
Abstract
Kaposi's sarcoma herpesvirus (KSHV)-encoded v-cyclin, a homolog of cellular cyclin D2, activates cellular CDK6, promotes G1-S transition of the cell cycle, induces DNA damage, apoptosis, autophagy and is reported to have oncogenic potential. Here we show that in vivo expression of v-cyclin in the B- and T-cell lymphocyte compartments results in a markedly low survival due to high penetrance of early-onset T-cell lymphoma and pancarditis. The v-cyclin transgenic mice have smaller pre-tumorigenic lymphoid organs, showing decreased cellularity, and increased proliferation and apoptosis. Furthermore, v-cyclin expression resulted in decreased amounts of CD3-expressing mature T-cells in the secondary lymphoid organs concurrent with alterations in the T-cell subpopulations of the thymus. This suggests that v-cyclin interferes with normal T-cell development. As the Notch pathway is recognized for its role in both T-cell development and lymphoma initiation, we addressed the role of Notch in the v-cyclin-induced alterations. Fittingly, we demonstrate induction of Notch3 and Hes1 in the pre-tumorigenic thymi and lymphomas of v-cyclin expressing mice, and show that lymphoma growth and viability are dependent on activated Notch signaling. Notch3 transcription and growth of the lymphomas was dependent on CDK6, as determined by silencing of CDK6 expression or chemical inhibition, respectively. Our work here reveals a viral cyclin-CDK6 complex as an upstream regulator of Notch receptor, suggesting that cyclins can play a role in the initiation of Notch-dependent lymphomagenesis.
Keywords:
Abbreviations
| Cdk6 | = | cyclin dependent kinase 6 |
| GSI | = | γ-secretase inhibitor |
| KS | = | Kaposi's sarcoma |
| KSHV | = | Kaposi's sarcoma herpesvirus |
| NICD1 | = | Notch1 intracellular domain |
| NICD3 | = | Notch3 intracellular domain |
| PEL | = | primary effusion lymphoma |
| PI | = | propidium iodide |
| v-cyclin | = | viral cyclin |
Introduction
Kaposi`s sarcoma herpesvirus (KSHV) belongs to the family of lymphotropic gammaherpesviruses and is an etiologic agent of 3 malignancies, namely Kaposi`s sarcoma (KS), primary effusion lymphoma (PEL) and some forms of multicentric Castleman`s Disease (MCD).Citation1-3 PELs are lymphomas with unique clinical, morphologic, and immunophenotypic features. They appear as pleural, pericardial or peritoneal effusions,Citation4 and show aggressive behavior with a median survival of only 6 months after diagnosis.Citation5 Although the majority of PELs exhibit an indeterminate (non B- non T-cell) immunophenotype, immunogenotypic studies have demonstrated both immunoblastic and plasma cell features defining a B-cell origin.Citation6 However, rare KSHV-associated T-cell lymphomas have also been documented, and PELs with an aberrant T-cell genotype or phenotype have been characterized.Citation7-9 CD138, also called syndecan-1 (Sdc1), is a marker of plasma cells and considered a specific marker for KSHV-associated primary effusion lymphomas (PEL),Citation10,11 and at least part of the KSHV-associated T-cell lymphomas have been shown to express it.Citation7
Abnormal Notch signaling is associated with KSHV-induced malignancies,Citation12 and KS tumors overexpress several Notch pathway components, including the ligands, Notch receptors and the target genes Hes-1 and Hey-1.Citation13,14 Moreover, Liu et al. have shown that exogenous expression of a plethora of KSHV encoded genes, namely LANA, vFLIP, RTA, vGPCR and vIL6 can induce expression of the Notch pathway components in HEK293T cells.Citation14 Activation of the Notch ligands Jagged1 and Dll4 by viral proteins vFLIP and vGPCR, has also been demonstrated in an endothelial cell model.Citation15 In addition, KSHV latency protein LANA-dependent accumulation of the active Notch1 intracellular domain (NICD1) in PEL cells was shown to result in increased proliferation.Citation16,17 Furthermore, primary KS tumor cells and experimental lesions in mice were sensitive to inhibition of Notch signaling by γ-secretase inhibitors (GSIs),Citation13,18 which block the proteolytic processing of the full-length Notch receptor to NICD. GSI treatment of the KSHV-infected PEL cell lines leads to G1 cell cycle arrest in vitroCitation16 and induces cell death in PEL xenografts in mice.Citation19 These data suggest that inhibition of activated Notch signaling could have therapeutic potential for both KS tumors and KSHV-induced PELs.
KSHV encodes a viral cyclin (v-cyclin, v-cyc), which is a cellular cyclin D2 homolog and associates with cellular cyclin dependent kinase 6 (CDK6).Citation20 v-cyclin is expressed both during KSHV latency and lytic replication phase. Binding of v-cyclin activates CDK6 to phosphorylate the retinoblastoma protein (pRb)Citation21 leading to accelerated S-phase entry in cultured cells.Citation22 Besides pRb, the v-cyclin-CDK6 complex phosphorylates a plethora of other substrates. These include substrates normally phosphorylated by the S-phase cyclin E-CDK2 complex including histone H1, CDC6, ORC1Citation23-25 and Bcl-2.Citation26 v-cyclin-CDK6 is insensitive to several cell cycle control mechanisms, thus deregulating the cell cycle. Evasion of the cell cycle control is at least partially attributed to the capacity of v-cyclin-CDK6 to phosphorylate and thereby inactivate the CDK inhibitors p27Kip1 and p21Cip1.Citation23,25,27-29 In addition, v-cyclin has recently been shown to promote KSHV-induced post-confluent growth and transformation in rat mesenchymal cells,Citation30 and to counteract the G1-arrest triggered by another latent viral protein, vFLIP, through NF-κB hyperactivation.Citation31 However, v-cyclin not only promotes proliferation, as its overexpression also induces a p53-dependent growth arrest and apoptosis,Citation26,32-34 activates the DNA damage response,Citation32 and increases autophagy.Citation35
The in vivo function of v-cyclin in the lymphocyte compartment has previously been addressed by expressing it as a transgene under the immunoglobulin heavy chain promoter/enhancer Eμ in a mixed CBA/C57BL/6 mouse background (Eμ-v-cyclin mice).Citation33,36 Expression of v-cyclin led to development of low penetrance (17%), late onset lymphomas, which was accelerated by p53 deficiency. Considering the multiple functions that have been assigned to v-cyclin in the in vitro cell culture studies,Citation22 the mild oncogenic phenotype in the Eμ-v-cyclin mice was quite surprising.Citation33,36 As the C57BL/6 background used in these studies is considered to be refractory to at least chemically induced tumors,Citation37 we crossbred the Eμ-v-cyclin mice from the mixed C57BL/6 background into ICR (CD1) and assessed the tumorigenic potential of v-cyclin in these mice. Our results show that v-cyclin expression in the ICR (CD1) mouse background leads to abnormal T-cell differentiation as well as early onset T-cell lymphomas in a vast majority of the animals. Furthermore, we show that v-cyclin induces Notch3 receptor expression in mouse pre-tumorigenic thymocytes, and that v-cyclin initiated T-cell lymphomas are dependent on both Cdk4/6 and Notch pathway activities.
Results
v-cyclin expression in thymocytes leads to high penetrance T-cell lymphomagenesis and pancarditis
Eμ-v-cyclin mice, initially generated in a mixed CBA/C57BL/6 mouse backgroundCitation33,36 were bred to the ICR (CD1) genetic background. The Kaplan-Meier analysis of the v-cyclin expressing ICR mice (ICR v-cyclin) revealed low survival (less than 5%) and early-onset disease starting at 1.5 months of age, while the disease-free survival of the non-transgenic ICR littermates (ICR wt) remained 100% during the follow-up period (). As this dramatically differed from the reported 83% survival of the CBA/C57BL/6-Eμ-v-cyclin mice,Citation33,36 we ruled out the possibility of mutations in the major tumor suppressors p53 or p19Arf by sequencing. The 10 Trp53 and 2 Cdkn2a exons containing most of the hot spot mutationsCitation38 were devoid of mutations in the ICR mice (data not shown). Moreover, when the ICR-Eμ-v-cyclin mice were backcrossed with C57BL/6 mice to generate C57BL/6-Eμ-v-cyclin mice (BL6 v-cyclin), the v-cyclin-associated disease phenotype was reverted to that observed in the original mixed background (Fig. S1A), suggesting that the decreased survival in v-cyclin mice was dependent on the ICR background. A comparison of the expression levels of v-cyclin in thymi of 5-week old mice in the 2 different backgrounds showed that v-cyclin expression was 2.5 to three-fold higher in ICR mice (Fig. S1B), which could partially contribute to the phenotype in ICR-Eμ-v-cyclin mice.
Figure 1. v-cyclin expression leads to T-cell lymphomas and pancardial inflammation. (A) Kaplan-Meyer survival charts of v-cyclin expressing (Eμ-v-cyclin, n = 40) and littermate control animals (wt, n = 28) in the ICR (CD1) mouse background. (B) Hematoxylin and eosin (H&E) stained sections of Eμ-v-cyclin lymphomas in i) thymus (low magnification), ii) thymus (high magnification), iii) liver, and iv) lung. (C) Immunohistochemistry of a representative Eμ-v-cyclin thymus lymphoma with antibodies against CD3 and B220. (D) FACS analysis of Eμ-v-cyclin lymphoma cell lines isolated from 3 lymphoma-bearing mice (v-cyc1, v-cyc2, v-cyc3) with the indicated combinations of antibodies against CD4 and CD8. T = thymus, S = spleen, LN = lymph node. (E) mRNA expression levels of CD138 in the cell lines in (D) compared and normalized to wt control thymus (n = 2). A thymic lymphoma cell line from an Eμ-Myc mouse (Eμ-Myc) was used as a control. (F) Sections of a representative Eμ-v-cyclin heart affected with pancarditis and stained with i) and ii) H&E, low and high magnification, respectively; and antibodies against iii) CD3 and iv) B220. Magnifications in (B); i, iii, iv: x10, ii: x40; in (C): x10 (insert x50). Scale bars in (F): i: 400 μm; ii, iii, and iv: 50 μm. Error bars in (E): s.e.m., n = 2. p-values: *P < 0.05, **P < 0.01, ***P < 0.001.
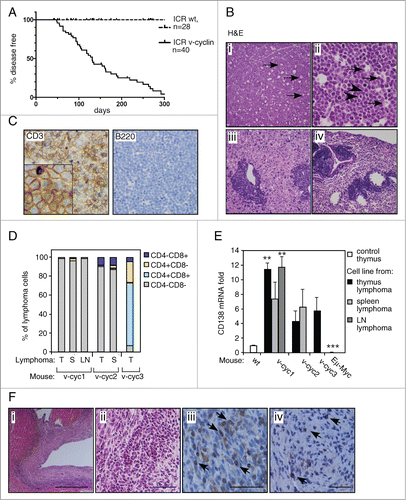
Examination of ICR-Eμ-v-cyclin mice by necropsy (n = 27) and histology (n = 13) showed that 74% of the diseased animals displayed signs of lymphoma (), mostly in the thymus and spleen (85% and 65% of lymphoma-bearing animals, respectively). The lymphomas showed diffuse proliferation of monotonous, intermediate sized lymphoid cells with numerous scattered tingible body macrophages (arrows) and mitotic figures (arrowheads) (, panels i and ii). Neoplastic lymphoid cells showed round to slightly irregular nuclei with finely clumped, dispersed chromatin and multiple basophilic nucleoli. In about a third of the lymphoma-bearing mice, the lymphoma had spread to liver and lungs, with tumor cells appearing around blood vessels or sinusoids in the liver and in perivascular and peribronchiolar spaces in the lung (, panels iii and iv). Tumor cells were occasionally detected in lymph nodes. All lymphomas examined by immunohistochemistry, IHC, (10/10) were composed of lymphomatous T-cells cells positive for CD3, a pan-T-cell marker recognizing the components of the TCR complex, and were negative for the B-cell marker B220 ().
We next established 6 cell lines from primary lymphomas isolated from different organs of 3 tumor-bearing animals. FACS analysis indicated variation in the CD4/CD8 status of the thymic lymphomas, with 2 of the animals containing tumors composed of mostly immature CD4/CD8 double negative (DN) T-cells (), and the third consisting primarily of CD4/CD8 double positive (DP) cells (). These immunophenotypes indicate an early T-cell stage and together with the histological blastic appearance, are consistent with a classification as lymphoblastic T-cell lymphomas. As also the rare KSHV-associated T-cell lymphomas have been shown to express the plasma cell marker CD138, we next analyzed the expression of CD138 mRNA in v-cyclin mouse lymphomas by qRT-PCR. Intriguingly, all thymic v-cyclin lymphoma cell lines showed a 10-fold higher expression of CD138 when compared to wt control thymus (). Control lymphoma cells isolated from Eμ-Myc thymic lymphoma showed no expression of CD138 (). This suggests that v-cyclin-induced lymphoma cells express markers of both immature T-cells, as well as plasma/PEL cells.
We noticed during follow-up that a large proportion (52%) of v-cyclin-expressing animals showed signs of insufficient heart function at young age (1–6 months). At necropsy, pancarditis was detected in all of these animals, with around half of them displaying no detectable lymphoma (). The inflammation suffused extensive areas of the hearts (, panels i and ii, H&E stainings) and, in all animals examined by IHC (6/6), primarily consisted of CD3-positive T-cells (, panel iii, examples of the CD3-positive cells are pointed by arrows) with occasional involvement of B220-positive B-cells (, panel iv, examples of the B220-positive cells are pointed by arrows). As this inflammatory state contributed to the poor survival of the ICR-Eμ-v-cyclin mice, and the T-cells were detected also in hearts of animals with no tumors, the data indicates that T-cell escape from the thymus or spleen is an early event and suggests that both T-cell function and immune system may be severely compromised in these animals.
v-cyclin is expressed and activates its kinase partner Cdk6 in vivo
We next analyzed expression of the transgene and observed that v-cyclin mRNA was expressed at almost 700-fold lower level in the pre-tumorigenic thymus than in the spleen leading to a markedly reduced protein expression ( and S2A). To assess if the lymphoma cells retained v-cyclin expression, we compared v-cyclin expression levels to those of pre-tumorigenic thymi and spleens by qRT-PCR. The transcripts of v-cyclin remained at similar low levels in the thymi and the thymic lymphomas, and were 50-fold further decreased in the thymic Eμ-v-cyclin lymphoma cell lines ( left panel). The splenic lymphomas exhibited significantly lower v-cyclin mRNA and protein expression levels than the pre-tumorigenic spleens, probably reflecting the changes in the cellular composition toward a higher T-cell fraction, which is in accordance with the elevated CD3 expression in the splenic lymphomas ( right panel and 2C). The splenic Eμ-v-cyclin lymphoma cell lines showed similar low v-cyclin expression levels as the thymic lymphoma cell lines ( right panel). No v-cyclin mRNA was detected in the control wt mice or Eμ-Myc cell line (data not shown).
Figure 2. v-cyclin transgene is expressed and functional in the splenocytes isolated from the Eμ-v-cyclin mice. qRT-PCR analysis of v-cyclin mRNA expression (A) in 5-week old Eμ-v-cyclin mouse pretumorigenic thymi (n = 4) and spleens (n = 3), and normalized to the thymus expression levels set to one and (B) in thymic and splenic lymphomas (n = 3 and n = 4, respectively) compared and normalized to the respective pre-tumorigenic organs of 5-week old mice (thymus n = 4, spleen n = 3). (C) Protein levels of FLAG-v-cyclin, Cdk6 and CD3 were analyzed by immunoblotting of total cell lysates prepared from isolated splenocytes of Eμ-v-cyclin (v-cyc), control mice (wt), and cells isolated from v-cyclin splenic lymphomas. γ-tubulin served as a loading control. (D) In vitro kinase assay using GST-Rb and Histone H1 as substrates. Prior to the kinase reaction, isolated splenocytes from Eμ-v-cyclin (v-cyc TG; +) and control mice (−) were immunodepleted with antibodies against Cdk2, Cdk4, and Cdk6 followed by immunoprecipitation of v-cyclin by anti-FLAG antibodies. Kinase activity was determined by autoradiography after SDS-PAGE (12%) followed by immunoblotting with antibodies against FLAG. Total cell extracts showing the input served as controls for the immunoprecipitated proteins. Error bars in (A), (B): s.e.m. p-values: *P < 0.05, **P < 0.01, ***P < 0.001.
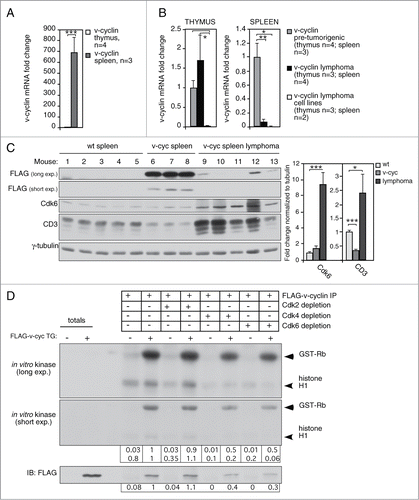
As the numerous in vitro studies have shown that v-cyclin mediates its known functions through activation of CDK6,Citation22 we next analyzed the levels of Cdk6 from pre-tumorigenic organs and v-cyclin lymphomas. In accordance with our recent study demonstrating NF-κB activation by v-cyclin-Cdk6,Citation39 2.3-fold higher Cdk6 protein levels were detected in the v-cyclin expressing thymi when compared to the littermate controls (Fig. S2A). Stabilization of Cdk6 by 1.7-fold was also observed in the pre-tumorigenic spleens ( and S2A-B) and especially clearly in splenic lymphomas (). Cdk6 was expressed at higher levels in the thymi than in the spleens irrespective of the v-cyclin transgene expression (Fig. S2A). This suggests that although v-cyclin is expressed at low levels in the thymus, it is possible that the v-cyclin mediated Cdk6 stabilization contributes to the stronger phenotypes observed in the thymus than spleen.
To confirm that the expressed v-cyclin was functional and capable to activate Cdk6 in our transgenic model, we isolated splenocytes from the Eμ-v-cyclin and control mice, and first individually immunodepleted Cdk2, Cdk4, or Cdk6 from the cell extracts (). Efficient immunodepletion of the individual Cdks was confirmed by immunoblotting with Cdk2, Cdk4 and Cdk6 antibodies (Fig. S2B). We then immunoprecipitated v-cyclin from the Cdk-depleted and control extracts and subjected the immunoprecipitates (IPs) to an in vitro kinase assay using GST-Rb and Histone H1 as substrates. Phosphorylation of both substrates was evident in the IPs of Eμ-v-cyclin splenocytes, whereas only a weak background signal could be detected in the long exposure of the filter when IPs were performed from wild type (wt) splenocytes (, data from one representative mouse of each genotype is shown, n = 3). However, depletion of Cdk6 or Cdk4, but not Cdk2, from the Eμ-v-cyclin splenocyte extracts resulted in a 2.5 to 3-fold decrease in both v-cyclin levels (detected by its FLAG-tag) and diminished phosphorylation of GST-Rb and Histone H1 by 50% and 80–99%, respectively (). As Cdk4 immunodepletion also reduced the levels of Cdk6 (50% depleted, Fig. S2B), it is possible that the detected reduction in v-cyclin levels and kinase activity in the Cdk4 depleted samples is actually due to the co-depletion of Cdk6 rather than specific v-cyclin-dependent Cdk4 activity.
v-cyclin-induced lymphomas are dependent on the Cdk4/6 activity
To address if the activity of v-cyclin cellular kinase partner Cdk6 was necessary for the growth of the Eμ-v-cyclin lymphoma cells, lymphoma cell lines isolated from 3 Eμ-v-cyclin and one control Eμ-Myc mice were treated with a Cdk4/6 kinase inhibitor PD0332991 (PD). The efficacy of PD treatment was first demonstrated by a 10-fold reduction in cyclin A protein levels after treatment (Fig. S3A), indicating a G1 arrest in the cell cycle due to Cdk4/6 inhibition. Interestingly, PD treatment resulted in a growth arrest in all Eμ-v-cyclin derived lymphoma lines examined (see one representative example in ). To investigate if the PD treatment compromised the viability of the lymphoma cells, we determined the number of dead cells by trypan blue exclusion. The low concentration PD treatment induced significant cell death in all Eμ-v-cyclin lymphomas, as there was a 10–50% increase in cell death after 48 hours of treatment (). In contrast, PD had no effect on cell viability of the Eμ-Myc derived, v-cyclin negative control lymphoma (2.5% cell death; ), suggesting that Cdk6 activity is required for the growth and survival of v-cyclin-induced lymphomas.
Figure 3. Inhibition of the CDK4/6 activity induces cell death in Eμ-v-cyclin lymphomas and BCBL-1 cells. (A) Number of live cells in a representative Eμ-v-cyclin lymphoma cell line left untreated or treated with vehicle control (DMSO) or 0.5 μM PD033291(PD) for 3 d (d0–d3). (B) Cell death in Eμ-v-cyclin lymphoma cell lines (v-cyc1, v-cyc2, v-cyc3) and a control Eμ-Myc thymic lymphoma cell line (Eμ-Myc), all treated with 0.5 μM PD for 2 d. The percentage of cell death in the corresponding vehicle (DMSO) treated samples is subtracted from the PD-treated samples as a background. LN = lymph node. (C) Number of live cells in BCBL-1 cells treated with vehicle control (DMSO) or 0.5 μM or 1 μM PD, for 3 d (d0–d3). (D) Cell death in BCBL-1 cells treated with 0.5 μM or 1 μM PD for 2 d. Background cell death in the corresponding vehicle (DMSO)-treated samples was subtracted as in (B). Error bars in (B) and (D): s.e.m., n = 2–4. p-values: *P < 0.05, **P < 0.01, ***P < 0.001.
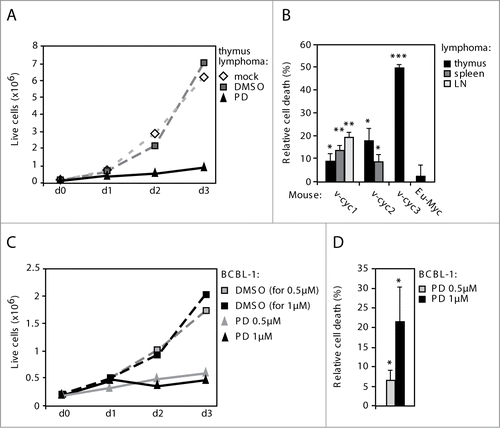
As v-cyclin has been shown to bind and activate CDK6 also in patient-derived KSHV-infected BCBL-1 PEL cells,Citation40 we next tested the effect of PD treatment on the growth and viability of BCBL-1 cells. Confirming its efficacy on the v-cyclin-dependent kinase activity, PD treatment led to a 50–90% (with 0.5 μM and 1 μM concentrations of PD, respectively) reduction in phosphorylation of a known v-cyclin-CDK6 substrate, pT199NPM (nucleophosmin)Citation41 (Fig. S3B) as well as a marked reduction in cyclin A levels (Fig. S3B). Importantly, the inhibitor treatment of BCBL-1 cells led to a growth arrest () and a dose-dependent induction of relative cell death by 7% and 22% (0.5 μM and 1 μM, respectively) () when compared to the vehicle control. These results demonstrate that both the v-cyclin-expressing mouse lymphoma cells and the patient-derived lymphoma cells are sensitive to CDK4/6 inhibition.
v-cyclin induces increased proliferation/apoptosis and an intra-S-phase block in pre-tumorigenic lymphoid organs
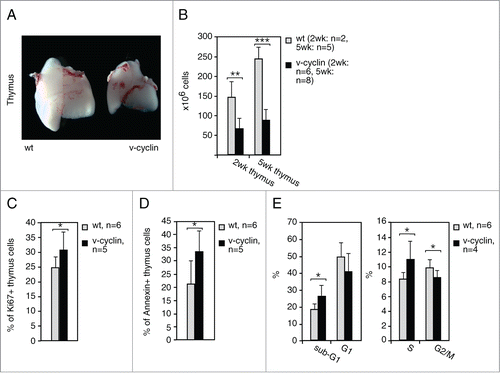
We next sought to investigate the effect of v-cyclin expression in the lymphocyte compartment prior to the onset of lymphomas. Macroscopic analysis of the lymphoid organs revealed that v-cyclin expressing animals had markedly smaller thymi, and a slight decrease in spleen size when compared to wt littermate controls (; Fig. S4A). This was due to decreased cellularity, as both the number of thymocytes and splenocytes in v-cyclin animals were significantly reduced by 55–65% () and 45–60% (Fig. S4B), respectively. As v-cyclin is known to be a strong inducer of the cell cycle, proliferationCitation22 as well as apoptosis,Citation26,32,34 isolated thymocytes were analyzed by FACS for proliferation and apoptosis. A moderate, but significant, increase in proliferation (Ki67) and apoptosis (Annexin V) was observed, suggesting an accelerated turnover of thymocytes in v-cyclin mice (). The cell cycle profiles (propidium iodide, PI) showed an increased proportion of both sub-G1 and S-phase cells (), reflecting increased apoptosis and induction of an intra-S-phase block by v-cyclin. These results suggest that v-cyclin elicits similar effects in T-cells in vivo as seen previously in a variety of in vitro models.Citation22,26,32
Figure 4. v-cyclin increases proliferation and apoptosis and induces an intra S-phase block in vivo. (A) Images of representative thymi from a littermate control (wt) and an Eμ-v-cyclin (v-cyclin) mouse at 5 weeks of age. (B) The number of thymic cells counted from 2-week and 5-week old littermate control (wt, n = 2 and n = 5, respectively) and Eμ-v-cyclin (v-cyclin, n = 6 and n = 8, respectively) mice. (C and D) Quantification of a FACS analysis for proliferation (Ki67 in (C)) and apoptosis (Annexin V in (D)) from the control (wt, n = 6) and v-cyclin (n = 5) thymocytes. Unstained thymocytes were used as background controls. (E) Quantification of a cell cycle analysis by FACS from PI-stained control (wt, n = 4) and v-cyclin (n = 6) thymocytes. Error bars in (B–E): s.d. p-values: *P < 0.05, **P < 0.01, ***P < 0.001.
v-cyclin interferes with development of lymphocytes
The histological comparison of lymphoid organs revealed that the spleens in the v-cyclin mice had a rather normal tissue architecture and similar numbers of B220 expressing B-cells as the non-transgenic controls (Fig. S4C). However, the number of CD3-positive T-lymphocytes was reduced both when analyzed by immunohistochemical staining (Fig. S4C) or by FACS using an anti-CD3 antibody (Fig. S4D). To address the differentiation state of T-cells, we performed multi-color FACS analysis of the isolated splenic T lymphocytes using anti-CD4 and -CD8 antibodies. This revealed that the numbers of mature T-cells, i.e. CD4 single positive (CD4+CD8-) and CD8 single positive cells (CD4-CD8+), were significantly diminished in the v-cyclin expressing spleens (Fig. S4D).
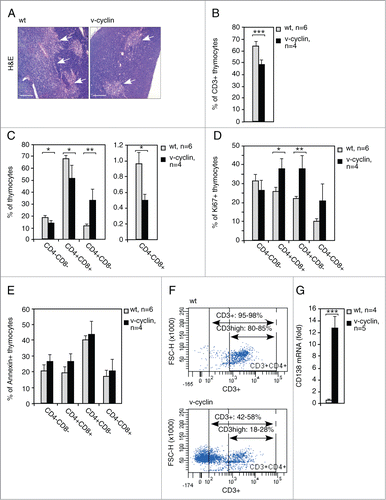
We next assessed T-cell maturation in the thymus of v-cyclin animals. Interestingly, histological stainings showed that the thymic medullae, which normally contain mature T-cells, were markedly smaller in v-cyclin mice (, thymic medullae are pointed by arrows). Fittingly, FACS analysis revealed a significant reduction in the amount of CD3-positive cells in v-cyclin-expressing thymi (65% in the wt vs. 50% in the v-cyclin, ). Next, we analyzed the distribution of T-cell subpopulations using anti-CD4 and -CD8 antibodies and FACS (). Among the v-cyclin thymocytes, the proportional numbers of CD4-CD8-, CD4+CD8+ and CD4-CD8+ cells were decreased, whereas the proportion of CD4+CD8- cells was increased if compared to the distribution of CD4- and CD8-expressing cells among the control thymocytes (). Simultaneous analysis of Ki67 and AnnexinV expression by FACS revealed that the extent of proliferation and apoptosis in these T-cell subpopulations was otherwise similar to those in the whole thymocyte population (), except that the number of Ki67 positive cells in the most immature, CD4-CD8- double negative population was moderately but not significantly decreased (). When single positive CD4+CD8- cells, whose relative amount was 2.8-fold- higher in the v-cyclin expressing thymi over the control, were analyzed in conjunction with anti-CD3, it appeared that they do not represent normal CD4+ T-helper cells as they lack the pan-T-cell marker CD3 expression (). Since the thymic lymphomas showed an interesting expression pattern of both T-cell markers and the plasma cell and PEL marker CD138, we decided to study the expression of CD138 also in the pre-tumorigenic thymi. Interestingly, an approximate ten-fold increase in the CD138 transcript was detected in v-cyclin expressing thymi when compared to littermate controls (). These data suggest that v-cyclin expression in thymus leads to a dramatic reduction of normal T-cell populations, shifting to cell populations not normally found in thymi at such high proportions.
Figure 5. v-cyclin expression alters the thymic morphology and interferes with T-cell differentiation. (A) Representative images of the H&E stained control (wt) and Eμ-v-cyclin (v-cyclin) thymi prior to the onset of lymphomagenesis. The thymic medullae are indicated by arrows. (B) Quantification of FACS analysis with anti-CD3 antibodies of the thymi from 5-week old littermate control (wt, n = 6) and Eμ-v-cyclin (v-cyclin, n = 4) mice. (C) The thymi in (B) stained with combinations of anti-CD4 and -CD8 antibodies and analyzed by FACS (quantifications are shown). Unstained thymocytes were used as background controls. (D and E) FACS analysis of cells positive for Ki67 (D) and AnnexinV (E) in the CD4- and CD8-expressing thymocyte subpopulations in (C). (F) Analysis of the CD3 status in the CD4+CD8+ cells of the thymi in (B). Percentages of the CD3+CD4+CD8+ and CD3high+CD4+CD8− cells, as well as representative examples of the FACS plots are shown. (G) qRT-PCR analysis of CD138 mRNA levels in the pre-tumorigenic thymi of 5-week old littermate control (wt, n = 4) and Eμ-v-cyclin (v-cyclin, n = 5) mice. Scale bars in (A): 200 μm. Error bars in (B): s.d.; in (C–E) and (G): s.e.m. p-values: *P < 0.05, **P < 0.01, ***P < 0.001.
Notch3 is induced in pre-tumorigenic thymi and thymic lymphomas of Eμ-v-cyclin mice
To gain further insight into the mechanisms driving lymphomagenesis in the ICR-Eμ-v-cyclin mice, we next addressed expression of the Notch pathway components due to its well-established role in T-cell development and lymphoma initiation.Citation42,43 Fittingly, a 3.4-fold increase of Notch3 receptor mRNA levels was observed in pre-tumorigenic thymi of the v-cyclin expressing animals as compared to the non-transgenic (wt) littermates () along with a further 1.9-fold upregulation in the thymic lymphomas (). Notably, expression of Notch1 mRNA was found to be modestly but not significantly downregulated in pre-tumorigenic organs of v-cyclin mice, but showed a clear induction in the thymic lymphomas (), suggesting that its induction occurs at a later stage of lymphomagenesis. The increase in Notch3 expression was also confirmed at the protein level by immunostaining of thymus sections from these mice (). To investigate if Notch3 induction leads to pathway activation, we analyzed the protein levels of the activated Notch3 intracellular domain (NICD3) and found it to be specifically expressed in the v-cyclin expressing thymi () as well as in 3 out of 4 of the v-cyclin expressing lymphomas (). In accordance with this, one of the Notch downstream targets, Hes1, was found to be significantly upregulated in the v-cyclin expressing thymi (2.9-fold, ) and thymic lymphomas (16.3-fold, ). Similar induction of another known Notch target, Hey1, was detected in the pre-tumorigenic v-cyclin expressing thymi, but the increase was not significant probably due to intrinsically low expression levels in these samples (). Multiple tested Notch targets were not expressed at all or expressed at very low levels in the pre-tumorigenic thymi (Hey2 and Hes5, data not shown) or thymic lymphomas (Hey1, Hey2 and Hes5, and data not shown). In line with the Notch pathway activation, cyclin D3, another downstream target of the Notch pathway, was found to be upregulated in the v-cyclin expressing spleens and splenic lymphomas ().
Figure 6. Notch signaling is induced in v-cyclin expressing pre-tumorigenic thymi and lymphomas. (A) Notch3 and Notch1 mRNA expression levels in the pre-tumorigenic thymi of 5-week old littermate control (wt, n = 5) and Eμ-v-cyclin (v-cyclin, n = 5) mice, and in Eμ-v-cyclin thymic lymphomas (n = 3). (B) Immunohistochemistry with anti-Notch3 antibody (red) and counterstained with Hoechst (blue) of the thymi in (A). Sections stained without the primary antibody (1Ab omitted) were used as a control. (C) Immunoblotting with antibodies against NICD3 from cell extracts prepared from pre-tumorigenic thymi of control (wt, n = 4) and Eμ-v-cyclin (v-cyclin, n = 4) mice, and from Eμ-v-cyclin thymic lymphomas (n = 4). γ-tubulin served as a loading control. (D) Notch pathway target Hes1 and Notch1 mRNA expression levels in the thymi in (A). (E) The membrane analyzed in was further immunoblotted with anti-cyclin D3 antibodies and the expression levels were analyzed in splenocytes of Eμ-v-cyclin (v-cyc) and control mice (wt), as well as lymphoma cells isolated from the v-cyclin spleens. Quantification of the signal normalized to γ-tubulin is shown in the lower panel. (F) qRT-PCR analysis of CDK6, NOTCH3, NOTCH1, HEY1, and HES1 in 293HEK cells transduced with lentiviruses expressing scrambled shRNA (scr) or 2 different shCDK6 (sh-CDK6_1 and sh-CDK6_2). Error bars in (A) and (D–F): s.e.m., n = 2–4. p-values: *P < 0.05, **P < 0.01, ***P < 0.001.
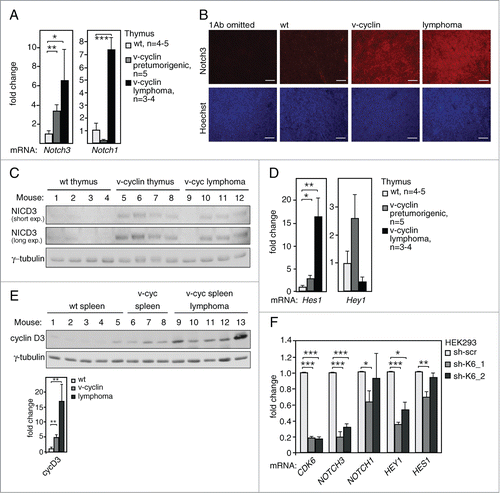
Notch3 transcription is regulated by CDK6
To gain more insight into the mechanism of v-cyclin-mediated induction of Notch3, we tried to establish several systems with transient and inducible v-cyclin expression in a variety of different cellular backgrounds. However, as v-cyclin has been demonstrated to induce apoptosis, cell cycle arrest, DNA damage and autophagy in a variety of cellular backgrounds,Citation26,32-35 it turned out to be challenging to establish a relevant and reliable cell model to study the molecular mechanism of Notch3 induction in v-cyclin-expressing cells. Therefore, we decided to turn our attention to the v-cyclin kinase partner CDK6 and assessed the role of CDK6 in regulation of NOTCH3 transcription. To this end, we silenced CDK6 in HEK293 cells by 2 different shRNA constructs against CDK6. Silencing of CDK6 was efficient (85% in HEK293 ) and led to a 70–80% inhibition in NOTCH3 mRNA levels (). This effect was specific for NOTCH3 as there was no consistent effect on NOTCH1 mRNA with these 2 constructs (). In addition, CDK6 silencing induced a significant downregulation of the Notch downstream target HEY1 (), and a minor effect on HES1, possibly reflecting that it is regulated more by NOTCH1 (). To address if inhibition of NOTCH3 transcription was CDK6-specific we silenced CDK4, another G1 cell cycle regulating kinase. Interestingly, efficient depletion of CDK4 expression (80%) had no effect on NOTCH3 expression, thus suggesting that regulation of NOTCH3 expression was specific for CDK6 (data not shown). Next, we tested whether the kinase activity of CDK6 was needed for NOTCH3 transcription by treating the HEK293 cells with PD inhibitor. The inhibition led to 40% reduction in NOTCH3 levels and significantly downregulated levels of the Notch pathway targets HEY1 and HES, but CDK6 and NOTCH1 levels were not affected (Fig. S5). This suggests that the Notch pathway activation by CDK6 is kinase-dependent.
Eμ-v-cyclin lymphomas are dependent on active Notch signaling
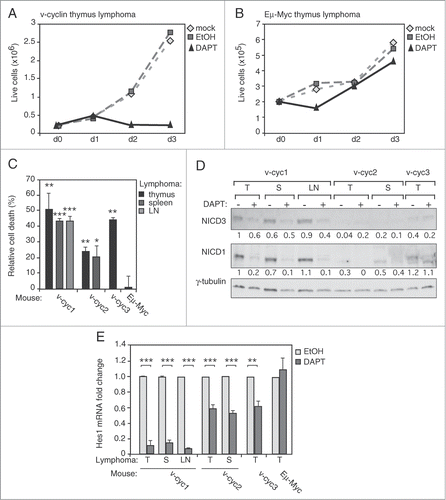
To assess if the activated Notch sustains growth and survival of v-cyclin lymphoma cells, we treated the cells with 10 μM gamma-secretase inhibitor DAPT for 72 hours. DAPT treatment resulted in a growth arrest in all 3 Eμ-v-cyclin thymic lymphoma lines examined (see a representative graph in ), but had no effect on growth of the control Eμ-Myc thymic lymphoma cells (example graph in ). DAPT also significantly increased cell death of v-cyclin lymphoma cells at 48 h () as indicated by a 25–55% increase in the number of dead cells when compared to vehicle treated cells. Similar results were obtained with lymphoma lines isolated from the spleens and lymph nodes (). DAPT treatment of the v-cyc1 lymphoma cells with initial high expression levels of the activated NICD3 and NICD1, led to a 20–54% and 80–90% reduction in their expression, respectively (). The inhibitory effect of DAPT on generation of NICD3 and NICD1 was less evident in the other lymphoma cell lines probably due to lower protein expression levels of the Notch receptors (). To further prove the efficacy of the inhibitor we perfomed qRT-PCR analysis which showed 50–90% inhibition of the Notch1 downstream target Hes1 mRNA after the DAPT treatment (). These data suggest that growth and viability of Eμ-v-cyclin lymphomas are dependent on activation of the Notch pathway.
Figure 7. Eμ-v-cyclin lymphoma cells are dependent on Notch signaling. (A) Number of live cells in a representative Eμ-v-cyclin lymphoma cell line left untreated (mock), or treated with a vehicle control (EtOH) or 10 μM DAPT for 3 d (d0–d3). (B) Number of live cells in control Eμ-Myc lymphoma cells treated as in (A). (C) Cell death in Eμ-v-cyclin lymphoma cell lines (v-cyc1, v-cyc2, v-cyc3) and in a control Eμ-Myc cell line (Eμ-Myc), all treated with 10μM DAPT for 2 d. Dead cells in the corresponding vehicle (EtOH) treated samples are subtracted from the DAPT-treated samples as a background. LN = lymph node. (D) Immunoblotting using anti-NICD3 and -NICD1 antibodies from extracts prepared from Eμ-v-cyclin lymphoma cell lines (v-cyc1, v-cyc2, v-cyc3) treated as in (C). Quantifications of the signals are shown under blots and are normalized to γ-tubulin, which serves as a loading control. T = thymus, S = spleen, LN = lymph node. (E) Hes1 mRNA levels analyzed by qRT-PCR in Eμ-v-cyclin lymphoma lines (v-cyc1, v-cyc2, v-cyc3) and in the control Eμ-Myc lymphoma line after vehicle (EtOH) or 10 μM DAPT treatment for one day. Expression levels are normalized to vehicle-treated Eμ-v-cyclin thymic lymphoma cells (v-cyc1). T = thymus, S = spleen, LN = lymph node. Scale bars in (B): 50 μm. Error bars in (C) and (E): s.e.m., n = 3. p-values: *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
T-cell development and lymphomagenesis are closely linked, and aberrations in the carefully regulated molecular events of T-cell differentiation can trigger T-cell lymphomagenesis. For example, expression of oncogenes at specific differentiation stages leads to developmental arrest and accumulation of immature T-lymphocytes.Citation42,44 In our mouse model, v-cyclin functions as an oncogene in the T-cells, leading to defects in their maturation and initiating early-onset T-cell lymphoma and T-cell autoimmunity. It is somewhat surprising that the v-cyclin mice have defects in thymic development and T-cells, although v-cyclin expression levels are dramatically lower in thymocytes than splenocytes. This suggests that v-cyclin expression targets molecules and pathways that are particularly important for the development of T-lymphocytes and T-cell lymphoma. These are likely mediated through activation of the v-cyclin cellular kinase partner CDK6. CDK6 is known to be highly active in T-cells and important for the orderly development of the thymus,Citation45-47 and of its cellular cyclin partners cyclin D3 shares unique functions in lymphocyte development.Citation48 Moreover, CDK6 expression levels are often increased in T-cell lymphomas,Citation49 and cyclin D3-CDK6 activity has been shown to be essential for T-cell lymphomas.Citation48 Fittingly, we see in this mouse model that v-cyclin expression in T-lymphocytes elevates both the Cdk6 and cyclin D3 expression levels and they are further upregulated in the lymphomas (, , S2A, S2B and ref.Citation39). As the v-cyclin expression levels are low at the lymphoma stage, it is possible that v-cyclin initiates the changes that affect the cellular targets and pathways important for T-cell development and lymphoma, but it is the elevated cyclin D3-CDK6 that is responsible for the proliferation and growth of the lymphomas.
Importantly, we show here that inhibition of CDK6 activity induces cell death in the Eμ-v-cyclin lymphoma cells and BCBL-1 PEL cells, suggesting that growth and viability of both lymphoma types are dependent on CDK6 activity. Appropriately, CDK4/6 inhibition has shown promising results in therapeutic targeting of both B- and T-cell lymphomas,Citation48,50 and CDK4/6 inhibitors are now in phase III clinical trials for estrogen receptor (ER)-positive breast cancer and chronic lymphocytic leukemia.Citation50,51 This implies that new generation CDK4/6 inhibitors may provide significant therapeutic potential in the near future, and our data suggests that testing these inhibitors in KSHV-positive patients with PELs may improve prognosis of this currently incurable disease.
Besides Cdk6, the Notch signaling pathway plays a critical role in T-cell development.Citation43 This is the first study to describe that KSHV v-cyclin expression activates Notch signaling through induction of Notch3 and Hes1 (). Notch signaling is a tightly regulated process with a recognized role in the initiation of T-cell acute lymphoblastic leukemia/lymphoma (T-ALL).Citation42,52 Several studies have confirmed that Notch is an important regulator of haematopoietic progenitor commitment to the T-cell lineage, and is involved in the DN to DP transition.Citation42 Activating mutations of NOTCH1 are found in more than 50% of the human T-ALL cases,Citation53 and several knock in/out animal models support its importance in T-ALL.Citation42 In addition, transgenic mice expressing the constitutively active Notch3 intracellular domain NICD3 have been shown to develop T-cell lymphomas,Citation54,55 indicating that also Notch3 activation can potentially initiate T-cell lymphoma. Therefore, the increased expression of Notch3, its intracellular activated form NICD3, and the Notch pathway targets Hes1, Hey1 and cyclin D3 are likely to play a role in the abnormal T-cell development and initiation of lymphomas in the Eμ-v-cyclin mice. Notably, these T-cell lymphomas are best classified as T-cell lymphoblastic lymphoma, a cancer of immature T-cells highly related to T-ALL. We further demonstrate that Notch3 transcription is dependent on CDK6 expression. Previous studies have established D-type cyclins as downstream effectors of Notch in lymphomagenesis.Citation56-58 Our work here reveals a cyclin-CDK complex as an upstream regulator of Notch receptor transcription, suggesting that also cellular cyclins may not only function as effectors but also have a role in the initiation of Notch-dependent lymphomagenesis.
Besides T-cell lymphoma, Notch signaling reportedly plays an important role in the pathogenesis of KSHV-associated malignancies.Citation12 Several Notch pathway components, including the Notch receptors themselves, are upregulated in KS.Citation13 However, the viral factors inducing the Notch receptors have remained mostly unknown. Here we identify v-cyclin as an additional KSHV viral protein engaging the Notch pathway through induction of a Notch receptor. Importantly, this renders v-cyclin-expressing lymphomatous T-cells dependent on the Notch activity, as chemical inhibition of the pathway resulted in cell death of v-cyclin-driven lymphoma cells. Our study thus reveals new insights in KSHV pathogenesis by linking expression of a viral oncogene to Notch pathway activation, and further supports the earlier reports showing that inhibition of Notch might serve as a good strategy in treating KSHV-induced malignancies.Citation13,16,18,19
It is clear that this mouse model, in which v-cyclin is expressed in isolation of the other viral factors, only covers a small subset of all v-cyclin functions. Yet, this study reveals that v-cyclin expression in vivo not only recapitulates several of its properties identified in cell culture models, but also provides novel insights into KS/PEL pathogenesis. Our data shows that, in an appropriate cellular context, v-cyclin can act as a true oncogene, giving rise to lymphoma when expressed even at low expression levels, without a requirement for additional initiator mutations in the common tumor suppressors. This is especially important as v-cyclin is a latent KSHV gene and, in contrast to the majority of the KSHV genes, expressed constitutively during the viral infection. In addition, here we show for the first time that KSHV v-cyclin modulates the Notch pathway via receptor up-regulation, and demonstrate that v-cyclin expression leads to dysregulated organ development due to inhibition of normal T lymphocyte maturation. Moreover, we report an abnormal induction of the plasma cell/ PEL marker CD138 in the v-cyclin-expressing pre-tumorigenic thymi and lymphomas, which resembles the aberrant T-cell phenotype of some rare cases of PELs.Citation7,8 Lastly, we also demonstrate that growth of these lymphomas can be inhibited with compounds blocking Notch or Cdk4/6, implying that simultaneous inhibition of these 2 targets could improve the currently poor therapeutic outcome of advanced PEL, and perhaps also KS.
Materials and Methods
Eμ-v-cyclin transgenic mice and isolation of mouse lymphocytes
The Eμ-v-cyclin transgenic mouse strain is previously described in refs.Citation33,36 These mice were bred into the outbred ICR (CD1) mouse background for at least 5 generations (ICR- Eμ-v-cyclin) or backcrossed to the inbred C57BL6 background for at least 5 generations (BL6- Eμ-v-cyclin), and maintained as described earlier.Citation33,36 Eμ-Myc mice were generated and maintained as previously described.Citation59 All mouse experiments were approved by Finnish National Animal Experiment Board (license numbers: ESLH-2005–03350/Ym-23, ESLH-2006–04075/Ym-23, ESLH-2009–02139/Ym-23). Thymi and spleens from 2- or 5.5-week old pre-tumorigenic Eμ-v-cyclin mice as well as tumors from lymphoma bearing mice were obtained from the sacrificed mice by dissection. Tissue was disaggregated by pressing through a 70-μm nylon mesh cell strainer (BD Falcon) in RPMI containing 10% FCS to obtain a single cell suspension. Splenic erythrocytes were eliminated by incubation for 5 min at room temperature in ACK buffer (155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA, pH 7.8). Thymic and splenic lymphocytes and lymphoma cells were pelleted and washed once with PBS before further use for FACS as well as RNA lysates. Lymphoma cells used for cell culture were frozen (1 × 107 cells/vial) or cultured in lymphoma medium described in.Citation60
Cell lines
Lymphoma cell lines isolated from Eμ-v-cyclin/ICR mice, as well as control Eμ-Myc thymic lymphoma cultured in lymphoma medium were propagated every second to fourth day, kept at cell density between 2 × 105 to 107 cells/ml and grown for at least 3 passages before further analysis. The BCBL-1 (NIH AIDS Reagent Program, catalog number 3233 from McGrath and Ganem) cells,Citation61 as wells as HEK293FT (obtained from Biomedicum Functional Genomics Unit, FuGU), HEK293A (a kind gift from Dr. Vähä-Koskela) and 293HEK-FlipIn cells (Invitrogen) were cultured as described previously.Citation27 All cells were cultured in a humidified 5% CO2 atmosphere at 37°C.
Lentivirus production and transductions
Lentiviral constructs containing scrambled shRNA (sh-scr: SHC005) and 2 different shCDK6s (sh-CDK6_1: TRCN0000039747, sh-CDK_2: TRCN0000194893) in pLKO.1 vector backbone were obtained from Biomedicum Functional Genomics Unit (FuGU) and the viruses were produced in HEK293FT cells as previously described.Citation32 The HEK293A and HEK293-FlipIn cells were transduced with concentrated viruses as described in.Citation32 The cell pellets for qRT-PCR analysis were collected 48 h after transductions.
Antibodies
In western blotting and immunoprecipitations following antibodies were used: FLAG (M2, Sigma), Cdk2 (sc-163, Santa Cruz), Cdk4 (sc-601, Santa Cruz), Cdk6 (#MS-451, Thermo Scientific), v-cyclin,Citation27 Cdk6 (C-21, Santa Cruz), CD3 (A0452, Dako), Cyclin D3 (sc-182, Santa Cruz), γ-tubulin (GTU-88, Sigma), Cyclin A (sc-596, Santa Cruz), (phosphoT199-NPM (Cell Signaling, #3541), NPM (32–5200, Invitrogen), NICD1 (ab8925, Abcam), NICD3 (sc-7424, Santa Cruz). Antibodies against CD3 (A0452, Dako), B220 (RA3-6B2, SouthernBiotech) and Notch3 (N5038, Sigma) were used to stain tissue sections. For FACS, pre-conjugated antibodies (all from BD PharMingen) against CD4 (PE, H129.9), CD8 (FITC, 53-5.8), CD3 (PE-Cy7, 145-2C11) and Ki-67 (PerCP-Cy5.5, B56) or Annexin V (APC) were used.
In vitro kinase reaction, immunoprecipitation and protein gel blotting
For immunoprecipitations, and their whole-cell lysate controls, cells were lysed in an ELB lysis buffer (50 mM Hepes (pH 7.4), 150 mM NaCl; 50 mM HEPES, pH 7.4; 0.1% Igepal; 5 mM EDTA; 2 mM DTT; 1 mM phenylmethylsulfonyl fluoride [PMSF]; 2 μg/mL leupeptin; 2 μg/mL pepstatin; and 1.5 μg/mL aprotinin). For immunoprecipitations 300–1000 μg of protein were used per sample. Immunodepletions were performed using antibodies against Cdk2, Cdk4 and Cdk6 followed by the immunoprecipitation against v-cyclin by using antibodies against v-cyclin or FLAG and in vitro kinase reaction by using GST-Rb and Histone H1 as substrates as described previously.Citation27 40 μg-75 μg of the cleared whole-cell extracts were analyzed by western blotting as described previously.Citation27 Antibody binding was visualized by enhanced chemiluminescence (ECL, Femto from Pierce or Sirius from Advansta).
Inhibitor assays
Mouse lymphoma cells and PEL cells at a starting density of 1–2 × 105 cells/ml were incubated for 72 h with DAPT (10 μM; Sigma) or PD0332991 (0.5–1 μM; Adooq Bioscience) or corresponding vehicle control (EtOH/DMSO). The number of live and dead cells were determined by trypan blue exclusion and counting with a TC10 Automated cell counter (Bio-rad) at 0 h, 24 h, 48 h and 72 h after adding the inhibitor. Cell pellets for analysis by real time quantitative PCR were collected at 24 h or 48 h and for WB analysis at 48 h.
Real time quantitative PCR
Total RNA was extracted using the RNeasy mini kit (Qiagen) or the NucleoSpin RNA II kit (Macherey Nagel). Transcript levels were measured by qRT-PCR using Taqman Gene Expression Assays (Applied Biosystems) with the following FAM-labeled primers for Notch3 (Mm00435270_m1), Hes1 (Mm01342805_m1), Hey1 (Mm00468865_m1), Gapdh (Mm03302249_g1), HEY1 (Hs00232618_m1) HES1 (Hs00172878_m1) or GAPDH (Hs03929097_g1) (Applied Biosystems) in the StepOnePlus Real Time PCR system (Applied Biosystems) or in the Lightcycler 480 (Roche). For unlabeled primers, reactions were done using the SYBR Green PCR mix (Fermentas) and QuantiTect Primer Assay against NOTCH3 (QT00003374, Qiagen) or the following primer sequences (forward and reverse):
v-cyclin: CGGACGTCACTTCCTTCTTG and CGCAGATCAAAGTCCGAAAC
Notch1: CCGTGTAAGAATGCTGGAACG and AGCGACAGATGTATGAAGACTCA
Gapdh: TCAACGACCCCTTCATTGAC and ATGCAGGGATGATGTTCTGG
CDK6: CCAGATGGCTCTAACCTCAGT and AACTTCCACGAAAAAGAGGCTT
NOTCH1: GAGGCGTGGCAGACTATCATGC and CTTGTACTCCGTCAGCGTGA
GAPDH: TCACCACCATGGAGAAGGCT and GCCATCCACAGTCTTCTGGG
The data was normalized to expression of the Gapdh/GAPDH cellular housekeeping gene.
Statistical analysis
For statistical analysis of the qRT-PCR data logarithmic values were converted to ddCt values (linear log2 scale values) and p values were calculated with a one-tailed unpaired Student's t test. The p values for FACS data were calculated directly from the data normalized to the appropriate control. *P < 0.05, **P < 0.01, ***P < 0.001.
964118_Supplementary_Materials.zip
Download Zip (2.8 MB)Acknowledgments
We thank Jenny Bärlund and Sari Tynkkynen for excellent technical assistance, and Markus Vähä-Koskela for critical comments and provided reagents.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Cesarman E, Chang Y., Moore, PS., Said, JW, Knowles, DM. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. New Eng J Med 1995; 332:1186-91; PMID:7700311; http://dx.doi.org/10.1056/NEJM199505043321802
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 1994; 266:1865-9; PMID:7997879; http://dx.doi.org/10.1126/science.7997879
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 1995; 86:1276-80; PMID: 7632932
- Green I, Espiritu E, Ladanyi M, Chaponda R, Wieczorek R, Gallo L, Feiner H. Primary lymphomatous effusions in AIDS: a morphological, immunophenotypic, and molecular study. Mod Pathol: An Off J U S Can Acad Pathol, Inc 1995; 8:39-45; PMID:7731940
- Boulanger E, Gérard L, Gabarre J, Molina JM, Rapp C, Abino JF, Cadranel J, Chevret S, Oksenhendler E Prognostic factors and outcome of human herpesvirus 8-associated primary effusion lymphoma in patients with AIDS. J Clin Oncol: OffJ Am Soc Clin Oncol 2005; 23:4372-80; PMID:15994147; http://dx.doi.org/10.1200/JCO.2005.07.084
- Klein U, Gloghini A, Gaidano G, Chadburn A, Cesarman E, Dalla-Favera R, Carbone A. Gene expression profile analysis of AIDS-related primary effusion lymphoma (PEL) suggests a plasmablastic derivation and identifies PEL-specific transcripts. Blood 2003; 101:4115-21; PMID:12531789; http://dx.doi.org/10.1182/blood-2002-10-3090
- Goto H, Kojima Y, Nagai H, Okada S. Establishment of a CD4-positive cell line from an AIDS-related primary effusion lymphoma. Int J Hematol 2013; 97:624-33; PMID:23605439; http://dx.doi.org/10.1007/s12185-013-1339-3
- Nepka C, Kanakis D, Samara M, Kapsoritakis A, Potamianos S, Karantana M, Koukoulis G. An unusual case of Primary Effusion Lymphoma with aberrant T-cell phenotype in a HIV-negative, HBV-positive, cirrhotic patient, and review of the literature. CytoJournal 2012; 9:16; PMID:22919423; http://dx.doi.org/10.4103/1742-6413.97766
- Said JW, Shintaku IP, Asou H, deVos S, Baker J, Hanson G, Cesarman E, Nador R, Koeffler HP. Herpesvirus 8 inclusions in primary effusion lymphoma: report of a unique case with T-cell phenotype. Arch Pathol Lab Med 1999; 123:257-60; 1232.0.CO;2 (1999); PMID:10086517; http://dx.doi.org/10.10430003-9985
- Carbone A, Cilia AM, Gloghini A, Capello D, Fassone L, Perin T, Rossi D, Canzonieri V, De Paoli P, Vaccher E, et al. Characterization of a novel HHV-8-positive cell line reveals implications for the pathogenesis and cell cycle control of primary effusion lymphoma. Leukemia 2000; 14:1301-9; PMID:10914556; http://dx.doi.org/10.1038/sj.leu.2401802
- Gaidano G, Gloghini A, Gattei V, Rossi MF, Cilia AM, Godeas C, Degan M, Perin T, Canzonieri V, Aldinucci D, et al. Association of Kaposi's sarcoma-associated herpesvirus-positive primary effusion lymphoma with expression of the CD138syndecan-1 antigen. Blood 1997; 90:4894-900; PMID:9389706
- Cheng F, Pekkonen P, Ojala PM. Instigation of Notch signaling in the pathogenesis of Kaposi's sarcoma-associated herpesvirus and other human tumor viruses. Future Microbiol 2012; 7:1191-205; PMID: 23030424; http://dx.doi.org/10.2217/fmb.12.95
- Curry CL, Reed LL, Golde TE, Miele L, Nickoloff BJ, Foreman KE. Gamma secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi's sarcoma tumor cells. Oncogene 2005; 24:6333-44; PMID: 15940249; http://dx.doi.org/10.1038sj.onc.1208783
- Liu R, Li X, Tulpule A, Zhou Y, Scehnet JS, Zhang S, Lee JS, Chaudhary PM, Jung J, Gill PS. KSHV-induced notch components render endothelial and mural cell characteristics and cell survival. Blood 2010; 115:887-95; PMID:19965636; http://dx.doi.org/10.1182/blood-2009-08-236745
- Emuss V, Lagos D, Pizzey A, Gratrix F, Henderson SR, Boshoff C. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathogens 2009; 5:e1000616; PMID: 19816565; http://dx.doi.org/10.1371/journal.ppat.1000616
- Lan K, Choudhuri T, Murakami M, Kuppers DA, Robertson ES. Intracellular activated Notch1 is critical for proliferation of Kaposi's sarcoma-associated herpesvirus-associated B-lymphoma cell lines in vitro. J Virol 2006; 80:6411-9; PMID:16775329; http://dx.doi.org/10.1128/JVI.00239-06
- Lan K, Verma SC, Murakami M, Bajaj B, Kaul R, Robertson ES. Kaposi's sarcoma herpesvirus-encoded latency-associated nuclear antigen stabilizes intracellular activated Notch by targeting the Sel10 protein. Proc Nat Acad Sci U S A 2007; 104:16287-92; PMID: 17909182; http://dx.doi.org/10.1073/pnas.0703508104
- Curry CL, Reed LL, Broude E, Golde TE, Miele L, Foreman KE. Notch inhibition in Kaposi's sarcoma tumor cells leads to mitotic catastrophe through nuclear factor-kappaB signaling. Mol Cancer Therap 2007; 6:1983-92; PMID:17604336; http://dx.doi.org/10.1158/1535-7163.MCT-07-0093
- Lan K, Murakami M, Bajaj B, Kaul R, He Z, Gan R, Feldman M, Robertson ES. Inhibition of KSHV-infected primary effusion lymphomas in NODSCID mice by gamma-secretase inhibitor. Cancer Biol Ther 2009; 8:2136-43; PMID:19783901; http://dx.doi.org/10.4161/cbt.8.22.9743
- Chang Y, Moore PS, Talbot SJ, Boshoff CH, Zarkowska T, Godden-Kent, Paterson H, Weiss RA, Mittnacht S. Cyclin encoded by KS herpesvirus. Nature 1996; 382:410; PMID:8684480; http://dx.doi.org/10.1038/382410a0
- Godden-Kent D, Talbot SJ, Boshoff C, Chang Y, Moore P, Weiss RA, Mittnacht S. The cyclin encoded by Kaposi's sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J Virol 1997; 71:4193-8; PMID:9151805
- Verschuren EW, Jones, N, Evan, GI. The cell cycle and how it is steered by Kaposi's sarcoma-associated herpesvirus cyclin. J Gen Virol 2004; 85:1347-61; PMID:15166416; http://dx.doi.org/10.1099/vir.0.79812-0
- Ellis M, Chew YP, Fallis L, Freddersdorf S, Boshoff C, Weiss RA, Lu X, Mittnacht S. Degradation of p27(Kip) cdk inhibitor triggered by Kaposi's sarcoma virus cyclin-cdk6 complex. EMBO J 1999; 18:644-53; PMID: 9927424; http://dx.doi.org/10.1093/emboj/18.3.644
- Laman H, Coverley D, Krude T, Laskey R, Jones N. Viral cyclin-cyclin-dependent kinase 6 complexes initiate nuclear DNA replication. Mol Cell Biol 2001; 21:624-35; PMID:11134348; http://dx.doi.org/10.1128/MCB.21.2.624-635.2001
- Mann DJ, Child ES, Swanton C, Laman H, Jones N. Modulation of p27(Kip1) levels by the cyclin encoded by Kaposi's sarcoma-associated herpesvirus. EMBO J 1999; 18:654-63; PMID:9927425; http://dx.doi.org/10.1093/emboj/18.3.654
- Ojala PM, Yamamoto K, Castaños-Vélez E, Biberfeld P, Korsmeyer SJ, Mäkelä TP. The apoptotic v-cyclin-CDK6 complex phosphorylates and inactivates Bcl-2. Nat Cell Biol 2000; 2:819-25; PMID:11056537; http://dx.doi.org/10.1038/35041064
- Sarek G, Jarviluoma A, Ojala PM. KSHV viral cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood 2006; 107:725-32; PMID:16160006; http://dx.doi.org/10.1182/blood-2005-06-2534
- Swanton C, Mann DJ, Fleckenstein B, Neipel F, Peters G, Jones N. Herpes viral cyclinCdk6 complexes evade inhibition by CDK inhibitor proteins. Nature 1997; 390:184-7; PMID:9367157; http://dx.doi.org/10.1038/36606
- Jarviluoma A, Child ES, Sarek G, Sirimongkolkasem P, Peters G, Ojala PM, Mann DJ. Phosphorylation of the cyclin-dependent kinase inhibitor p21Cip1 on serine 130 is essential for viral cyclin-mediated bypass of a p21Cip1-imposed G1 arrest. Mol Cell Biol 2006; 26:2430-40; PMID:16508017; http://dx.doi.org/10.1128/MCB.26.6.2430-2440.2006
- Jones T, Ramos da Silva S, Bedolla R, Ye F, Zhou F, Gao SJ. Viral cyclin promotes KSHV-induced cellular transformation and tumorigenesis by overriding contact inhibition. Cell Cycle 2014; 13:845-58; PMID: 24419204; http://dx.doi.org/10.4161/cc.27758
- Zhi H, Zahoor MA, Shudofsky AM, Giam CZ. KSHV vCyclin counters the senescence G1 arrest response triggered by NF-kappaB hyperactivation. Oncogene 2014;doi: 10.1038/onc.2013.567; PMID:24469036
- Koopal S, Furuhjelm JH, Järviluoma A, Jäämaa S, Pyakurel P, Pussinen C, Wirzenius M, Biberfeld P, Alitalo K, Laiho M, et al. Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathogens 2007; 3:1348-60; PMID: 17907806; http://dx.doi.org/10.1371/journal.ppat.0030140
- Verschuren EW, Klefstrom J, Evan GI, Jones N. The oncogenic potential of Kaposi's sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell 2002; 2:229-41; PMID:12242155; http://dx.doi.org/10.1016/S1535-6108(02)00123-X
- Ojala PM, Tiainen M, Salven P, Veikkola T, Castaños-Vélez E, Sarid R, Biberfeld P, Mäkelä TP. Kaposi's sarcoma-associated herpesvirus-encoded v-cyclin triggers apoptosis in cells with high levels of cyclin-dependent kinase 6. Cancer Res 1999; 59:4984-9; PMID: 10519412
- Leidal AM, Cyr DP, Hill RJ, Lee PW, McCormick C. Subversion of autophagy by Kaposi's sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 2012; 11:167-80; PMID: 22341465; http://dx.doi.org/10.1016/j.chom.2012.01.005
- Verschuren EW, Hodgson JG, Gray JW, Kogan S, Jones N, Evan GI. The role of p53 in suppression of KSHV cyclin-induced lymphomagenesis. Cancer Res 2004; 64:581-9; PMID:14744772; http://dx.doi.org/10.1158/0008-5472.CAN-03-1863
- Meuwissen R, Berns A. Mouse models for human lung cancer. Genes Dev 2005; 19:643-64; PMID: 15769940; http://dx.doi.org/10.1101/gad.1284505
- Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol 2007; 9:573-80; PMID: 17417627; http://dx.doi.org/10.1038/ncb1571
- Buss H, Handschick K, Jurrmann N, Pekkonen P, Beuerlein K, Müller H, Wait R, Saklatvala J, Ojala PM, Schmitz ML, et al. Cyclin-dependent kinase 6 phosphorylates NF-kappaB P65 at serine 536 and contributes to the regulation of inflammatory gene expression. PloS One 2012; 7:e51847; PMID: 23300567; http://dx.doi.org/10.1371/journal.pone.0051847
- Jarviluoma A, Koopal S, Rasanen S, Makela TP, Ojala PM. KSHV viral cyclin binds to p27KIP1 in primary effusion lymphomas. Blood 2004; 104:3349-54; PMID:15271792; http://dx.doi.org/10.1182/blood-2004-05-1798
- Sarek G, Järviluoma A, Moore HM, Tojkander S, Vartia S, Biberfeld P, Laiho M, Ojala PM. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathogens 2010; 6:e1000818; PMID:20333249; http://dx.doi.org/10.1371/journal.ppat.1000818
- Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev. Immunol 2008; 8:380-90; PMID:18421304; http://dx.doi.org/10.1038/nri2304
- Ciofani M, Zuniga-Pflucker JC. The thymus as an inductive site for T lymphopoiesis. Ann Rev Cell Dev Biol 2007; 23:463-93; PMID:17506693; http://dx.doi.org/10.1146/annurev.cellbio.23.090506.123547
- Crist WM, Shuster JJ, Falletta J, Pullen DJ, Berard CW, Vietti TJ, Alvarado CS, Roper MA, Prasthofer E, Grossi CE. Clinical features and outcome in childhood T-cell leukemia-lymphoma according to stage of thymocyte differentiation: a pediatric oncology group study. Blood 1988; 72:1891-7; PMID:3058229
- Grossel MJ, Hinds PW. From cell cycle to differentiation: an expanding role for cdk6. Cell Cycle 2006; 5:266-70; PMID:16410727; http://dx.doi.org/10.4161/cc.5.3.2385
- Grossel MJ, Hinds PW. Beyond the cell cycle: a new role for Cdk6 in differentiation. J Cell Biochem 2006; 97:485-93; PMID:16294322; http://dx.doi.org/10.1002/jcb.20712
- Hu MG, Deshpande A, Schlichting N, Hinds EA, Mao C, Dose M, Hu GF, Van Etten RA, Gounari F, Hinds PW. CDK6 kinase activity is required for thymocyte development. Blood 2011; 117:6120-31; PMID:21508411; http://dx.doi.org/10.1182/blood-2010-08-300517
- Sawai CM, Freund J, Oh P, Ndiaye-Lobry D, Bretz JC, Strikoudis A, Genesca L, Trimarchi T, Kelliher MA, Clark M, et al. Therapeutic targeting of the cyclin D3:CDK46 complex in T cell leukemia. Cancer Cell 2012; 22:452-65; PMID:23079656; http://dx.doi.org/10.1016/j.ccr.2012.09.016
- Chilosi M, Doglioni C, Yan Z, Lestani M, Menestrina F, Sorio C, Benedetti A, Vinante F, Pizzolo G, Inghirami G. Differential expression of cyclin-dependent kinase 6 in cortical thymocytes and T-cell lymphoblastic lymphomaleukemia. Am J Pathol 1998; 152:209-17; PMID:9422538
- Leonard JP, LaCasce AS, Smith MR, Noy A, Chirieac LR, Rodig SJ, Yu JQ, Vallabhajosula S, Schoder H, English P, et al. Selective CDK46 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 2012; 119:4597-607; PMID:22383795; http://dx.doi.org/10.1182/blood-2011-10-388298
- Guha M. Blockbuster dreams for Pfizer's CDK inhibitor. Nat Biotechnol 2013; 31:187; PMID:23471056; http://dx.doi.org/10.1038/nbt0313-187a
- Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest 2012; 122:3398-406; PMID:23023710; http://dx.doi.org/10.1172/JCI61269
- Weng AP, Ferrando AA, Lee W, Morris JP 4th, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004; 306:269-71; PMID:15472075; http://dx.doi.org/10.1126/science.1102160
- Bellavia D, Campese AF, Alesse E, Vacca A, Felli MP, Balestri A, Stoppacciaro A, Tiveron C, Tatangelo L, Giovarelli M, et al. Constitutive activation of NF-kappaB and T-cell leukemialymphoma in Notch3 transgenic mice. EMBO J 2000; 19:3337-48; PMID:10880446; http://dx.doi.org/10.1093/emboj/19.13.3337
- Bellavia D, Campese AF, Checquolo S, Balestri A, Biondi A, Cazzaniga G, Lendahl U, Fehling HJ, Hayday AC, Frati L, et al. Combined expression of pTalpha and Notch3 in T cell leukemia identifies the requirement of preTCR for leukemogenesis. Proc Nat Acad Sci U S A 2002; 99:3788-93; PMID:11891328; http://dx.doi.org/10.1073/pnas.062050599
- Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, Signoretti S, Look AT, Kung AL, von Boehmer H, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell 2012; 22:438-51; PMID:23079655; http://dx.doi.org/10.1016/j.ccr.2012.09.015
- Joshi I, Minter LM, Telfer J, Demarest RM, Capobianco AJ, Aster JC, Sicinski P, Fauq A, Golde TE, Osborne BA. Notch signaling mediates G1S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood 2009; 113:1689-98; PMID:19001083; http://dx.doi.org/10.1182/blood-2008-03-147967
- Sicinska E, Aifantis I, Le Cam L, Swat W, Borowski C, Yu Q, Ferrando AA, Levin SD, Geng Y, von Boehmer H, et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 2003; 4:451-61; PMID:14706337; http://dx.doi.org/10.1016/S1535-6108(03)00301-5
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985; 318:533-8; PMID:3906410
- Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002; 1:289-98; PMID:12086865; http://dx.doi.org/10.1016/S1535-6108(02)00047-8
- Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 1996; 2:342-6; PMID:8612236; http://dx.doi.org/10.1038/nm0396-342