proliferation, differentiation, apoptosis and survival.Citation3-5 During neural differentiation, profound mitochondrial morphologic and metabolic alterations occur to assure successful differentiation.Citation6-9 The undifferentiated stage is usually associated with low mitochondrial oxygen consumption, needed to maintain the proliferative capacity,Citation10 while aerobic metabolism, in association with increased reactive oxygen species (ROS) production, is crucial for the differentiation process. Citation7,11These subtle alterations in the redox state of NSCs affect not only mitochondrial DNA (mtDNA) damage repair, but also mitochondrial maturation and function.Citation5,11 Interestingly, it has been demonstrated that mitochondria-generated ATP and high mitochondrial reactive metabolites of oxygen, such as ROS, may also regulate cell cycle progression.Citation12 In fact, cell differentiation usually implicates cell cycle exit and irreversible proliferative arrest. In contrast, cell proliferation and concomitant DNA replication require significant energetic supply, where a compromise between both cell cycle machinery and metabolism is a prerequisite for efficient cell division. Differentiation-induced cell cycle arrest occurs in G1 phase and is mediated by upregulation of cyclin-dependent kinase (cdk) inhibitory proteins, such as p21 and p27, and by activation of the retinoblastoma protein family.Citation13,14
Importantly, mitochondrial apoptosis-associated events were recently shown to be crucial during the early stages of NSC differentiation. We have previously demonstrated that, under neural differentiation conditions, p53 is actively translocated to mitochondria, and attenuates mitochondrial oxidative stress, mitochondrial loss, apoptosis-associated events, such as cytochrome c release and mitophagy. The protective effect of mitochondrial p53 appeared to be partially dependent on manganese superoxide dismutase (MnSOD).Citation15 Moreover, a tight control of mitochondrial damage during differentiation was also shown to be important for cell fate determination. Indeed, mtDNA damage might be the primary signal for NSC commitment to the astroglial lineage, and lack of neurogenesis seen during repair of neuronal injury.Citation11 In this respect, 8-oxoguanine DNA glycosylase (OGG1), an enzyme responsible for DNA repair, was shown to play a critical role in both mtDNA damage repair and NSC survival, as OGG1 expression is associated to higher neurogenic potential.Citation16
The endogenous bile acid ursodeoxycholic acid (UDCA), an FDA-approved molecule widely used in treatment of liver diseases, was shown to be a potent inhibitor of apoptosis by preventing mitochondria membrane instability, Bax-induced mitochondrial membrane perturbation and inhibition of cytochrome c release.Citation17–20 Notably, its taurine-conjugated form, TUDCA, has been shown to be an orally bioavailable and central nervous system penetrating agent.Citation21 TUDCA plays an important neuroprotective role in animal models of acute stroke and Huntington's disease, by regulating mitochondrial apoptosis and oxidative stress, while also improving survival and function of cell transplants in Parkinson disease.Citation21–25 UDCA has been used also in clinical trials for amyotrophic lateral sclerosis.Citation26 TUDCA properties in preserving mitochondrial function, together with the key role of mitochondria in regulating NSC fate, provide a new framework to further explore the use of this bile acid in the treatment of neurological disorders with neurogenesis deficit.
Here we sought to investigate the role of TUDCA in modulating NSC fate, and explore its potential mechanisms of action. Our results revealed that TUDCA prevents differentiation-induced mitochondrial alterations, including mitochondrial membrane depolarization, cytochrome c release, ROS production, and ATP depletion, thus increasing self-renewal and cell cycle progression of NSCs, as well as directing differentiating NSCs toward the neuronal lineage. Importantly, cyclosporin A (CsA) or oligomycin A (OligA), inhibitors of the mtROS scavenger enzyme MnSOD and ATP synthase, respectively, abrogated TUDCA effects indicating that NSC fate regulation by the bile acid is dependent on mtROS and ATP regulation.
Results
TUDCA prevents differentiation-induced mitochondrial stress
We have shown that mitochondrial apoptosis-associated events are typical of early mouse NSC differentiation.Citation15 In addition, we have previously demonstrated that TUDCA is a strong inhibitor of apoptosis by preventing mitochondrial apoptotic signaling, including mitochondrial membrane perturbations and cytochrome c release.Citation4,25 Therefore, we investigated the potential effect of TUDCA during differentiation-induced mitochondrial stress, as well as its efficacy in regulating NSC differentiation. For that, a NSC line was cultured and allowed to differentiate for 1 to 24 h, in an optimized neuronal differentiation-inducing medium, as previously described.Citation15 TUDCA prevented mitochondrial membrane depolarization occurring at early stages of neural differentiation, such as at 6 h, as assessed by a significant increase in DiOC6(3) mitochondrial incorporation (P < 0.01) (). Cells were also incubated with MitoSOXTM Red reagent, which exhibits red fluorescence when oxidized by superoxide, hence allowing the detection and quantification of mtROS. As expected, mtROS production increased at 1 h of neural differentiation (P < 0.01). However, in TUDCA-treated cells, mtROS levels decreased significantly, when compared with differentiated control cells (P < 0.01) (). We then evaluated the efficacy of TUDCA in modulating mitochondrial release of cytochrome c during NSC differentiation, and found a marked reduction of cytochrome c release at 6 h, when compared to control differentiating cells (at least P < 0.05) (). The relative purity of mitochondrial and cytosolic extracts was controlled using GAPDH and VDAC antibodies, respectively. Since mitochondrial translocation of p53 was shown to induce mitochondrial survival at early stages of NSC differentiation,Citation15 we also determined the effect of TUDCA treatment on p53 mitochondrial levels after 6 h of NSC differentiation induction. Curiously, TUDCA significantly decreased p53 translocation to the mitochondria, when compared to differentiating cells (P < 0.01) (). The relative purity of mitochondrial p53 fractionation was controlled using Lamin B1 antibody, which indicated the absence of nuclear contamination in mitochondrial extracts.
Figure 1. TUDCA modulation of NSC differentiation-induced mitochondrial alterations. Mouse NSCs were expanded, induced to differentiate in the presence or absence of TUDCA, and then collected for flow cytometry, immunoblotting and quantitative real-time PCR, as described in Materials and Methods. (A) Representative histogram (left) and quantification data (right) of DiOC6(3)-positive cells in self-renewal or at 6 h of differentiation evaluated by flow cytometry. (B) Representative histogram (left) and quantification data (right) of mtROS levels in self-renewal or at 1 h of differentiation, evaluated by FACS, using MitoSOXTM Red reagent. (C) Representative immunoblots of cytochrome c (top) and corresponding densitometry analysis (bottom) in both mitochondria and cytosolic extracts, during self-renewal or at 6 h of differentiation. The mitochondrial and cytosolic fractionation was monitored by the presence of VDAC and GAPDH endogenous protein levels. (D) Representative immunoblots of p53 in mitochondrial extracts (top) and respective quantification data (bottom), in self-renewal or at 6 h of differentiation. Results were normalized to endogenous VDAC protein levels, and nuclear contamination was assessed using Lamin B1 antibody. (E) Real-time PCR analysis of relative mtDNA copy number in self-renewal or at 24 h of differentiation. (F) Representative quantification data of ATP levels in self-renewal or at 24 h of differentiation. Results are expressed as mean ± SEM fold-change for at least 3 different experiments. *P < 0.01 and §P < 0.05 from undifferentiated cells; ‡P < 0.01 and †P < 0.05 from cells treated with TUDCA alone.
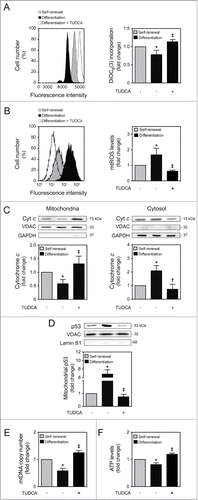
Finally, to explore differences in mitochondrial viability and function after TUDCA treatment, mtDNA content and ATP production were evaluated throughout NSC differentiation, in the presence or absence of TUDCA. The results obtained by real-time PCR experiments revealed that TUDCA reverted the decrease in mtDNA copy number observed at 24 h of NSC differentiation (P < 0.01) (). Notably, at this time of differentiation, our results also revealed a significant drop in ATP levels when compared to the undifferentiated cells (P < 0.01). Nevertheless, TUDCA restored ATP levels that were lost with differentiation-induced mitochondrial stress (P < 0.01) (). Given the well-established survival role of taurine in several biological processes including anti-oxidation, radioprotection, detoxification and proliferation,Citation27–29 we evaluated the role of taurine in modulating mtROS and ATP production levels. Taurine had no significant effect on both mtROS and ATP levels, indicating that TUDCA function is not dependent on its taurine-conjugated moiety (Fig. S1). These findings indicate that TUDCA prevents mitochondrial membrane-damaging and biogenesis alterations associated with early-stage mouse NSC differentiation.
TUDCA regulates cell cycle and proliferation of NSCs
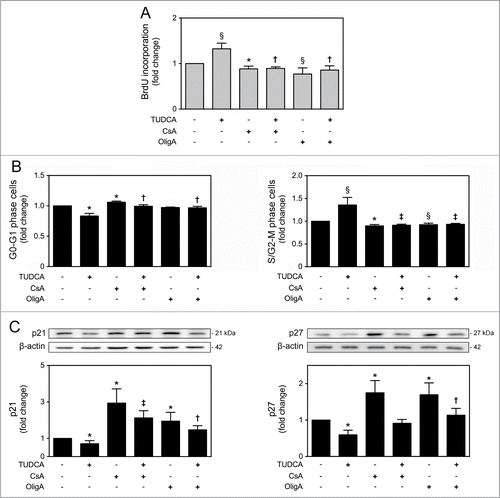
It has been recently recognized that mitochondria may also regulate cell cycle progression and cell proliferation, as a retrograde signal.Citation2,12 In fact, it appears that mitochondrial oxidative rate has to remain depressed for cell proliferation.Citation30 To further explore the impact of TUDCA on NSC fate, we next investigated the effect of this bile acid in regulating cell cycle progression and proliferation of NSCs. Cell proliferation was determined by fluorescence-activated cell sorting analysis (FACS) of bromodeoxyuridine (BrdU) incorporation in undifferentiated NSCs treated or untreated with TUDCA. Interestingly, our results showed a significant increase in BrdU incorporation after TUDCA incubation (P < 0.01) (). To clarify whether TUDCA enhances NSC proliferation by modulating cell cycle progression, we next investigated cell cycle phases throughout this process. As expected, 24 h after induction of NSC differentiation, a significant increase of cells in G0-G1 phase was accompanied by a decrease in S/G2-M phases (P < 0.01). In contrast, TUDCA treatment decreased cells in G0-G1 phase, and markedly increased S/G2-M phases (P < 0.01) ().
Figure 2. TUDCA modulation of cell cycle and proliferation of NSCs. NSCs were expanded, induced to differentiate in the presence or absence of TUDCA, and then collected for flow cytometry and immunoblotting, as described in Materials and Methods. (A) Representative histogram of BrdU incorporation by flow cytometry (left) and quantification data (right) in self-renewal conditions, 24 h after cell treatments. (B) Representative quantification data of G0-G1 and S/G2-M phase cells in self-renewal or at 24 h of differentiation. (C) Representative immunoblots of p21 (top left) and p27 (top right) in total extracts and respective quantification data (bottom right or left), in self-renewal or at 6 h of differentiation. Results were normalized to endogenous β-actin protein levels, and are expressed as mean ± SEM fold-change for at least 3 different experiments. *P < 0.01 from undifferentiated cells; ‡P < 0.01 from cells treated with TUDCA alone.
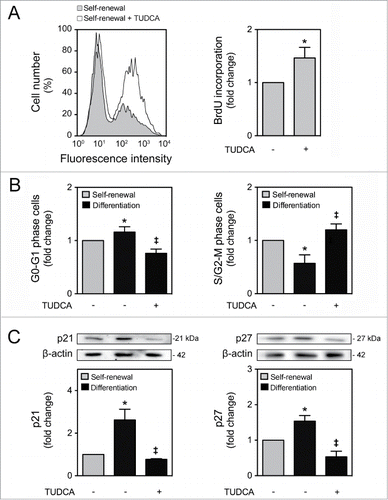
p21 and p27 are cdk inhibitors that play a major role in cell cycle control, promoting cell cycle arrest, not allowing the entrance into S phase and leading to cell cycle exit.Citation31,32 Since differentiation has been associated with a reduction in cdk-activity at G1, caused by an increase of its inhibitors, p21 and p27,Citation18 we next evaluated the expression of these regulatory proteins, during NSC differentiation, as well as in the presence of TUDCA. p21 and p27 expression levels significantly increased after 6 h of differentiation (P < 0.01). More importantly, when differentiating NSCs were treated with TUDCA, p21 and p27 expression dramatically decreased (P < 0.01) (). Our data suggest that TUDCA increases proliferation of NSCs by delaying cell cycle arrest, through inhibition of p21 and p27 levels.
We then addressed the impact of TUDCA in NSC ability to self-renew, by evaluating the potential regulation of Sox2 stemness marker expression. As expected, the endogenous expression of the neural progenitor Sox2 was significantly decreased in cells that start to differentiate (). On the other hand, TUDCA did not impact on Sox2 expression under propagation conditions. Indeed, it is expected that the percentage of growth factors in undifferentiation culture medium assures the right conditions for NSC self-renewal, leaving less room for the regulatory contribution of TUDCA. However, in differentiation conditions, the effect of TUDCA in delaying the decrease in Sox2 expression was clearly evident, when compared with the control, thus suggesting that this bile acid maintains the self-renewing potential of NSCs even during differentiation context (). In addition, symmetrical and asymmetrical divisions were analyzed through the cell pair assay by culturing NSCs in low percentage of growth factors, in the presence or absence of the bile acid, and evaluating Sox2 expression in mitotic cell pairs through immunocytochemistry. In accordance with increased BrdU incorporation, higher levels of symmetrical divisions were detected after 24 h of TUDCA incubation, when compared with control cells (P < 0.01) ( and D). Given the well-established anti-apoptotic role of TUDCA in several biological contexts, we also evaluated the effect this bile acid on NSC viability at 24 h of neural differentiation. Surprisingly, measurement of Annexin-V- and PI-negative cells by FACS analysis, revealed no significant alterations in cell death or cell viability during early stages of NSC differentiation after TUDCA treatment ().
Figure 3. TUDCA modulation of self-renewal and NSC pool. NSCs were expanded and induced to differentiate, in the presence or absence of TUDCA, and then collected for immunoblotting, immunocytochemistry counting and flow cytometry, as described in Materials and Methods. (A) Representative immunoblot of Sox2 in total extracts in self-renewal or at 6 h and 24 h of differentiation. Results were normalized to endogenous β-actin protein levels. (B) Representative histograms of Sox2-positive cells in self-renewal or at 6 and 24 h of differentiation, in the presence or absence of TUDCA, evaluated by flow cytometry. (C) Representative quantification data of paired cells Sox2-positive and -negative, in low EGF/bFGF containing medium, 24 h after TUDCA treatment. (D) Representative microscopy images of undifferentiated NSC pairs labeled with Hoechst 33258 and anti-Sox2 antibody, 24 h after TUDCA treatment. (E) Quantification of NSC viability, by measuring Annexin-V- and PI-negative cells using flow cytometry, in self-renewal or at 24 h of differentiation. (F) Representative bright-field images (left) and quantification of viable cells (right), after 24 h of differentiation. Scale bar, 5 μm and 10 μm (zoom). Results are expressed as mean ± SEM fold-change for at least 3 different experiments. *P < 0.01 from undifferentiated cells; ‡P < 0.01 from cells treated with TUDCA alone.
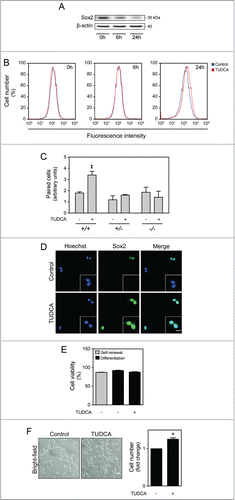
Finally, since TUDCA promotes self-renewal and cell cycle progression without involving cell death per se, the contribution of this bile acid to increase the NSC pool was investigated. For that, the number of cells was quantified in NSCs in the presence or absence of TUDCA, 24 h after induction of differentiation. Curiously, our results revealed a significant increase in cell number in TUDCA-treated NSCs when compared with untreated differentiating cells (P < 0.01) (). Therefore, TUDCA not only promotes self-renewal and cell cycle progression, but also contributes to an increase of the NSC pool.
TUDCA-regulated NSC mitochondrial alterations are dependent on mtROS and ATP regulation
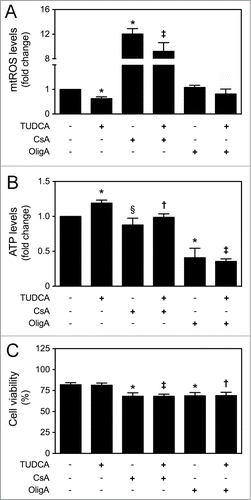
In the past few years, mitochondria have been ascribed as important regulators of cellular fate, not only by controlling energy generation, but also by modulating cell cycle for development and growth.Citation30 The mitochondrial regulation of proliferation and differentiation potential is dependent on a very precise control of mitochondrial metabolism, biogenesis and dynamics, with ROS and ATP acting as 2 major players.Citation33 To further explore whether TUDCA-regulated NSC fate was dependent on its mitochondrial effects, both mtROS and ATP levels were modulated in the presence of TUDCA. For that, CsA, inhibitor of mtROS scavenger MnSOD, and OligA, inhibitor of F1F0-ATP synthase complex, were used. As expected, a marked increase in mtROS levels was observed in the presence of CsA alone, when compared to control (P < 0.01) (Fig. S1). On the other hand, ATP levels significantly decreased in the presence of OligA, when compared to control (P < 0.01) (Fig. S1). More importantly, CsA and OligA treatments rescued TUDCA-induced decrease in mtROS production (P < 0.01) and increase in ATP levels throughout differentiation (P < 0.01), respectively (). Finally, determination of Annexin-V- and PI-negative cells by FACS analysis, revealed a significant decrease in the number of viable cells incubated with CsA or OligA for 24 h (P < 0.01). In fact, increased mtROS and decreased ATP levels, through incubation with CsA or OligA, respectively, reduced cell viability of differentiating NSCs, when compared with control (P < 0.01) or TUDCA treatment alone (at least P < 0.05) (). Thus, these results indicate that TUDCA-regulated mitochondrial alterations and NSC survival might involve regulation of mtROS and ATP levels.
Figure 4. TUDCA modulation of NSC differentiation-induced mitochondrial alterations is dependent on mtROS and ATP regulation. NSCs were expanded, induced to differentiate for 1 h and 24 h, in the presence or absence of TUDCA and/or CsA or OligA, and then collected for flow cytometry and luminescence detection, as described in Materials and Methods. (A) Representative quantification data of mtROS levels after 1 h of differentiation, using MitoSOXTM Red reagent. (B) Representative quantification data of ATP levels at 24 h of differentiation, using Mitochondrial ToxGloTM assay. (C) Representative quantification of NSC viability by measuring Annexin-V- and PI-negative cells using flow cytometry, at 24 h of differentiation. Results are expressed as mean ± SEM fold-change for at least 3 different experiments. *P < 0.01 and §P < 0.05 from non treated cells (control); ‡P < 0.01 and †P < 0.05 from cells treated with TUDCA alone.
TUDCA-regulated NSC proliferation and cell cycle progression are dependent on mtROS and ATP regulation
To further characterize the mechanisms by which TUDCA increases proliferation of NSCs, we reevaluated TUDCA effects on cell fate, in the presence of CsA or OligA. In fact, treatments with CsA (P < 0.01) and OligA (P < 0.05) markedly decreased BrdU incorporation of NSCs when compared with control cells, corroborating the idea that high ROS and diminished ATP impair cell cycle progression.Citation34,35 More importantly, our results showed that TUDCA was ineffective in the presence of both inhibitors, indicating that TUDCA-regulated NSC proliferation is indeed dependent on mtROS and ATP regulation (P < 0.05) (). Moreover, the effect of TUDCA in cell cycle progression was also determined by FACS analysis, following modulation of mtROS and ATP levels. After incubation with both CsA or OligA, we found a significant increase of cells in G0-G1 phase accompanied by a marked decrease of cells in S/G2-M phases, when compared to control (at least P < 0.05). Interestingly, inhibitors of the mtROS scavenger and ATP synthase were able to rescue the TUDCA-mediated S/G2-M phase progression (P < 0.01) (). In fact, recent evidence demonstrated that p21 and p27 might also be controlled by mitochondrial retrograde signals, including mtROS and ATP levels.Citation12 In this regard, incubation of NSCs with CsA or OligA, resulted in a marked increase of p21 and p27 levels when compared with untreated cells (P < 0.01), which reflects the effects of increased mtROS and decreased ATP levels in promoting cell cycle arrest. However, co-incubations of NSCs with CsA or OligA, and TUDCA, abolished the bile acid effect in reducing p21 and p27 levels (at least P < 0.05) of differentiating cells (). Therefore, these results suggest that TUDCA may increase proliferation of NSCs by retarding cell cycle arrest, through inhibition of p21 and p27 levels, which in turn is mediated by mtROS and ATP regulation.
Figure 5. TUDCA modulation of cell cycle and proliferation of NSCs is dependent on mtROS and ATP regulation. NSCs were expanded, induced to differentiate up to 24 h in the presence or absence of TUDCA and/or CsA or OligA, and then collected for flow cytometry and immunoblotting, as described in Materials and Methods. (A) Representative quantification data of BrdU incorporation in self-renewal conditions, 24 h after cell treatments. (B) Representative quantification of G0-G1 and S/G2-M phase cells. (C) Representative immunoblots of p21 (top left) and p27 (top right) in total extracts and respective quantification data (bottom right or left), at 6 h of differentiation. Results were normalized to endogenous β-actin protein levels, and are expressed as mean ± SEM fold-change for at least 3 different experiments. *P < 0.01 and §P < 0.05 from non treated cells (control); ‡P < 0.01 and †P < 0.05 from cells treated with TUDCA alone.
TUDCA-mediated mitochondrial protection favors neuronal cell fate determination of NSCs
mtDNA integrity was shown to be essential for mitochondrial maturation and cell fate determination during NSC differentiation. mtDNA damage has already been associated to higher astrogliogenesis and lack of neurogenesis during neural injury repair.Citation11,16 On the other hand, cell division speed may also influence the neurogenesis process. In fact, anti-proliferative genes-mediated changes in G1 phase length were shown to directly regulate the differentiation of neural precursors. Several studies have reported that timing of cell cycle has an impact on the relative rates of gliogenesis and neurogenesis.Citation36–39 Therefore, we explored the potential role of TUDCA in directing NSCs toward neuronal fate by assessing the ratio of neurons versus glial cells. Cells were grown under optimized neuronal differentiation-inducing conditions for 24 h, and the expression of neuronal (βIII-tubulin) and glial (glial fibrillary acidic protein, GFAP) markers was assessed by FACS analysis, in the presence or absence of TUDCA, as well as in the presence of mtROS and ATP modulators. Notably, our results revealed that TUDCA significantly increased the ratio of neurons vs. glial cells, when compared with control (P < 0.01). The incubation with mtROS and ATP modulators, in turn, significantly decreased this ratio, when compared with control (P < 0.01). More importantly, our results showed that increased mtROS levels and reduction of ATP levels by CsA and OligA, respectively, markedly reduced TUDCA effects on NSC fate determination (P < 0.01) (), suggesting that TUDCA shifts NSC differentiation by regulating mitochondrial stress and function. These results were also corroborated by immunocytochemistry, using fluorescence microscopy ().
Figure 6. TUDCA mediates neuronal rather than astroglial conversion of NSCs. NSCs were expanded and induced to differentiate for 24 h in the presence or absence of TUDCA and/or CsA or OligA. Cells were then collected for flow cytometry and immunocytochemistry, as described in Materials and Methods. (A) Flow cytometry analysis of the ratio between βIII-tubulin- and GFAP-positive cells cultured in optimized neuronal differentiation-inducing medium. (B) Representative images of immunofluorescence detection of cells labeled with anti- βIII-tubulin and anti-GFAP antibodies. Nuclei were stained with Hoechst 33258. Scale bar, 10 μm. (C) Flow cytometry analysis of the ratio between βIII-tubulin- and GFAP-positive cells cultured in optimized glial differentiation-inducing medium. Results are expressed as mean ± SEM fold-change for at least 3 different experiments. *P < 0.01 from non treated cells (control); ‡P < 0.01 and †P < 0.05 from cells treated with TUDCA alone.
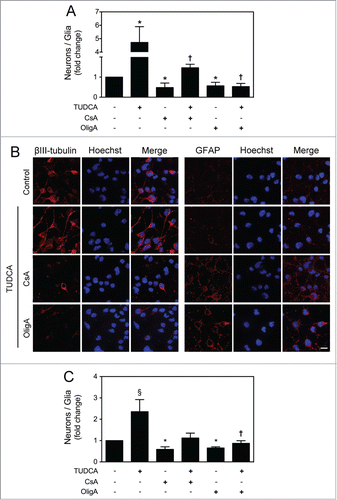
Finally, to evaluate whether TUDCA was still able to modulate NSC fate determination in non-neurogenic conditions, NSCs were grown under optimized glial differentiation-inducing conditions for 24 h, to hamper neuronal commitment, and TUDCA effect was re-evaluated. FACS analysis of βIII-tubulin and GFAP expression showed that TUDCA-treated cells still have a higher proportion of neurons, when compared with control (P < 0.01) (). CsA or OligA markedly increased the proportion of glial-differentiating cells when compared with control (P < 0.01), and were able to revert the bile acid effect on NSC fate determination. Thus, our results support a role for TUDCA in directing NSCs to a neuronal lineage, and underline the importance of mtROS and ATP in this regulatory mechanism.
Discussion
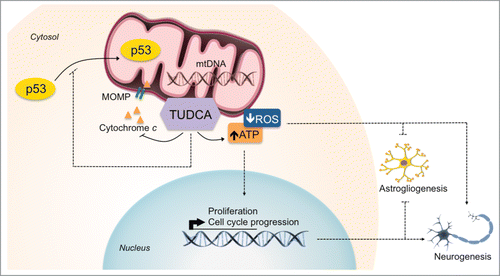
We have recently shown that apoptosis-associated mitochondrial events, including ROS production, mitochondrial membrane permeabilization, cytochrome c release and mitophagy are associated with early stages of NSC differentiation, without involving cell death per se.Citation15 In addition, the endogenous bile acid, TUDCA, has been identified as a general modulator of apoptosis by preventing mitochondrial membrane instability, Bax-induced mitochondrial membrane permeabilization and cytochrome c release in different cell types.Citation17,18 However, its potential effect in regulating NSC fate has never been explored. In this study, we identified a novel role for TUDCA in enhancing both proliferation and neuronal conversion potential of NSCs. TUDCA prevented differentiation-induced mitochondrial alterations, without influencing cell death, while increasing self-renewal and proliferation levels of NSCs. Importantly, the mechanisms by which TUDCA modulates cell fate were shown to be dependent on mtROS and ATP regulation, ultimately favoring NSC commitment to a neuronal fate ().
Figure 7. TUDCA mediates mitochondria-cell cycle retrograde signals to regulate NSC fate. The NSC modulatory properties of TUDCA result from inhibition of differentiation-induced mitochondrial apoptotic events by the bile acid, and from subsequent decrease in ROS and ATP mitochondrial levels. This, in turn, contributes for the enhancement of both NSC proliferation and neuronal rather than astroglial conversion of differentiating NSCs. Importantly, TUDCA-mediated effects in increasing NSC pool and lineage determination occur in a mitochondrial redox state- and ATP-dependent manner. Interactions that were described, suggested or shown indirectly are depicted.
TUDCA is neuroprotective in several animal models of neurodegeneration and improves survival and function of mesencephalic cell transplants in Parkinson disease.Citation21–23,Citation25 In a 3-nitropropionic acid (3-NP) rat model of Huntington's disease, TUDCA prevents mitochondrial outer membrane permeabilization (MOMP), inhibits cytochrome c release, and modulates downstream apoptotic events, such as caspase activation and poly ADP-ribose polymerase (PARP) cleavage.Citation17 Furthermore, TUDCA was also shown to inhibit bilirubin- and Aβ-induced Bax translocation, MOMP and subsequent cytochrome c release, in isolated mitochondria of both neuronal and glial cells.Citation40,41
Based on the fact that mitochondrial integrity and function is crucial for the life-long activity of NSCs,Citation16,42 and that disturbances in mitochondrial signaling have been largely associated with age-related decrease in neuroplasticity and neurogenesis,Citation6,8,43 we decided to investigate the potential role of TUDCA in increasing NSC activity.
We began to explore the effect of this bile acid on differentiation-induced mitochondrial alterations, and demonstrated that transient ROS production, mitochondrial membrane depolarization and cytochrome c release were significantly prevented by TUDCA at 6 hours of NSC differentiation. In fact, it is well established that increased mitochondrial activity is a prerequisite for the differentiation process.Citation44 During neural differentiation, profound mitochondrial morphologic and metabolic alterations occur to assure successful differentiation,Citation6-9,Citation11 leading to increased oxidative stress and mitochondrial damage. On the other hand, differentiation-mediated mitochondrial oxidative stress was shown to trigger mitophagy, a selective type of autophagy characterized by the engulfment of dysfunctional mitochondria, as a protective mechanism.Citation45 Of note, we have also found that TUDCA rescued the decrease in mitochondrial mass and ATP levels observed after induction of the differentiation process. These data are in accordance with our previous observation revealing a decrease of mitochondrial mass at early stages of neural differentiation,Citation15 as well as with other studies reporting the protective effect of TUDCA on mitochondria integrity.Citation17,21,25
Intriguingly, mitochondrial translocation of p53, already described as a possible protective mechanism of neural differentiation-induced mitochondrial stressCitation15 and also detected in our differentiation conditions, was not observed in differentiating cells treated with TUDCA. This may be explained by the fact that p53 mitochondrial translocation was only shown to occur after the first signs of differentiation-induced mitochondrial damage.Citation15 It is possible that mitochondrial defense against ROS injury by TUDCA might dismiss p53 mitochondrial translocation as a backup mechanism of mitochondrial protection. Indeed, the effect of the bile acid in inhibiting both the increase of ROS and the depletion of ATP was very pronounced since differentiation-induced changes in mtROS and ATP were completely abolished by TUDCA. Thus, it is not surprise that p53 mitochondrial translocation, usually triggered by differentiation-mediated mitochondrial stress, was also totally repressed by the bile acid.
Additionally, TUDCA treatment resulted in profound changes of NSC cycle progression. In fact, it thus appear that the relieve of mitochondrial stress by bile acid exposure allowed NSCs to re-enter the cell cycle, as detected by increased BrdU incorporation and decreased expression of cell cycle arrest-related proteins, p21 and p27. By evaluating cell cycle, our results disclosed that TUDCA incubation elicited a marked increase in S phase with a subsequent reduction in NSC population at G1 phase. In fact, other studies have already reported that short G1 is typical of cells in expansion. For instance, fast cell cycles were shown to be characteristic of pluripotent embryonic stem cells in early stages of animal body development, followed by short lengthening during gastrulation, to finally reach differentiation of the 3 germ layers.Citation38,46,47 Our results are in agreement by showing increased self-renewing potential of NSCs in the presence of TUDCA. Further, TUDCA-induced NSC proliferation was associated with augmented symmetric divisions, self-renewal and NSC number. It has been shown that distribution of cyclin D2 mRNA is associated with the maintenance of stemness state during NSC divisions.Citation48 Therefore, it is possible that TUDCA might also affect the distribution of cyclin D2 in daughter cells during NSC proliferation. Importantly, although TUDCA treatment did not affect cell survival, it ultimately increased the number of cells capable of differentiating. Of note, this TUDCA effect might prove useful for in vitro expansion of NSCs, but also for a more efficient increase of the in vivo NSC pool. Therapeutic strategies pushing NSC asymmetric divisions and neurogenesis are thought to contribute to a faster loss of the NSC pool, hence compromising the regenerative capacity of the brain. On the other hand, the self-renewing process becomes scarce with aging and the regenerative properties of aged stem cells are progressively deteriorated in ways that tissue repair and maintenance become inefficient.Citation49–51 Indeed, systemic, cellular and molecular alterations induced by aging have been shown to interfere with the neurogenic niche, at the level of NSC proliferation, their fate, neuronal survival, and subsequent integration in neural circuitry.Citation52 In this respect, and given the fact that TUDCA is an orally bioavailable and central nervous system-penetrating molecule,Citation21 it would be interesting to also test in the future the effects of this bile acid in vivo.
To go deeper into the molecular mechanism by which TUDCA enhances the NSC pool in vitro, namely whether it could be dependent on its mitochondrial effects, we decided to re-evaluate TUDCA effects following mtROS and ATP modulation. Importantly, anti-oxidant and proliferative effects, as well as maintenance of mitochondrial metabolism and function of NSCs, by TUDCA, were dependent on mtROS and ATP modulation. In fact, several reports have already supported the idea that distinct mitochondrial retrograde signals control cell cycle checkpoints and that low levels of ROS might function as intracellular messengers of G1-S cell cycle arrest.Citation53,54 Curiously, it has been demonstrated that mitochondrial dysfunction activates two retrograde signals resulting in cell cycle modulation. The decrease in ATP production is usually followed by downregulation of cyclin E, while increased ROS production induces upregulation of p27.Citation12,33 Accordingly, studies in Drosophila have also demonstrated that decreased levels of ATP result in mitochondrial checkpoints in late G1 and the concomitant arrest of energetically impaired cells.Citation55
We then investigated whether TUDCA-induced effects on mitochondria and cell cycle of differentiating cells could influence NSC lineage determination. And if so, whether this shift in differentiation potential would be caused by mtROS and ATP modulation of TUDCA. According with our expectations, TUDCA induced a significant increased in the ratio of β-III tubulin- to GFAP-positive cells throughout differentiation either in optimized neuronal or glial differentiation-inducing conditions. The mechanism by which TUDCA mediates neuronal rather than astroglial conversion appeared to involve the regulation of mtROS or ATP, since we did not observed this shift in the presence of CsA or OligA. These results are in agreement with the idea that increased levels of mitochondrial damage are associated with elevated astrogliogenesis and decreased neurogenesis,Citation11,16 and also with other studies showing the impact of the length of cell cycle in NSC lineage determination. In fact, G1 stage was recently shown to influence cell fate determination of neural precursors, leading to the hypothesis that longer cell cycles allowed the accumulation of factors necessary for cell fate changes to occur.Citation38,56 Furthermore, others have also demonstrated that glial cells are formed after neurons and that astrocytes take more time to re-enter cell cycle than neurons.Citation57 These conclusions were made after several experiments using cell cycle inhibitors, which demonstrated that the increasing expression of p27 in neural progenitor cells led to cell cycle exit and glial terminal differentiation.Citation36
Finally, given the involvement of Bcl-2 family elements in the determination of neuronal versus glial lineagesCitation58 and the well-established role of TUDCA in upregulating Bcl-2 and Bcl-XL,Citation23,59 it will be important to clarify whether this bile acid might also increase neuronal conversion by raising the expression levels of anti-apoptotic Bcl-2 family members. In addition, it is also possible that TUDCA might interfere with other regulators of NSC lineage specification, such as REST and CoREST. Of note, the profiles of REST and CoREST target genes were demonstrated to be largely non-overlapping between neuronal subtypes and astroglial lineage species.Citation60 In fact, further insights into the regulatory network of TUDCA during NSC differentiation could shed light on the molecular processes leading to a more efficient production of neurons during developmental, neurodegenerative, and aging processes.
In conclusion, the present study identifies a novel role for TUDCA as a modulator of long-term proliferation and neuronal conversion of NSC, and underlines its mechanism of action by showing the contribution of mitochondria redox state and ATP levels on the TUDCA-regulated NSC fate. This novel mechanism of TUDCA action suggests new intervention methods for preventing aged-associated loss of NSCs and for treating neurological disorders associated with deficits in neuronal density.
Materials and Methods
Ethics statement
The mouse NSC line used in this study was obtained from Dr. Smith's Laboratory, University of Cambridge, Cambridge, UK,Citation61 and provided by Dr. Henrique, Universidade de Lisboa, Lisbon, Portugal. The Animal Ethical Committee at the Faculty of Pharmacy, University of Lisbon, Portugal waived the need for approval.
Cell culture
Neural stem cells were derived from 14.5-dpc mouse fetal forebrain. This cell line was established using a method that produces pure cultures of adherent NSC, which continuously expand by symmetrical division and are capable of tripotential differentiation.Citation62–64 NSCs were grown in monolayer and routinely maintained, as previously described.Citation15,65 Neural differentiation was performed by first platting NSCs in undifferentiation medium onto uncoated tissue culture plastic dishes at 6.5 × 105 cells/mL for 24 h, and changing the culture medium to an optimized neuronal differentiation-inducing medium, comprising Euromed-N medium supplemented with 10 ng/mL bFGF, 0.5% N-2 supplement, 1% B27 supplement (17504–044; InvitrogenTM, Life Technologies Corporation) and 1% penicillin-streptomycin, or an optimized glial differentiation-inducing medium, comprising 1% fetal bovine serum (FBS, 10082147; InvitrogenTM, Life Technologies Corp.) instead of B27 supplement.
Cellular treatments
Cells were treated with chemical compounds upon medium change, allowed to differentiate for different times up to 24 h, and then collected for flow cytometry, immunoblotting, immunofluorescence and quantitative real-time polymerase chain reaction (PCR) assays. Concentrations of 100 μM tauroursodeoxycholic acid (TUDCA, T0266; Sigma-Aldrich Corporation) were routinely added to NSC. As a proof-control that TUDCA effect was not dependent of its taurine-conjugated group, 100 μM of taurine (T0625; Sigma-Aldrich Corp.) was also added to cells. Moreover, 0.5 μM N-acetyl-L-cysteine (NAC, T7250; Sigma-Aldrich Corp.), a well-established anti-oxidant, was used to compare TUDCA anti-oxidant effect in this cellular context.
To inhibit the mtROS scavenger, MnSOD, NSCs were treated with 10 μM CsA (30024; Sigma-Aldrich Corp.) for 1, 6 or 24 h before harvest. CsA induces nitration of MnSOD in tyrosine 34 residues, leading to MnSOD functional inactivation.Citation66,67 Since MnSOD is an important antioxidant that neutralizes superoxides at the mitochondrial compartment, its inhibition causes increased ROS, oxidative mtDNA damage as well as inactivation of respiratory and Krebs cycle enzymes.Citation68,69 Finally, inhibition of ATP synthesis was achieved by treating NSCs with 5 μg/mL of OligA (sc-201551; Santa Cruz Biotechnology, Inc.) for 1, 6 or 24 h before harvest. OligA inhibits F1F0 complex of the ATP synthase by blocking its proton channel (F0 subunit) and, consequently, proton conductance through the inner membrane, thus inhibiting both ATP synthesis and ATP hydrolysis.Citation70–72
Measurement of mitochondrial transmembrane potential
Mitochondrial permeabilization was determined as the retention of the dye 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3), D273; Molecular Probes®, Life Technologies Corp.). NSCs were loaded with 50 nM DiOC6(3) for 30 min at 37°C and resuspended in DPBS with 2% FBS. The emission of green fluorescence was analyzed by FACS using LSR FortessaTM (Becton, Dickinson and Company). Data were statistically evaluated using FlowJo software (Tree Star, Inc.).
Mitochondrial ROS detection
For mtROS quantification, cells were pre-treated with 5 μM MitoSOXTM Red mitochondrial superoxide indicator (M36008; Molecular Probes®, Life Technologies Corp.) in Hank's balanced salt solution (HBSS, 24020; Gibco®, Life Technologies Corp.) at 37°C for 10 min. NSCs were then washed twice with Ca2+- and Mg2+-free PBS (Gibco®, Life Technologies Corp.), collected with accutase and resuspended in DPBS with 2% FBS. Emission of red fluorescence was analyzed in live cells by cytometric analysis using Guava EasyCyte 5HT® (Merck Millipore Corporation). Data were statistically evaluated using GuavaSoft® software (Merck Millipore Corp.).
Immunoblotting
Levels of cytochrome c, p53, p21, p27 and Sox2 were determined by Western blot analysis of total, mitochondrial and cytosolic protein extracts, obtained as previously described.Citation15 Briefly, 50–80 μg of protein extracts were separated on 12 or 15% sodium dodecyl sulfate-polyacrylamide electrophoresis gels (SDS-PAGE) and then subjected to immunoblotting using primary mouse monoclonal antibodies reactive to cytochrome c (556433; BD Biosciences PharMingen), p53 (2524; Cell Signaling Technology®, Inc.), p27 (sc-1641; Santa Cruz Biotechnology, Inc.) and Sox2 (MAB2018; R&D Systems® Inc.); primary rabbit polyclonal antibodies reactive to p21 (sc-397-G; Santa Cruz Biotechnology, Inc.). Blots were subsequently incubated with secondary antibodies conjugated with anti-mouse and anti-rabbit IgG conjugated with horseradish peroxidase (Bio-Rad Laboratories) and anti-goat IgG-HRP (sc-2020; Santa Cruz Biotechnology, Inc.) for 2 h at room temperature. Finally, membranes were processed for protein detection using Immobilon (Merck Millipore Corp.). VDAC (4866; Cell Signaling Technology®, Inc.), GAPDH (sc-32233; Santa Cruz Biotechnology, Inc.), β-actin (A5441; Sigma-Aldrich Corp.) and Lamin B1 (ab16048; Abcam®) antibodies were used as loading controls and/or to monitor the purity of mitochondrial and cytosolic fractionation.
Quantification of mtDNA copy number
Total cellular DNA was isolated using the QiaAmp DNA Mini Kit (51304; Qiagen), following manufacturer's protocols. Subsequently, quantitative real-time PCR was run in the ABI 7300 (Applied Biosystems®, Life Technologies Corp.) sequence detection system. The relative mtDNA copy number was calculated based on the standard curve and the ratio of the amount of mtDNA vs. 18S for each sample, as previously described.Citation15,73 The values were first expressed as percentage of total input and converted to fold-change over control.
ATP measurements
For ATP production detection, cells were plated, treated and then collected for processing according to the manufacturer's instruction of the Mitochondrial ToxGlo™ assay Kit (G8001; Promega Company). ATP levels are measured by adding the ATP Detection Reagent, resulting in cell lysis and generation of a luminescent signal proportional to the amount of ATP present. Emission of luminescence was detected using the GloMax® 96 Microplate Luminometer (Promega Co.). Data were statistically evaluated using the Instinct® Software (Promega Co.).
Proliferation index
Proliferation levels were determined by BrdU incorporation analysis using the APC BrdU Flow Kit (BD Biosciences PharMingen). Of note, BrdU is a synthetic nucleoside thymidine analog that is incorporated into newly synthesized DNA during the S phase of the cell cycle.Citation74 Cells were plated in undifferentiation medium at 3.25 × 105 cells/mL for 24 h, in the presence or absence (control) of TUDCA. BrdU was added to the culture medium 21 h after cell treatments, and cells were re-incubated for additional 3 h. The emission of red fluorescence was later analyzed by FACS using LSR FortessaTM (Becton, Dickinson and Co.). Data were statistically evaluated using FlowJo software (Tree Star, Inc.).
The percentage of cells in G1, S and G2-M phases of the cell cycle was also evaluated by FACS. First, cells were synchronized by double-thymidine block. Ten hours after plating, 2 mM thymidine (Sigma-Aldrich Corp.) was added to the culture media for additional 14 h. Cells were then released from the first thymidine block by removing culture media, washing 3 times with PBS, and adding fresh undifferentiation medium, without thymidine. Ten hours later, cells were submitted to a second thymidine block. TUDCA was added to the medium, with no thymidine, at the end of the second block. Cells were grown for additional 24 h, prior to cell collection with accutase. Subsequently, cells were washed with PBS, fixed in 80% ethanol at 4°C overnight, and then stained with 5 μg/mL propidium iodide (PI) (Sigma-Aldrich Corp.) for DNA contents. Data were statistically evaluated using ModFit software (Verity Software House Inc.).
Finally, the number of cells was evaluated by cell counting, using the counting chamber of a hemacytometer. After 24 h in differentiation medium, in the presence or absence (control) of TUDCA, cells were collected with accutase and washed with PBS. Cell suspension was diluted 1:500 in a solution of 0.4% Trypan Blue (T8154; Sigma-Aldrich Corp.) and then gently applied between the coverslip and the chamber, for counting.
Evaluation of self-renewal: Sox2 cell pair assay
NSCs were plated in uncoated tissue culture plastic 12-well plates, at a density of 6400 cells/cm2. After seeding, NSCs cells were grown in low EGF/bFGF containing medium supplemented or not (control) with 100 μM TUDCA for 24 h. Cells were then fixed in paraformaldehyde (4%, w/v) in PBS for 30 min at 4°C and then processed for immunocytochemistry against Sox2. In fact, the expression of Sox2, a transcription factor essential for maintaining self-renewal and pluripontency, tends to disappear in dividing cells that start to differentiate.Citation75,76 The number of progenitor pairs undergoing proliferative or differentiative cell divisions was determined by counting 60 pairs of cells, in control conditions or with TUDCA, for at least 3 different experiments.Citation77,78
Immunocytochemistry
For visualization of paired symmetrical and asymmetrical divisions in NSCs, fixed cells were incubated with primary mouse monoclonal antibody reactive to Sox2 (MAB2018; R&D Systems® Inc.) at a dilution of 1:100, overnight at 4°C. After 2 washes, secondary DyLight 488 conjugated anti-mouse antibody (35502; Thermo Fisher Scientific Inc.) diluted 1:100 was added to cells for 2 h at room temperature. For evaluation of NSC differentiation, fixed cells were incubated with primary mouse monoclonal antibodies reactive to βIII-tubulin (MMS-435P, TUJ1; Covance) and GFAP (MAB360; Merck Millipore Corp.), at a dilution of 1:500 and 1:200, respectively, overnight at 4°C. This was followed by 2 h incubation with secondary anti-mouse Alexa Fluor® 568 (A-10037; InvitrogenTM, Life Technologies Corp.) diluted 1:200. Mouse NSC nuclei were stained with Hoechst 33258 (861405; Sigma-Aldrich Corp.) at 50 μg/ml in PBS, for 3 min at room temperature. Samples were mounted using Mowiol (81381; Sigma-Aldrich Corp.). The resulting fluorescent signals were imaged using fluorescence microscopy assessments performed with a Zeiss AX10 microscope (Carl Zeiss, Corp.), equipped with a 40× plan-apochromat objective and a Leica DFC490 camera (Leica Microsystems). Images were processed using ImageJ.
Evaluation of cell death and viability
Viability of NSCs was assessed by staining NSCs with the Annexin-V-APC apoptosis detection kit (88–8007; eBioscience, Inc.), according to manufacturer's instructions. This kit allows the determination of phosphatidylserine exposure as well as the inclusion of the viability dye propidium iodide (PI). Samples were then analyzed by FACS using LSR FortessaTM (Becton Dickinson). Data were statistically evaluated using FlowJo software (Tree Star, Inc.).
Analyses of NSC differentiation
For detection of Sox2, βIII-tubulin and GFAP expression levels, cells were collected and processed as previously described.Citation15 Subsequently, cells were washed and incubated for 30 min with mouse primary antibodies reactive to Sox2 (MAB2018; R&D Systems® Inc.), βIII-tubulin (MMS-435P, TUJ1; Covance) and GFAP (MAB360; Merck Millipore Corp.) at a dilution of 1:100, 1:500 and 1:100, respectively. Cells were then washed twice and incubated with appropriate secondary anti-mouse antibodies conjugated to DyLight 488 (35502; Thermo Fisher Scientific Inc.) at a dilution of 1:100, for 30 min. Finally, cells were analyzed by FACS using LSR FortessaTM (Becton Dickinson). Data were statistically evaluated using FlowJo software (Tree Star, Inc.).
Densitometry and statistical analysis
The relative intensities of protein bands were analyzed using the Quantity One Version 4.6.3 densitometric analysis program (Bio-Rad Laboratories). Results from different groups were compared using the Student's t test, 2-way ANOVA or one-way ANOVA followed by Bonferroni's or Dunnett's multiple comparison tests. Values of P < 0.05 were considered statistically significant. All statistical analysis was performed with GraphPad Prism 5 software (GraphPad Software, Inc.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to thank Dr. Domingos Henrique (Instituto de Medicina Molecular, Universidade de Lisboa, Lisbon, Portugal) for providing and assisting in establishing the NS cell line culture conditions. We would also like to thank all members of the laboratory for insightful discussions.
Funding
This work was supported by grants PTDC/SAU-NMC/117877/2010, PEst-OE/SAU/UI4013/2011, and PTDC/BIM-MED/0251/2012, and by fellowships SFRH/BD/68368/2010 (JMX) and SFRH/BD/80060/2011 (ALM) from Fundação para a Ciência e Tecnologia, Portugal.
Supplemental Material
Supplemental data for this article can be found on the publisher's website.
962951_Supplementary_Materials.zip
Download Zip (256 KB)References
- Koussevitzky S, Nott A, Mockler TC, Hong F, Sachetto-Martins G, Surpin M, Lim J, Mittler R, Chory J. Signals from chloroplasts converge to regulate nuclear gene expression. Science 2007; 316:715-9; PMID:17395793; http://dx.doi.org/10.1126/science
- Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell 2004; 14:1-15; PMID:15068799; http://dx.doi.org/10.1016/S1097-2765(04)00179-0
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol 2006; 16:R551-60; PMID:16860735; http://dx.doi.org/10.1016/j.cub.2006.06.054
- Nagley P, Higgins GC, Atkin JD, Beart PM. Multifaceted deaths orchestrated by mitochondria in neurones. Biochim Biophys Acta 2010; 1802:167-85; PMID:19751830; http://dx.doi.org/10.1016/j.bbadis.2009.09.004
- Parker GC, Acsadi G, Brenner CA. Mitochondria: determinants of stem cell fate? Stem Cells Dev 2009; 18:803-6; PMID:19563264; http://dx.doi.org/10.1089/scd.2009.1806.edi
- Kole AJ, Annis RP, Deshmukh M. Mature neurons: equipped for survival. Cell Death Dis 2013; 4:e689; PMID:23807218; http://dx.doi.org/10.1038/cddis.2013.220
- Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, Wu H, Kornblum HI. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2011; 8:59-71; PMID:21211782; http://dx.doi.org/10.1016/j.stem.2010.11.028
- Panchision DM. The role of oxygen in regulating neural stem cells in development and disease. J Cell Physiol 2009; 220:562-8; PMID:19441077; http://dx.doi.org/10.1002/jcp.21812
- Voloboueva LA, Lee SW, Emery JF, Palmer TD, Giffard RG. Mitochondrial protection attenuates inflammation-induced impairment of neurogenesis in vitro and in vivo. J Neurosci 2010; 30:12242-51; PMID:20844120; http://dx.doi.org/10.1523/JNEUROSCI.1752-10.2010
- Facucho-Oliveira JM, Alderson J, Spikings EC, Egginton S, St John JC. Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J Cell Sci 2007; 120:4025-34; PMID:17971411; http://dx.doi.org/10.1242/jcs.016972
- Wang W, Osenbroch P, Skinnes R, Esbensen Y, Bjoras M, Eide L. Mitochondrial DNA integrity is essential for mitochondrial maturation during differentiation of neural stem cells. Stem Cells 2010; 28:2195-204; PMID:20954243; http://dx.doi.org/10.1002/stem.542
- Owusu-Ansah E, Yavari A, Mandal S, Banerjee U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat Genet 2008; 40:356-61; PMID:18246068; http://dx.doi.org/10.1038/ng.2007.50
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995; 81:323-30; PMID:7736585; http://dx.doi.org/10.1016/0092-8674(95)90385-2
- Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol 1998; 18:753-61; PMID:9447971
- Xavier JM, Morgado AL, Sola S, Rodrigues CM. Mitochondrial translocation of p53 modulates neuronal fate by preventing differentiation-induced mitochondrial stress. Antioxid Redox Signal 2014; 21:1009-24; PMID:24329038; http://dx.doi.org/10.1089/ars.2013.5417
- Wang W, Esbensen Y, Kunke D, Suganthan R, Rachek L, Bjoras M, Eide L. Mitochondrial DNA damage level determines neural stem cell differentiation fate. J Neurosci 2011; 31:9746-51; PMID:21715639; http://dx.doi.org/10.1523/JNEUROSCI.0852-11.2011
- Rodrigues CM, Stieers CL, Keene CD, Ma X, Kren BT, Low WC, Steer CJ. Tauroursodeoxycholic acid partially prevents apoptosis induced by 3-nitropropionic acid: evidence for a mitochondrial pathway independent of the permeability transition. J Neurochem 2000; 75:2368-79; PMID:11080188; http://dx.doi.org/10.1046/j.1471-4159.2000.0752368.x
- Sola S, Aranha MM, Rodrigues CM. Driving apoptosis-relevant proteins toward neural differentiation. Mol Neurobiol 2012; 46:316-31; PMID:22752662; http://dx.doi.org/10.1007/s12035-012-8289-2
- Lazaridis KN, Gores GJ, Lindor KD. Ursodeoxycholic acid 'mechanisms of action and clinical use in hepatobiliary disorders’. J Hepatol 2001; 35:134-46; PMID:11495032; http://dx.doi.org/10.1016/S0168-8278(01)00092-7
- Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 2002; 36:525-31; PMID:12198643; http://dx.doi.org/10.1053/jhep.2002.36088
- Keene CD, Rodrigues CM, Eich T, Chhabra MS, Steer CJ, Low WC. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington's disease. Proc Natl Acad Sci U S A 2002; 99:10671-6; PMID:12149470; http://dx.doi.org/10.1073/pnas.162362299
- Duan WM, Rodrigues CM, Zhao LR, Steer CJ, Low WC. Tauroursodeoxycholic acid improves the survival and function of nigral transplants in a rat model of Parkinson's disease. Cell Transplant 2002; 11:195-205; PMID:12075985
- Rodrigues CM, Sola S, Nan Z, Castro RE, Ribeiro PS, Low WC, Steer CJ. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc Natl Acad Sci U S A 2003; 100:6087-92; PMID:12721362; http://dx.doi.org/10.1073/pnas.1031632100
- Sola S, Aranha MM, Steer CJ, Rodrigues CM. Game and players: mitochondrial apoptosis and the therapeutic potential of ursodeoxycholic acid. Curr Issues Mol Biol 2007; 9:123-38; PMID:17489439
- Oveson BC, Iwase T, Hackett SF, Lee SY, Usui S, Sedlak TW, Snyder SH, Campochiaro PA, Sung JU. Constituents of bile, bilirubin and TUDCA, protect against oxidative stress-induced retinal degeneration. J Neurochem 2011; 116:144-53; PMID:21054389; http://dx.doi.org/10.1111/j.1471-4159.2010.07092.x
- Parry GJ, Rodrigues CM, Aranha MM, Hilbert SJ, Davey C, Kelkar P, Low WC, Steer CJ. Safety, tolerability, and cerebrospinal fluid penetration of ursodeoxycholic Acid in patients with amyotrophic lateral sclerosis. Clin Neuropharmacol 2010; 33:17-21; PMID:19935406; http://dx.doi.org/10.1097/WNF.0b013e3181c47569
- Huxtable RJ. Physiological actions of taurine. Physiol Rev 1992; 72:101-63; PMID:1731369
- Hernandez-Benitez R, Pasantes-Morales H, Saldana IT, Ramos-Mandujano G. Taurine stimulates proliferation of mice embryonic cultured neural progenitor cells. J Neurosci Res 2010; 88:1673-81; PMID:20029963
- Shivaraj MC, Marcy G, Low G, Ryu JR, Zhao X, Rosales FJ, Goh EL. Taurine induces proliferation of neural stem cells and synapse development in the developing mouse brain. PLoS One 2012; 7:e42935; PMID:22916184; http://dx.doi.org/10.1371/journal.pone.0042935
- Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC. Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal 2012; 16:1150-80; PMID:21967640; http://dx.doi.org/10.1089/ars.2011.4085
- Kato JY, Matsuoka M, Polyak K, Massague J, Sherr CJ. Cyclic AMP-induced G1 phase arrest mediated by an inhibitor (p27Kip1) of cyclin-dependent kinase 4 activation. Cell 1994; 79:487-96; PMID:7954814; http://dx.doi.org/10.1016/0092-8674(94)90257-7
- Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res 2005; 65:3980-5; PMID:15899785; http://dx.doi.org/10.1158/0008-5472.CAN-04-3995
- Finkel T, Hwang PM. The Krebs cycle meets the cell cycle: mitochondria and the G1-S transition. Proc Natl Acad Sci U S A 2009; 106:11825-6; PMID:19617546; http://dx.doi.org/10.1073/pnas.0906430106
- Alenzi FQ, Alenazi BQ, Ahmad SY, Salem ML, Al-Jabri AA, Wyse RK. The haemopoietic stem cell: between apoptosis and self renewal. Yale J Biol Med 2009; 82:7-18; PMID:19325941
- Mandal S, Lindgren AG, Srivastava AS, Clark AT, Banerjee U. Mitochondrial function controls proliferation and early differentiation potential of embryonic stem cells. Stem Cells 2011; 29:486-95; PMID:21425411; http://dx.doi.org/10.1002/stem.590
- Durand B, Raff M. A cell-intrinsic timer that operates during oligodendrocyte development. Bioessays 2000; 22:64-71; PMID:10649292; http://dx.doi.org/10.1002/(SICI)1521-1878(200001)22:1<64::AID-BIES11>3.0.CO;2-Q
- Ohnuma S, Harris WA. Neurogenesis and the cell cycle. Neuron 2003; 40:199-208; PMID:14556704; http://dx.doi.org/10.1016/S0896-6273(03)00632-9
- Salomoni P, Calegari F. Cell cycle control of mammalian neural stem cells: putting a speed limit on G1. Trends Cell Biol 2010; 20:233-43; PMID:20153966; http://dx.doi.org/10.1016/j.tcb.2010.01.006
- Solter M, Locker M, Boy S, Taelman V, Bellefroid EJ, Perron M, Pieler T. Characterization and function of the bHLH-O protein XHes2: insight into the mechanisms controlling retinal cell fate decision. Development 2006; 133:4097-108; PMID:17008450; http://dx.doi.org/10.1242/dev.02567
- Rodrigues CM, Sola S, Silva R, Brites D. Bilirubin and amyloid-beta peptide induce cytochrome c release through mitochondrial membrane permeabilization. Mol Med 2000; 6:936-46; PMID:11147571
- Sola S, Castro RE, Laires PA, Steer CJ, Rodrigues CM. Tauroursodeoxycholic acid prevents amyloid-beta peptide-induced neuronal death via a phosphatidylinositol 3-kinase-dependent signaling pathway. Mol Med 2003; 9:226-34; PMID:15208744; http://dx.doi.org/10.2119/2003-00042.Rodrigues
- Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J 2012; 31:1336-49; PMID:22354038; http://dx.doi.org/10.1038/emboj.2012.38
- Cheng A, Hou Y, Mattson MP. Mitochondria and neuroplasticity. ASN Neuro 2010; 2:e00045; PMID:20957078; http://dx.doi.org/10.1042/AN20100019
- Facucho-Oliveira JM, St John JC. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev 2009; 5:140-58; PMID:19521804; http://dx.doi.org/10.1007/s12015-009-9058-0
- Tolkovsky AM. Mitophagy. Biochim Biophys Acta 2009; 1793:1508-15; PMID:19289147; http://dx.doi.org/10.1016/j.bbamcr.2009.03.002
- O'Farrell PH, Stumpff J, Su TT. Embryonic cleavage cycles: how is a mouse like a fly? Curr Biol 2004; 14:R35-45; PMID:14711435; http://dx.doi.org/10.1016/j.cub.2003.12.022
- Pauklin S, Vallier L. The cell-cycle state of stem cells determines cell fate propensity. Cell 2013; 155:135-47; PMID:24074866; http://dx.doi.org/10.1016/j.cell.2013.08.031
- Tsunekawa Y, Osumi N. How to keep proliferative neural stem/progenitor cells: a critical role of asymmetric inheritance of cyclin D2. Cell Cycle 2012; 11:3550-4; PMID:22895110; http://dx.doi.org/10.4161/cc.21500
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006; 443:448-52; PMID:16957738; http://dx.doi.org/10.1038/nature05091
- Behrens A, van Deursen JM, Rudolph KL, Schumacher B. Impact of genomic damage and ageing on stem cell function. Nat Cell Biol 2014; 16:201-7; PMID:24576896; http://dx.doi.org/10.1038/ncb2928
- Piccin D, Morshead CM. Potential and pitfalls of stem cell therapy in old age. Dis Model Mech 2010; 3:421-5; PMID:20504968; http://dx.doi.org/10.1242/dmm.003137
- Artegiani B, Calegari F. Age-related cognitive decline: can neural stem cells help us? Aging (Albany NY) 2012; 4:176-86; PMID:22466406
- Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem 2001; 11:173-86; PMID:11509825; http://dx.doi.org/10.1159/000047804
- Deng X, Gao F, May WS Jr. Bcl2 retards G1/S cell cycle transition by regulating intracellular ROS. Blood 2003; 102:3179-85; PMID:12855558; http://dx.doi.org/10.1182/blood-2003-04-1027
- Mandal S, Guptan P, Owusu-Ansah E, Banerjee U. Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev Cell 2005; 9:843-54; PMID: 16326395; http://dx.doi.org/10.1016/j.devcel.2005.11.006
- Lange C, Huttner WB, Calegari F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 2009; 5:320-31; PMID:19733543; http://dx.doi.org/10.1016/j.stem.2009.05.026
- Qian X, Shen Q, Goderie SK, He W, Capela A, Davis AA, Temple S. Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron 2000; 28:69-80; PMID:11086984; http://dx.doi.org/10.1016/S0896-6273(00)00086-6
- Sola S, Morgado AL, Rodrigues CM. Death receptors and mitochondria: Two prime triggers of neural apoptosis and differentiation. Biochim Biophys Acta 2013; 1830:2160-6; PMID:23041071; http://dx.doi.org/10.1016/j.bbagen.2012.09.021
- Castro RE, Sola S, Ramalho RM, Steer CJ, Rodrigues CM. The bile acid tauroursodeoxycholic acid modulates phosphorylation and translocation of bad via phosphatidylinositol 3-kinase in glutamate-induced apoptosis of rat cortical neurons. J Pharmacol Exp Ther 2004; 311:845-52; PMID:15190125; http://dx.doi.org/10.1124/jpet.104.070532
- Qureshi IA, Gokhan S, Mehler MF. REST and CoREST are transcriptional and epigenetic regulators of seminal neural fate decisions. Cell Cycle 2010; 9:4477-86; PMID:21088488; http://dx.doi.org/10.4161/cc.9.22.13973
- Silva J, Chambers I, Pollard S, Smith A. Nanog promotes transfer of pluripotency after cell fusion. Nature 2006; 441:997-1001; PMID:16791199; http://dx.doi.org/10.1038/nature04914
- Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, Sun Y, Sanzone S, Ying QL, Cattaneo E, et al. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol 2005; 3:e283; PMID:16086633; http://dx.doi.org/10.1371/journal.pbio.0030283
- Pollard SM, Conti L, Sun Y, Goffredo D, Smith A. Adherent neural stem (NS) cells from fetal and adult forebrain. Cereb Cortex 2006; 16 Suppl 1:i112-20; PMID:16766697; http://dx.doi.org/10.1093/cercor/bhj167
- Glaser T, Pollard SM, Smith A, Brustle O. Tripotential differentiation of adherently expandable neural stem (NS) cells. PLoS One 2007; 2:e298; PMID:17356704; http://dx.doi.org/10.1371/journal.pone.0000298
- Santos DM, Xavier JM, Morgado AL, Sola S, Rodrigues CM. Distinct regulatory functions of calpain 1 and 2 during neural stem cell self-renewal and differentiation. PLoS One 2012; 7:e33468; PMID:22432027; http://dx.doi.org/10.1371/journal.pone.0033468
- MacMillan-Crow LA, Crow JP, Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 1998; 37:1613-22; PMID:9484232; http://dx.doi.org/10.1021/bi971894b
- Redondo-Horcajo M, Romero N, Martinez-Acedo P, Martinez-Ruiz A, Quijano C, Lourenco CF, Movilla N, Enriquez JA, Rodriguez-Pascual F, Rial E, et al. Cyclosporine A-induced nitration of tyrosine 34 MnSOD in endothelial cells: role of mitochondrial superoxide. Cardiovasc Res 2010; 87:356-65; PMID:20106845; http://dx.doi.org/10.1093/cvr/cvq028
- Holley AK, Dhar SK, St Clair DK. Manganese superoxide dismutase vs. p53: regulation of mitochondrial ROS. Mitochondrion 2010; 10:649-61; PMID:20601193; http://dx.doi.org/10.1016/j.mito.2010.06.003
- Bakthavatchalu V, Dey S, Xu Y, Noel T, Jungsuwadee P, Holley AK, Dhar SK, Batinic-Haberle I, St Clair DK. Manganese superoxide dismutase is a mitochondrial fidelity protein that protects Polgamma against UV-induced inactivation. Oncogene 2012; 31:2129-39; PMID:21909133; http://dx.doi.org/10.1038/onc.2011.407
- Devenish RJ, Prescott M, Boyle GM, Nagley P. The oligomycin axis of mitochondrial ATP synthase: OSCP and the proton channel. J Bioenerg Biomembr 2000; 32:507-15; PMID:15254386; http://dx.doi.org/10.1023/A:1005621125812
- Symersky J, Osowski D, Walters DE, Mueller DM. Oligomycin frames a common drug-binding site in the ATP synthase. Proc Natl Acad Sci U S A 2012; 109:13961-5; PMID:22869738; http://dx.doi.org/10.1073/pnas.1207912109
- Pagliarani A, Nesci S, Ventrella V. Modifiers of the oligomycin sensitivity of the mitochondrial F1F0-ATPase. Mitochondrion 2013; 13:312-9; PMID:23597783; http://dx.doi.org/10.1016/j.mito.2013.04.005
- Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, Shadel GS. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J Clin Invest 2007; 117:2723-34; PMID:17786248; http://dx.doi.org/10.1172/JCI31604
- Nowakowski RS, Lewin SB, Miller MW. Bromodeoxyuridine immunohistochemical determination of the lengths of the cell cycle and the DNA-synthetic phase for an anatomically defined population. J Neurocytol 1989; 18:311-8; PMID:2746304; http://dx.doi.org/10.1007/BF01190834
- Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron 2003; 39:749-65; PMID:12948443; http://dx.doi.org/10.1016/S0896-6273(03)00497-5
- Thiel G. How Sox2 maintains neural stem cell identity. Biochem J 2013; 450:e1-2; PMID:23445224; http://dx.doi.org/10.1042/BJ20130176
- Xapelli S, Agasse F, Sarda-Arroyo L, Bernardino L, Santos T, Ribeiro FF, Valero J, Braganca J, Schitine C, de Melo Reis RA, et al. Activation of type 1 cannabinoid receptor (CB1R) promotes neurogenesis in murine subventricular zone cell cultures. PLoS One 2013; 8:e63529; PMID:23704915; http://dx.doi.org/10.1371/journal.pone.0063529
- Shen Q, Zhong W, Jan YN, Temple S. Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development 2002; 129:4843-53; PMID:12361975