
Abstract
Orphan receptors comprise nearly half of all members of the nuclear receptor superfamily. Despite having broad structural similarities to the classical estrogen receptors, estrogen-related receptors (ERRs) have their own unique DNA response elements and functions. In this study, we focus on 2 ERRβ splice variants, short form ERRβ (ERRβsf) and ERRβ2, and identify their differing roles in cell cycle regulation. Using DY131 (a synthetic agonist of ERRβ), splice-variant selective shRNA, and exogenous ERRβsf and ERRβ2 cDNAs, we demonstrate the role of ERRβsf in mediating the G1 checkpoint through p21. We also show ERRβsf is required for DY131-induced cellular senescence. A key novel finding of this study is that ERRβ2 can mediate a G2/M arrest in response to DY131. In the absence of ERRβ2, the DY131-induced G2/M arrest is reversed, and this is accompanied by p21 induction and a G1 arrest. This study illustrates novel functions for ERRβ splice variants and provides evidence for splice variant interaction.
Introduction
A growing field of research focuses on the emerging role of alternative splice variants in disease progression. Alternative splicing occurs in almost 95% of all mammalian genesCitation1 and represents an important source of functional diversity for the proteome.Citation2 Alternative splicing can dramatically alter the protein profile of a cell, and defects in splicing regulation have been directly linked to a variety of human diseases: β+-thalassemia,Citation3,4 spinal muscular atrophy,Citation5 acute myeloid leukemia,Citation6 hepatocellular carcinoma,Citation7 glioblastoma,Citation8 and others.
Alternative splicing affects proteins of all functional classes, including nuclear receptors.Citation9-11 Orphan receptors comprise nearly half of all members of the nuclear receptor superfamily.Citation12 These transcription factors apparently lack endogenous ligands, but their constitutive activity can be modulated by natural products, synthetic ligands, or the binding of coregulatory proteins. Estrogen-related receptors (ERRs) have broad structural similarity to canonical estrogen receptors α and β (ERα, ERβ) though they cannot bind estrogen, and are well-established transcriptional regulators of mitochondrial biogenesis and function, including fatty acid oxidation, oxidative phosphorylation, and the tricarboxylic acid cycle.Citation13
ERRβ is required for proper placental formation in mice,Citation14,15 and its conditional deletion in either the whole animal or specifically in neural progenitor cells increases lean body mass, energy expenditure, and feeding frequency by altering stress response signaling through the hypothalamic-pituitary-adrenal axis.Citation16,17 Frame-shift and point mutations mapping to the DNA- and ligand-binding domains of ERRβ at the DFNB35 locus are associated with autosomal recessive hearing loss.Citation18
The murine Esrrb gene produces a single confirmed mRNA encoding a protein of 433 amino acids, but in humans there exist 2 additional alternatively spliced forms of ESRRB with potentially distinct biological functions.Citation19-21 Short form ERRβ (ERRβsf) uses an intronic stop codon after exon 9 and is >90% homologous to mouse and rat ERRβ. ERRβ2 has an extended carboxyl-terminus encoded by exons 10, 11, and part of 12, while ERRβ-Δ10 splices exon 9 to exon 11 and includes all of exon 12. Due to a frame shift that occurs during alternative splicing, ERRβ-Δ10 and ERRβ2 each have a unique F domain which is absent in ERRβsf.Citation19
Exogenous expression of ERRβsf has transcription-dependent tumor suppressor activities that engage the G1 checkpoint in prostate cancer cell lines.Citation22 However, the molecular function(s) of endogenous ERRβsf, or the ERRβ2 and ERRβ-Δ10 splice variants in other tumor types remain unknown. Here, we evaluated a synthetic small molecule activator of ERRβ (DY131)Citation23,24 in cellular models of glioblastoma multiforme (GBM), where 2 ERRβ splice variants (ERRβsf and ERRβ2) are expressed. We found that this agonist induces cell death in cancer, but not non-transformed lines, and that apoptotic cell death in response to DY131 requires mutation or loss/silencing of p53. Using splice variant-selective shRNAs we determined that ERRβ splice variants have opposing functions in cell cycle regulation. In A172 cells, suppression of ERRβsf, but not ERRβ2, inhibits cell death and G1 arrest. Silencing of ERRβ2 abrogates a novel DY131-induced G2/M arrest and cell death in T98G cells, while suppression of ERRβsf enhances the arrest in G2/M. Lastly, we demonstrate that DY-mediated cellular senescence requires ERRβsf but is p53-independent. These results are the first to describe a function for endogenous ERRβ2 and reveal a novel interplay between ERRβ splice variants, which has broad implications for cell cycle control.
Results
DY131 inhibits cellular proliferation, induces cell death and prevents colony formation in cancer cells, but not in non-transformed control cells
Exogenous expression of ERRβsf inhibits the growth of prostate cancer cells,Citation22 but the molecular function(s) of endogenous ERRβsf, or the other splice variants of this receptor, remain unknown. To address this, we used the acyl hydrazone DY131 (DY), a synthetic agonist of ERRβ,Citation23 to activate endogenous ERRβ in 2 glioblastoma multiforme (GBM) cell lines. We cultured A172, T98G (GBM) and HFF1 cells (non-transformed human foreskin fibroblasts) in the presence of DY for up to 14 d and stained total DNA with crystal violet at various time points to measure the effects of DY on cellular proliferation (). DY selectively impaired growth in both cancer cell lines, but not in HFF1 cells. To determine whether these results were due to cytotoxic (cell death) versus cytostatic (cell cycle arrest) effects, cells were treated with DY for 24 h and we measured the fraction of dead cells by positive propidium iodide staining of fragmented DNA (subG1) (). DY induced cell death in the cancer lines, but not in HFF1 cells. Similar to our proliferation assay results, T98G cells were more sensitive to DY treatment than A172 cells. We also tested whether DY could prevent colony formation in cancer cell lines (). DY significantly impaired A172 and T98G cells’ ability to form colonies. We further verified that these differences in DY-induced cell death were not attributable to variations in basal proliferation rates between the 2 cancer cell lines ().
Figure 1. DY131 inhibits cellular proliferation, induces cell death and prevents colony formation in cancer cells, but not in non-transformed cells. (A) A crystal violet assay staining total DNA (measured by absorption at 550 nm) of A172, T98G, and HFF1 to show growth rates in the presence or absence of DY at indicated concentrations. (B) Fraction of cells containing fragmented DNA (PI positive) 24 h after DY treatment determined by flow cytometry (n = 3, one-way ANOVA). (C) Representative images and quantification of a colony formation assay. Cells were seeded on day 0, treated with indicated DY on day 1 before drug was washed out on day 2. Plates were stained and colonies counted on day 10 (n = 4, one-way ANOVA). (D) Crystal violet assay demonstrating basal cellular growth rates through a 14d assay. (E) Basal ERRβ and ERRγ protein expression. Lanes labeled Δ10, β2, and SFβ2 contain whole cell lysate from T98G cells transiently transfected with the indicated cDNA to demonstrate endogenous splice variant specificity of the ERRβ antibodies. (F) Fraction of T98G cells containing fragmented DNA (PI positive) 24 h after indicated drug treatments determined by flow cytometry (n = 3). (*P < 0.05 ** P < 0.01 ***P < 0.001).
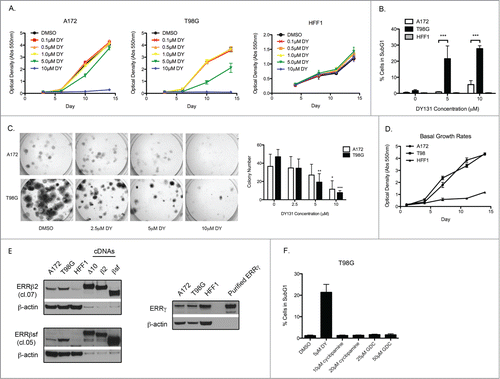
DY is also an agonist for ERRgamma (ERRγ),Citation23 which is 77% identical to ERRβsf and whose exogenous expression can also inhibit the growth of prostate cancer cells.Citation25 The precise mechanism by which DY enhances the constitutive transcriptional activity of these orphan nuclear receptors is not known, though a related compound (GSK4716) increases the overall stability of the ERRγ ligand-binding domain in thermal stability assays.Citation26 We therefore measured basal, endogenous expression of ERRβ and ERRγ protein in our cell lines () alongside positive controls generated by exogenous expression of cDNAs encoding specific splice variants (ERRβ), or purified protein (ERRγ). Two commercially available antibodies from R&D Systems preferentially detect endogenous ERRβ2 (500 amino acids, predicted molecular weight = 55.6 kDa) and ERRβsf splice variants (433 amino acids, predicted molecular weight = 48.0 kDa) in A172 and T98G cells (cl.07 and cl.05, respectively). Under exogenous expression conditions, cl.07 and cl.05 can each detect both variants. Endogenous expression of the third splice variant (ERRβ-Δ10, 508 amino acids, predicted molecular weight = 56.2 kDa) is not detected in these cells. Nontransformed HFF1 cells express very low levels of all ERRβ splice variants. By contrast, ERRγ expression is robust in both the GBM and non-transformed cell lines.
Recently, DY has been shown to have “off-target” effects on primary cilia formation through inhibition of the G-protein coupled receptor Smoothened.Citation27 To test whether the observed DY cytotoxicity was attributable to Smoothened inhibition, we treated T98G cells with 2 known Smoothened inhibitors, cyclopamineCitation28 and GDC-0449Citation29 (). We observed no cell death with either compound, suggesting that the DY-induced cell death phenotype is unlikely to involve Smoothened.
DY131 mediates cell cycle arrest
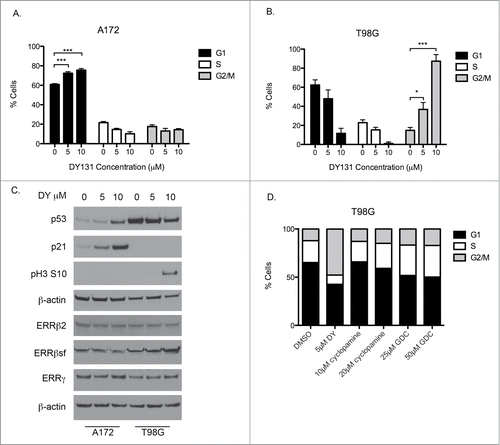
Given the anti-proliferative effects of DY and the difference in p53 status between A172 (p53 wild type, wt) and T98G (p53 mutant, mut) cells, we examined whether these effects were also accompanied by a cell cycle arrest. In A172 (p53 wt) cells, we found DY induced a G1 arrest after 24 h (). Interestingly, the same treatment in T98G (p53 mut) cells caused a G2/M arrest (). We then identified specific G1 (p53 and p21) and G2/M (phospho-H3ser10) protein markers to confirm cell cycle arrest signaling in each cell line (). A172 (p53 wt) cells, which arrest in G1, showed a corresponding induction of 2 major G1 checkpoint regulators: p53 and its downstream target, p21. In T98G (p53 mut) cells, we did not observe an induction of G1 checkpoint mediators, but DY induced phosphorylation of histone H3 at serine 10, previously shown to be a specific phosphorylation site during prophase and important for chromatin condensation.Citation30,31 These data suggest DY induces a cell cycle arrest specifically in mitosis in p53 mutant T98G cells. We also observed no change in ERRβsf, ERRβ2 or ERRγ at the protein level in DY-treated cells (). To verify the cell cycle arrest phenotypes were not due to Smoothened inhibition by DY, we treated T98G cells with 2 Smoothened inhibitors and compared their cell cycle profiles to the profile induced by DY (). Neither cyclopamine nor GDC-0449 caused any G2/M arrest; however an increase in S-phase was observed.
Figure 2. DY131-mediated cell cycle arrest differs between p53 wild type and p53 mutant GBM cells. (A) Cell cycle profile of p53 wild type A172 cells 24 h after DY treatment determined by flow cytometry (n = 3, one-way ANOVA). Corresponding subG1 data from same assay shown in . (B) Cell cycle profile of p53 mutant T98G cells 24 h after DY treatment determined by flow cytometry (n = 3, one-way ANOVA). Corresponding subG1 data from same assay shown in . (C) Protein expression for p53, p21, phospho-H3 ser10, ERRβ2, ERRβsf and ERRγ in A172 and T98G cells after 24 h DY treatment. (D) T98G cell cycle profile 24 h after indicated drug treatments determined by flow cytometry (n = 3). (*P < 0.05 **P < 0.01 ***P < 0.001).
Loss of p53 function promotes DY131 mediated apoptosis
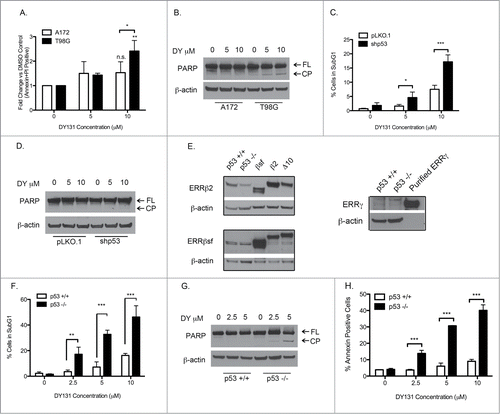
To understand how DY causes cell death in A172 (p53 wt) and T98G (p53 mut) cells, we first determined whether cells were undergoing apoptosis. After 24 h of DY treatment, T98G cells showed a significant increase in annexin and PI double-positive cells, whereas the A172 cells did not (). To confirm these data, we examined PARP cleavage and observed a similar result; DY induced PARP cleavage in T98G cells, but not A172 cells (). To test whether the magnitude of cell death and induction of apoptosis were dependent on p53, we stably silenced wild type p53 in A172 cells using lentiviral delivery of shRNA.Citation32 We then measured the level of subG1 () and PARP cleavage () after DY treatment. p53 knockdown significantly increased the amount of DY-induced cell death at 5 μM and 10 μM, and caused PARP cleavage at 10 μM. Because A172 and T98G cells have molecular differences other than p53 status, and PARP cleavage in A172-shp53 cells was not as robust as in parental T98G (p53 mut) cells, we used a second model to more directly test p53's involvement in DY-mediated apoptosis: the RKO isogenic p53 null (p53−/−) system,Citation33 where both p53 alleles have been deleted by targeted homologous recombination. First, we verified that DY protein targets (ERRβ and ERRγ) were detectably expressed in these cell lines (). Parental RKO cells and the p53−/− variant express the ERRβ2 and ERRβsf splice variants, but weak-to-undetectable levels of ERRγ. Importantly, RKO-p53−/− cells showed a significantly higher percentage of cell death than the RKO-p53+/+ parental cells when treated with DY (). Similar to our GBM p53 wild type and p53 mutant pair, the RKO-p53−/− cells also showed a DY-mediated induction of PARP cleavage whereas the RKO-p53+/+ cells did not (). Finally, annexin staining showed DY induced apoptosis in the RKO-p53−/−, but not the RKO-p53+/+ cells () Taken together, these 3 model systems support the conclusion that cells lacking wild type p53 (by mutation, silencing, or deletion) die by apoptosis when treated with DY.
Figure 3. Loss of wild type p53 function promotes DY131-mediated apoptosis. (A) Fold change of Annexin V and PI double positive cells after 24 h DY treatment relative to DMSO control determined by flow cytometry (n = 3, 2-way ANOVA). (B) Protein expression of PARP (full length (FL) and cleavage product (CP)) 24 h after DY treatment. (C) Percentage of A172-pLKO.1 and -shp53 stable cells in subG1 24 h after DY treatment determined by flow cytometry (n = 3, 2-way ANOVA). (D) Protein expression of PARP in A172-pLKO.1 and -shp53 stable cells. (E) Basal protein expression of ERRβ2 (cl.07), ERRβsf (cl.05) and ERRγ in RKO isogenic mutants. Lanes labeled Δ10, β2, and SFβ2 contain whole cell lysate from T98G cells transiently transfected with the indicated cDNA. (F) Fraction of RKO cells in subG1 after 24 h DY treatment determined by flow cytometry (n = 3, 2-way ANOVA). G, PARP protein expression in RKO isogenic mutants 24 h post-DY treatment. H, Percentage of Annexin V positive RKO cells after 18 h DY treatment (n = 3, 2-way ANOVA). (*P < 0.05 **P < 0.01 ***P < 0.001).
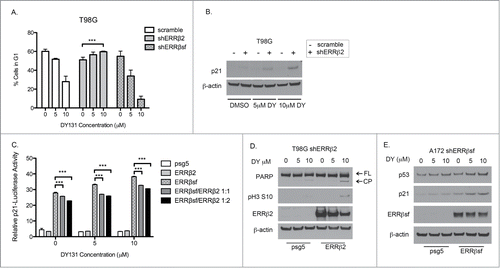
ERRβ2 knockdown reverses DY-induced apoptosis and G2/M cell cycle arrest in p53 mutant T98G cells
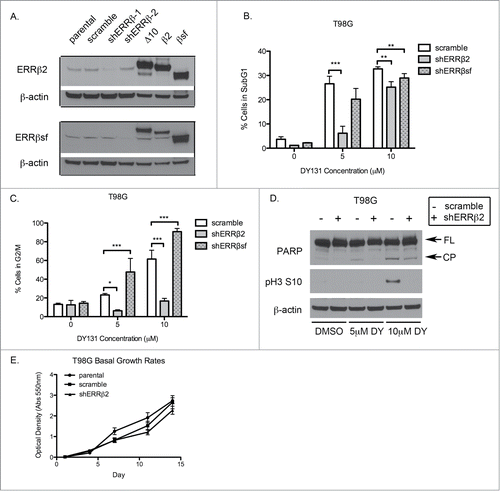
DY is a synthetic agonist for both ERRβ and ERRγ that, thus far, we have demonstrated to cause cell cycle arrest, inhibit cellular proliferation, and induce death specifically in cancer cells. Although DY has been reported to antagonize Smoothened, our data do not support a role for this “off-target” activity in contributing to the observed cytotoxic and cytostatic effects of DY, since these are not recapitulated by more specific Smoothened inhibitors (). To test whether DY-induced growth inhibition and cell death is mediated by ERRβ, we stably transduced p53 mutant T98G cells with lentiviral vectors containing 2 different shRNAs against ESRRB (shERRβ-1 and shERRβ-2), or a scrambled control. We compared these stable cell lines to positive controls for each of the 3 ERRβ splice variants (lanes 5–7) to assess ERRβ knockdown at the protein level (). Interestingly, each shRNA preferentially targeted a different ERRβ splice variant (ERRβsf was silenced by shERRβ-2 and ERRβ2 by shERRβ-1). In these modified T98G cells, knockdown of ERRβ2 significantly reduced cell death caused by DY treatment (). Knockdown of ERRβsf also showed a modest reduction in cell death, but only in the presence of 10 μM DY (). Strikingly, silencing of ERRβ2 but not ERRβsf, completely reversed the DY-mediated G2/M arrest in T98G cells (). These data are further supported by the reversal of protein signaling indicative of apoptosis (PARP cleavage) and G2/M arrest (phospho-H3 ser10) in the shERRβ2 cells (). We also compared the basal growth rates of the T98G stable cells to parental T98G cells () and saw no difference in proliferation, confirming these results were not due to a fundamental change in cellular proliferation resulting from stable infection. Altogether, these data support a role for ERRβ2 in regulation of cell cycle arrest in mitosis as well as apoptotic cell death in p53 mut T98G cells.
Figure 4. ERRβ2 knockdown reverses DY131-mediated cell death and G2/M arrest in T98G cells. (A) ERRβsf(cl.05) and ERRβ2 (cl.07) protein expression in T98G shERRβ stable cells. Lanes labeled Δ10, β2, and SFβ2 contain whole cell lysate from T98G cells transiently transfected with the indicated cDNA. (C) Percentage of T98G shERRβ stable cells in subG1 after 24 h DY treatment determined by flow cytometry (n = 3, 2-way ANOVA). (C) Percentage of T98G shERRβ stables cells in G2/M after 24 h DY treatment determined by flow cytometry (n = 3, 2-way ANOVA). (D) PARP and phospho-H3 ser10 protein expression in control cells compared to shERRβ2 cells after 24 h DY treatment. (E) Crystal violet assay staining total DNA (measured by absorption at 550 nm) to determine basal growth rates of T98G parental, stable scramble control and shERRβ2 stable cells. (*P < 0.05 **P < 0.01 ***P < 0.001).
ERRβsf knockdown reverses DY-induced cell death, p21 induction and G1 cell cycle arrest in p53 wild type A172 cells
We used the same approach to silence ERRβ2 and ERRβsf in A172 cells as discussed above for T98G cells. In A172 cells, stable knockdown of ERRβsf reduced DY-mediated cell death, whereas silencing of ERRβ2 did not (). Similarly, stable knockdown of ERRβsf reversed the G1 arrest caused by DY () and reduced the induction of p53 and p21 (). We confirmed ERRβ2 knockdown in A172 by shERRβ-1 at the protein level (). Finally, we examined the basal growth rates of the A172 stable cell lines and again found no significant difference (). Because ERRγ is also a target of DY and has been shown to activate p21 causing a G1 arrest in a prostate cancer model,Citation25 we transiently knocked down ERRγ in A172 cells () to determine whether ERRγ knockdown impacted the DY-mediated G1 arrest observed (). ERRγ knockdown caused no change to the DY-induced G1 arrest measured in A172 cells. These data demonstrate that ERRβsf, but not ERRγ, activates a G1 checkpoint through p53 and/or p21 in A172 cells.
Figure 5. ERRβsf knockdown reverses DY131-mediated cell death and G1 arrest in A172 cells. (A) Fold change vs DMSO control of A172 shERRβ stable cells in subG1 after 24 h DY treatment determined by flow cytometry (n = 3, 2-way ANOVA).(B) Fraction of A172 shERRβ cells in G1 24 h after DY treatment determined by flow cytometry (n = 3, one-way ANOVA). (C) Protein expression of p53, p21 and ERRβsf in A172 stable scramble control cells compared to A172 shERRβsf stable cells. Densitometric values for the ratio of the indicated proteins to β-actin are normalized to the level of control expression in lane 1. (D) Western blot of A172 shERRβ stable cell lines using ERRβ-cl. 07 antibody to demonstrate ERRβ2 knockdown by shERRβ-1. Lanes labeled Δ10, β2, and SFβ2 contain whole cell lysate from T98G cells transiently transfected with the indicated cDNA. (E) Crystal violet assay staining total DNA (measured by absorption at 550 nm) to determine basal growth rates of A172 parental, stable scramble control and shERRβ stable cells. (F) Western blot of A172 ERRγ protein expression 72 h after scramble or shERRγ transient infection. (G) Percentage of A172 shERRγ cells in G1 after 24 h DY treatment determined by flow cytometry (n = 3) (*P < 0.05 **P < 0.01 ***P < 0.001).
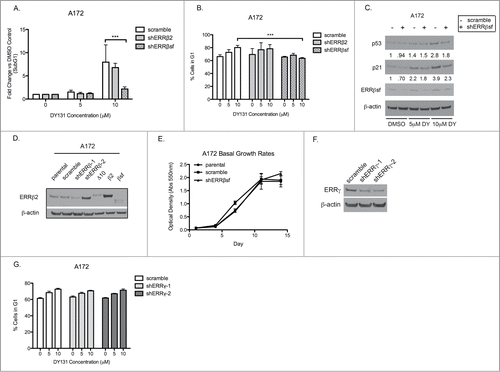
ERRβ isoform, not p53 status, determines phase of DY131-mediated cell cycle arrest
We next sought to determine if p53, in addition to its role in apoptosis, also contributes to DY-induced cell cycle arrest. We observed a significant reduction in the level of G1 arrest in DY-treated A172-shp53 cells relative to the A172-pLKO.1 controls (). However, A172-shp53 cells still underwent a dose-dependent G1 arrest in response to DY, and knockdown of p53 protein did not abolish the p21 induced by DY (). This may suggest that DY-activated ERRβsf can induce p21 independent of p53, which would be consistent with work by Yu et al.,Citation22 who show in a prostate cancer model that exogenous expression of ERRβsf causes G1 arrest via direct transcriptional upregulation of p21. However, the observed G1 arrest and p21 induction in A172 shp53 cells could also be due to incomplete p53 knockdown. We therefore tested the ability of DY to activate the p21 promoter in wild type and p53-silenced A172 cells, in the presence or absence of p53 response elementsCitation34 (). Deletion of one (p21-2) or both (p21-4) p53 response elements dramatically reduced basal p21 promoter activity when compared to the full-length promoter (p21-0) in A172-pLKO.1 cells. However, DY still showed a dose-dependent increase in p21-activity on all 3 contructs, regardless of p53 binding ability. In A172-shp53 cells, p53 knockdown reduced the basal activity of both p53 response element-containing promoters to the same level as the p53 response element-deleted promoter, and all 3 p21 promoter constructs showed the same magnitude of DY-mediated activation. Altogether, these data imply that p53 is not the sole contributor to DY-mediated G1 arrest and p21 induction. Further evidence against a role for p53 status in dictating DY-mediated cell cycle arrest is that p53+/+ and p53−/− isogenic variant RKO cells both arrested in G2/M () and showed increased phosphorylation of histone H3 at serine 10 (). We still observed an induction of p53 and p21 in the RKO-p53+/+ cells, even though there was not a corresponding G1 arrest.
Figure 6. ERRβ isoform determines phase of cell cycle arrest, not p53 status. (A) Percentage of A172-pLKO.1 and -shp53 stable cells in G1 determined by flow cytometry (n = 3, 2 way ANOVA) after 24 h DY treatment. (B) Protein expression of p53 and p21 in A172-pLKO.1 and -shp53 24 h after DY treatment. (C) p21 promoter reporter assay. Schematic of the p21 promoter constructs used containing both p53 binding sites (p21-0), deletion of one p53 binding site (p21-2) or deletion of both p53 binding sites (p21-4) (top). A172 pLKO.1 and -shp53 stable cells transfected with indicated p21 promoter deletion constructs (24 h) and treated with DY131 (20 h). (D) Cell cycle profile of RKO p53 wild type cells (n = 3, one-way ANOVA) after 24 h treatment determined by flow cytometry. (E) Cell cycle profile of RKO p53 null isogenic mutant (n = 3, one-way ANOVA) after 24 h treatment determined by flow cytometry. (F) Protein expression of p53, p21 and phospho-H3 ser10 in RKO isogenic mutants 24 h after DY treatment. (*P < 0.05 **P < 0.01 ***P < 0.001).
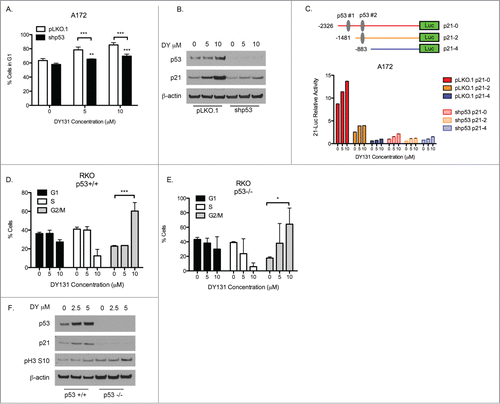
Our data strongly suggest that the ERRβ splice variants have different functions in cell cycle regulation. In p53 mutant T98G cells, silencing ERRβ2 but not ERRβsf, completely reversed the DY-mediated G2/M arrest (, light gray bars). Interestingly, this G2/M arrest reversal was accompanied by a G1 arrest () and p21 expression () not previously observed in T98G parental cells. These data imply a potential dominant inhibitory role for ERRβ2, where in the absence of this splice variant, ERRβsf-mediated p21 induction and G1 arrest now occur in the presence of mutant p53, consistent with the ability of ERRβsf to regulate p21 directly.Citation22 To confirm that this G1 arrest was not driven by ERRγ, we transiently knocked down ERRγ (Fig. S1A) in T98G-shERRβ2 cells and observed no change in G1 arrest (Fig. S1B). Moreover, silencing of ERRβsf in T98G cells led to an enhanced G2/M arrest (, speckled bars), suggesting that removal of ERRβsf from the system permits amplified signaling through ERRβ2, leading to a stronger arrest in G2/M after treatment with DY. To directly test the potential dominant inhibitory role of ERRβ2 on ERRβsf, we measured p21 promoter activity in cells transfected with either or both receptors. Exogenous expression of ERRβsf activated p21 and this activation was enhanced by DY, whereas exogenous ERRβ2 did not affect p21 activity (). Furthermore, ERRβsf-induced p21 activity was significantly reduced in a dose-dependent manner by co-transfected ERRβ2. Together, these data demonstrate that ERRβ splice variant interaction is a critical component of DY-mediated cell cycle regulation.
Figure 7. ERRβ2 inhibits ERRβsf activation of p21. (A) Fraction of T98G shERRβ stable cells in G1 determined by flow cytometry (n = 3, one-way ANOVA). (B) Corresponding p21 protein expression in T98G shERRβ2 stable cells 24 h after DY treatment. (C) p21 promoter reporter assay. HeLa cells co-transfected with psg5, ERRβsf and/or ERRβ2 (24 h) and then treated with DY (20 h). Experiment was performed in triplicate (2-way ANOVA). (D) Protein expression of PARP, ERRβ2 and phospho-H3 ser10 in T98G-shERRβ2 cells transfected with the shRNA-resistant ERRβ2 plasmid (28 h) and treated with DY (24 h). (E) Protein expression of p53, p21 and ERRβsf in A172-shERRβsf cells transfected with the shRNA-resistant ERRβsf plasmid (28 h) and treated with DY (24 h). (*P < 0.05 **P < 0.01 ***P < 0.001).
To confirm the specific function of ERRβ2 and ERRβsf in DY-mediated G2/M and G1 arrest, respectively, we rescued ERRβ-silenced cells by transient transfection of plasmids encoding shRNA-resistant versions of the appropriate splice variant. Exogenous expression of ERRβ2 in T98G-shERRβ2 cells rescued both the apoptotic response and G2/M arrest phenotype in DY-treated cells (). Similarly, exogenous expression of ERRβsf in A172-shERRβsf cells restored p53 and p21 induction by DY (). Taken together, these data illustrate a dynamic interplay between the cell cycle regulatory functions of these ERRβ splice variants, and demonstrate for the first time that ERRβ2 specifically drives G2/M arrest.
ERRβsf induces cellular senescence independent of p53
Thus far, we and othersCitation22 have demonstrated that ERRβsf can cause G1 arrest and induce p21. Given the critical role for p21 in cellular senescence,Citation35 either downstream of p53 or in a p53-independent manner, we asked whether DY could also induce senescence associated β-galactosidase in p53 wt A172 cells (, quantified in Fig. S2A). We detected a dose-dependent relationship for cellular senescence caused by DY. DY did not induce senescence in p53 mut T98G cells (). To establish that this phenotype required ERRβsf expression, we performed the same assay in our A172-shERRβsf stable cells, and found that knockdown of ERRβsf significantly reduced the level of senescence caused by 5 μM DY ( and S2B).
Figure 8. ERRβsf induces cellular senescence independent of p53 status. (A) SA-β-galactosidase assay in A172 parental cells after 24 h DY treatment. (B) SA-β-galactosidase assay in T98G parental cells after 24 h DY treatment. (C) SA-β-galactosidase assay comparing A172-scramble and –shERRβsf cells after 24 h DY treatment. (D) SA-β-galactosidase assay in RKO isogenic mutant cells after 24 h DY treatment. (E) SA-β-galactosidase assay comparing A172-pLKO.1 and -shp53 cells after 24 h DY treatment.

To determine whether p53 plays any role in the observed senescent phenotype, we first measured senescence in RKO-p53+/+ and RKO-p53−/− cells treated with DY (). Despite clear p53 and p21 induction in RKO-p53+/+ cells following DY exposure (), these cells did not undergo senescence. Furthermore, when we assayed our A172-shp53 cells, we saw no reduction in DY-induced cellular senescence ( and S2C), possibly because of the remaining p21 induction (). Collectively, these data demonstrate that DY-mediated cellular senescence requires ERRβsf, and imply that while p53 is neither necessary nor sufficient for the senescent phenotype, p21 may be required.
Discussion
Exogenous overexpression studies with one of the 3 alternatively spliced forms of ERRβ have suggested a role for this receptor in growth inhibition and cell cycle arrest in prostate cancer, but the molecular function(s) of endogenous ERRβ splice variants in this and other tumor types remain unknown. Here, using a synthetic, small molecule activator of ERRβ, we demonstrate novel cellular functions for the ERRβ2 and ERRβsf splice variants that have broad implications for cell death and cell cycle control.
We used stable transduction of 2 different ESRRB-targeted shRNAs to selectively silence ERRβ2 and ERRβsf in A172 and T98G cells. However, the target sequences for both shRNAs are present in both splice variants. Local structure of target mRNA is known to contribute to the efficiency of RNA interference. shERRβ-2 targets a sequence in the ESRRB gene close to the end of exon 9, where use of an intronic stop codon yields ERRβsf but conventional splicing produces ERRβ2. Using Mfold,Citation36 we demonstrate that the predicted secondary structure in this region differs notably between the 2 splice variants (Fig. S3), with the shERRβ-2 target site taking a more favored loop structureCitation37,38 in ERRβsf (where silencing is observed) vs. a less favored stem-like structure in ERRβ2 (where silencing is not observed). Structural differences between splice variants do not explain ERRβ2-selective silencing by shERRβ-1 since the predicted secondary structures for the target sequence of this shRNA are the same in both. Additional factors such as tertiary structure, pre-mRNA binding proteins, and/or loading of shRNAs into the RISC complex may contribute to this selectivity.Citation39
The tumor suppressor p53 is widely considered to positively regulate apoptosis, particularly in cancer where restoration of p53 function is an ongoing therapeutic challenge.Citation40 However, using 3 different models, we comprehensively show that loss of wild type p53 function, coupled with a reduction in or the absence of p21 induction, is required for DY-mediated apoptosis. p21 has been shown to suppress both p53-dependent and p53-independent apoptosis, although the precise mechanisms remain unclear.Citation41,42 Our data are consistent with a cytoprotective role for p21. In cell lines where DY induces p21 expression - A172 (), RKO-p53+/+ () and T98-shERRβ2 () – apoptosis does not occur. In contrast, where DY fails to induce a p21 response – parental T98G and RKO-p53−/− cells - or the p21 induction is dampened (A172-shp53 cells), there is significantly enhanced apoptosis when compared to p21-inducing cells ().
Table 1. Summary results of the cytoprotective role of p21 in preventing apoptosis, independent of p53 status
Our data suggest that DY-mediated activation of ERRβsf and the subsequent G1 arrest are required for cellular senescence that is independent of p53. Cell enlargement and senescence-associated β–galactosidase staining are only induced in G1-arresting A172 cells, not in G2/M-arresting RKO cells, both of which are p53 wt (). In addition, silencing of ERRβsf in A172 cells significantly reduces the senescent phenotype at 5 μM DY treatment, while silencing of p53 does not (). Although p53 and its upstream regulator ARF are often considered the predominant inducers of cellular senescence, a number of p53-independent mechanisms can contribute to this tumor suppressive mechanism,Citation35 and the CDKN2A/B locus that encodes ARF shows homozygous deletion in A172 cells.Citation43 Direct regulation of p21 by ERRβsf could be responsible for DY-mediated senescence, given that many ARF/p53-independent senescence mechanisms still rely on p21, and that p21 upregulation in response to DY is prevented by ERRβsf silencing () vs. modestly inhibited by knockdown of p53 () in A172 cells. However, RKO-p53+/+ cells, which do not senesce, still show induction of p21 in response to DY, so we must consider that i. pro-senescence signaling downstream of p21 is fundamentally different in RKO cells, or ii. ERRβsf drives a p21-independent senescence pathway. The latter mechanism is more likely, given that others have shown p21-associated senescence can occur in RKO-p53+/+ cells.Citation44
The most novel finding of our study is that ERRβ2 can mediate G2/M arrest in response to DY. Silencing of ERRβ2 but not ERRβsf in T98G cells blocks G2/M arrest (), and this is rescued by exogenous expression of shRNA-resistant ERRβ2 (). To our knowledge, this is the first demonstrated function for endogenous ERRβ2. Our data also suggest that ERRβ2 is dominant-inhibitory for ERRβsf. In T98G cells, silencing of ERRβ2 suppresses DY-mediated G2/M arrest and apoptosis (), which now permits G1 arrest and p21 induction (). Furthermore, exogenous expression of ERRβ2 directly inhibits ERRβsf-mediated p21 activity in a reporter assay competition experiment (). Results based on splice variant dominance are summarized in . Splice variants often have opposing roles (e.g., pro-survival BclXL vs. pro-apoptotic BclXS)Citation45 and/or serve as dominant-negative inhibitors of each other (e.g., Ets-1 p51 and p27).Citation46 This is also true of nuclear receptor splice variants, including other orphan receptors such as the pregnane X receptor (PXR),Citation10 where the dominant-inhibitory variant of PXR has a deletion in the ligand-binding domain. By comparison, ERRβ2 differs from ERRβsf by the presence of 68 additional amino acids at the extreme carboxyl-terminus (F domain) of the receptor. In ligand-regulated receptors like ERβ, amino acid changes in the F domain have been shown to inhibit hormone-induced transcriptional activity.Citation47
Table 2. Summary of ERRβ splice variant dominance and resulting phenotype
We propose that ligand-activated ERRβ2 promotes G2/M arrest through non-transcriptional mechanisms. Zhou et al.Citation19 have reported that exogenous ERRβ2 is largely cytoplasmic in interphase COS-1 cells, with only 30% of cells showing predominantly nuclear staining. This is in direct contrast to ERRβsf, which is >90% nuclear. The presence of 2 pools of ERRβ2 – one nuclear, one cytoplasmic – supports a scenario in which nuclear ERRβ2 can repress ERRβsf transcriptional activity, while cytoplasmic ERRβ2 participates in novel protein/protein interactions and signaling events via the unique carboxyl-terminal F domain. Within this region there is a proline-rich sequence that forms a consensus binding site for the src homology 3 (SH3) domains of c-Src and the actin binding protein cortactin (both of which have key roles in mitosis). There is also a strong consensus sequence for the substrates of the mitotic cyclin-dependent kinase Cdk1/CDC2, an important mediator of the G2/M transition. While these features may not independently determine why ERRβ2 is dominant over ERRβsf in T98G, but not A172 cells, differences in other putative binding partners and regulatory kinases are likely essential for splice variant dominance in different cell contexts. Indeed, exploiting the role of novel splice variants in disease is an emerging, viable therapeutic strategy.Citation48 In this study, we focused on understanding the function of ERRβ splice variants in the context of glioblastoma multiforme (GBM), where 2 ERRβ splice variants (ERRβsf and ERRβ2) are expressed and where alterations in splicing factor abundance can promote tumorigenicity by attenuating the formation of tumor suppressive splice variants.Citation8
Methods and Materials
Cell lines, culturing conditions, and reagents
A172 and T98G cells were provided by Dr. Todd Waldman (Lombardi Comprehensive Cancer Center (LCCC), Georgetown University, Washington, DC). HEK293T and HeLa cells were acquired from the LCCC Tissue Culture Shared Resource. HFF1 cells were obtained from Dr. Louis Weiner (LCCC, Georgetown University, Washington, DC). RKO cells and variants were provided by Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD). All cells tested negative for Mycoplasma spp. contamination, and were maintained in a humidified incubator with 95% air: 5% carbon dioxide. A172, T98G, and T98G stable cell lines were fingerprinted by the LCCC Tissue Culture Shared Resource to verify their authenticity using the standard 9 STR loci and Y-specific amelogenin. A172, T98G HeLa and HEK293T cells were grown in IMEM supplemented with 10% FBS. HFF1 cells were grown in high glucose Dulbecco's Modified Eagles Medium (DMEM; Thermo Scientific) supplemented with 15% FBS.
G418 was purchased from the LCCC Tissue Culture Shared Resource and used at a final concentration of 1.2 mg/ml for A172- and T98G-shERRβ stable cells. Puromycin was purchased from Life Technologies (http://www.lifetechnologies.com/order/catalog/product/A1113803) and used at a final concentration of 2 μg/ml for A172-shp53 stable cells. DY131 (Tocris Bioscience, http://www.tocris.com/dispprod.php?ItemId = 132020#.U43-Fy_yuM4) was dissolved in dimethyl sulfoxide (DMSO), stored as 10 mM stocks at −20°C, and used at the concentrations indicated. Hexadimethrine bromide (polybrene) was purchased from Sigma (http://www.sigmaaldrich.com/catalog/product/sigma/h9268?lang=enandregion=US). Cyclopamine provided by Dr. Insoo Bae (LCCC, Georgetown University, Washington, DC) and GDC-0449 was purchased from Selleckchem (http://www.selleckchem.com/products/GDC-0449.html).
Cell cycle analysis
Cells were seeded at a density of 75,000–100,000 cells per well in 6-well plastic tissue culture dishes on day 0. The following day cells were treated with vehicle or drug. On day 2, floating cells were collected. Adherent cells were trypsinized and added to the collected, floating cells. Cells were pelleted by centrifuging for 5 min at 1,000 RPM. Media was aspirated and cells were washed once with cold 1× PBS and centrifuged again. PBS was aspirated and the cells were then fixed in 75% ethanol. SubG1 (propidium iodide (PI) staining) and cell cycle (DNA content) analyses were performed by the LCCC Flow Cytometry and Cell Sorting Shared Resource.
Cell proliferation assay
Cells were seeded at a density of 1,000 cells per well in 5, 96-well plastic tissue culture dishes per cell line on day 0. On day 1, one plate was stained with crystal violet (Sigma, http://www.sigmaaldrich.com/catalog/product/sigma/c0775?lang=enandregion=US) (untreated). The remaining plates were dosed with vehicle or DY at the specified concentrations. Plates were re-dosed every 72 h and stained on days 3, 6, 10 and 14. For staining, plates were rinsed 1× with 1× PBS to remove excess cellular debris. After, 100 μl of 0.5% crystal violet in 25% methanol was added to each well and incubated at 4C for 10 min. The stain was then removed and the plate was rinsed 4–6× with diH2O to remove excess stain. The plates were left to air-dry at least overnight. On day 15, all plates were rehydrated with a 0.1 M sodium citrate buffer solution in 50% ethanol and read at an absorbance of 550 nm.
Colony formation assay
Cells were seeded at a density of 250 (A172) or 200 (T98G) cells per well in a 12-well plastic tissue culture dish on day 0. On day 1, cells were treated with the indicated doses of DY. The drug was removed on day 2, cells were washed 1× with 1× PBS before returning the cells to their normal culture media (in the absence of DY) for the remainder of the assay. Media was changed one time throughout the assay. Wells were stained on day 10 with 1 ml crystal violet (as above) and left to dry overnight before counting colonies. Images were taken on a Nikon SMZ1500 fluorescence stereoscope at 0.375× magnification.
Immunoblotting
Cells were lysed in modified radioimmunoprecipitation assay (RIPA) bufferCitation49 supplemented with CompleteMini protease inhibitor (http://lifescience.roche.com/shop/products/complete-mini-3271372-1) and PhosSTOP phosphatase inhibitor tablets (http://lifescience.roche.com/shop/products/phosstop) (Roche Applied Science). Polyacrylamide gel electrophoresis and protein transfer were performed as described previously.Citation49,50 Membranes were blocked in 5% nonfat dry milk buffer, unless otherwise noted, and incubated overnight at 4°C with primary antibodies for: PARP (1:1000, http://www.cellsignal.com/products/primary-antibodies/9542), phospho-H3 serine 10 (1:1000, http://www.cellsignal.com/product/productDetail.jsp?productId= 3377) (all from Cell Signaling), p53 (1:1000, Millipore, http://www.millipore.com/catalog/item/05-224) p21 (1:300, Santa Cruz Biotechnology, http://www.scbt.com/datasheet-756-p21-h-164-antibody.html), ERRβ (1:1000, clone H6707 (cl.07) http://www.rndsystems.com/Products/PP-H6707-00) and 1:500 clone H6705 (cl.05) http://www.rndsystems.com/Products/PP-H6705-00), R&D Systems manufactured by Perseus Proteomics), ERRγ (1:100, Abcam, http://www.abcam.com/estrogen-related-receptor-gamma-antibody-ab82319.html). ERRγ purified protein (transcript variant 2) was purchased from Origene (http://www.origene.com/protein/TP312143.aspx). As a loading control, all membranes were re-probed with β–actin primary antibody (1:10,000, Sigma, http://www.sigmaaldrich.com/catalog/product/sigma/a5316?lang=enandregion=US) for ≥1 hour at room temperature. Horseradish peroxidase-conjugated secondary antibodies (1:5000, GE Healthcare Life Sciences, http://www.gelifesciences.com/webapp/wcs/stores/servlet/catalog/en/GELifeSciences/products/AlternativeProductStructure_16827/25005173#) and enhanced chemiluminescent detection HyGLO™ Quick Spray Chemiluminescent (Denville, http://www.denvillescientific.com/node/1213) were used for detection as decribed in.Citation50
Annexin V assay
On day 0, cells were seeded at a density of 100,000 cells per well in 6-well plastic tissue culture dishes. Cells were treated with DY on day 1 for 18 h (RKO) or 24 h (A172 and T98G). Floating cells were collected. Adherent cells were trypsinized and added to the collected, floating cells. Cells were pelleted by centrifuging for 5 min at 1,000 RPM. Media was aspirated and cells were washed once with 1× PBS and centrifuged again. PBS was aspirated and cells were washed once with 500 μl binding buffer (BioLegend, http://www.biolegend.com/annexin-v-binding-buffer-5162.html) and centrifuged. Binding buffer was aspirated and 5 μl of Annexin V antibody conjugated with FITC (BioLegend, http://www.biolegend.com/fitc-annexin-v-5161.html) was added to the cell pellets and lightly vortexed. Samples were incubated for 15 min at room temperature, in the dark, before adding 400 μl of binding buffer. PI was added and levels of FITC and PI were measured by the LCCC Flow Cytometry and Cell Sorting Shared Resource.
Lentiviral shRNA and stable cell lines
Short hairpin RNA (shRNA) directed against human TP53 (pLKO-p53-shRNA-941) and the empty pLKO.1 vector were provided by Dr. Todd Waldman.Citation32 Customized shRNAs directed against ERRβ, ERRγ and the control scrambled inserts in psiLv-mU6 (ERRβ) and psiLv-U6 (ERRγ) were purchased from Genecopeia. The lentiviral helper plasmids for pLKO viral packaging, pHR′8.2ΔR and pCMV-VSV-G, were provided by Dr. Chunling Yi (LCCC, Georgetown University, Washington, DC). To prepare viral stocks, HEK293T cells were seeded at a density of 1.5 million cells per 100 mm plastic tissue culture dish. Packaging cells were triply transfected using Lipofectamine LTX and Plus reagent (Life Technologies, http://www.lifetechnologies.com/us/en/home/life-science/protein-expression-and-analysis/transfection-selection/lipofectamine-ltx-reagent.html) and the following ratios of plasmids: 4 μg pLKO-p53-shRNA-941 or empty pLKO.1, 3 μg pHR′8.2ΔR and 2 μg pCMV-VSV-G. For production of shERRβ-containing virus, cells were co-transfected with shRNA or scrambled control plasmids using the Lenti-Pac FIV Expression Packaging Kit (Genecopoeia, http://www.genecopoeia.com/product/lentiviral-packaging-kit-cells/#order) according to manufacturer's instructions. Media was changed the next day. Supernatant was collected 48 h post-transfection, centrifuged, aliquoted and stored at −80°C. Knockdown was assessed at the protein level by immunoblotting.
shp53: CACCATCCACTACAACTACAT
shERRβ-1: TGAGGACTACATCATGGAT
shERRβ-2: TGCAGCACTTCTATAGCGT
shERRγ−1: GGATGATGGTAGAGCAATA
shERRγ−2: GTTAAGAGGTGTAATCTAA
ERRβ expression constructs
ERRβsf (murine ERRβ, >90% homology to human ERRβsf) was initially purchased from Addgene (plasmid #40798).Citation51 The insert was amplified by PCR, purified using a GE Illustra GFX kit (http://www.gelifesciences.com/webapp/wcs/stores/servlet/productById/en/GELifeSciences/28903470), digested with EcoRI and BamHI restriction enzymes (Promega, http://www.promega.com/products/product-subcategory-search-results/?f=caf9d273-9242-48c5-877e-7c42c91b23c7ands=title) and cloned into the recipient pSG5 vector that was also digested with EcoRI and BamHI using standard molecular biology techniques. Proper insertion was confirmed by automated DNA sequencing (Genewiz) and this plasmid has been re-deposited at Addgene (http://www.addgene.org/52188/). The ERRβ2 and ERRβ-Δ10 splice variants were synthesized and cloned into pSG5 by Genewiz with codon optimization to confer resistance to shERRβ-1 and shERRβ-2, and have also been deposited at Addgene (http://www.addgene.org/52186/ and http://addgene.org/52187/).
Transfection of ERRβsf and ERRβ2
Cells were seeded at a density of 100,000 (T98G) or 150,000 (A172) cells per well in 6-well plastic tissue culture dishes on day 0. On day 1, cells were transfected using either jetPRIME (T98G, Polyplus, http://www.polyplus-transfection.com/2009/08/jetprime%C2%AE/) or Lipofectamine LTX and Plus reagent (A172, Life Technologies, http://www.lifetechnologies.com/us/en/home/life-science/protein-expression-and-analysis/transfection-selection/lipofectamine-ltx-reagent.html) according to manufacturer's instructions. After 4 h, transfection complexes were removed and media was added containing either DMSO or indicated concentrations of DY. Protein was harvested 24 h post-DY treatment.
Dual-luciferase reporter assay
Cells were seeded at 75,000 cells per well in 24-well plastic tissue culture dishes on day 0. On day 1, cells were triply-transfected with 139 ng of reporter plasmid (p21-luc http://www.addgene.org/21723/,Citation34 360 ng of expression plasmid (psg5, ERRβsf and/or ERRβ2), and 1 ng of pRL-SV40-Renilla (https://www.promega.com/products/reporter-assays-and-transfection/reporter-vectors-and-cell-lines/prl-renilla-luciferase-control-reporter-vectors/) using jetPRIME () or 499 ng of reporter plasmid (p21-0 http://www.addgene.org/21723/, p21-2 http://www.addgene.org/21724/, p21-4 http://www.addgene.org/21725/)Citation34 and 1 ng of pRL-SV40-Renilla (). Four hours post-transfection, cells were treated with specified concentrations of DY131. On day 3, 18–20 h after DY treatment, cells were harvested using the dual-luciferase reporter assay system (https://www.promega.com/products/reporter-assays-and-transfection/reporter-assays/dual_luciferase-reporter-assay-system/) according to the manufacturer's instructions. Luciferase activity was normalized to Renilla activity. The experiment was performed in triplicate.
Senescence assay
Cells were seeded at 15,000–20,000 cells per well in 12-well plastic tissue culture dishes on day 0. The following day cells were treated with vehicle or drug and then a senescence associated β-galactosidase staining kit (Cell Signaling, http://www.cellsignal.com/product/productDetail.jsp?productId = 9860) was used 24 h post-treatment to detect cellular senescence. Images were taken on an Olympus IX-71 inverted epifluorescence microscope at 20× magnification in brightfield. The percentage of SA- β-gal positive cells was calculated for 5–12 fields per experiment (∼100 cells per field).
Image analysis and statistics
NIH Image J (http://rsbweb.nih.gov/ij/) was used to perform densitometry. Statistics were performed using GraphPad Prism software 5.0. Analyses used in this study include one-way ANOVA followed by Tukey's multiple comparisons posttest or 2-way ANOVA followed by Bonferroni's posttest. In all figures, data are presented as the mean ± standard deviation (SD) unless otherwise specified. Statistical significance is defined by a P value of ≤0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
972886_Supplementary_Material.zip
Download Zip (8.3 MB)Acknowledgments
We gratefully acknowledge Drs. Todd Waldman and Bert Vogelstein for providing the glioblastoma and RKO isogenic cell lines, respectively. We would also like to thank Drs. Christopher Albanese, Maria Laura Avantaggiati, Insoo Bae, Stephen Byers, Karen Creswell, G. Ian Gallicano, Michael Johnson, Jung-Sik Kim, Susette Mueller, Jeffrey Toretsky, Louis Weiner, Anton Wellstein, Ronit Yarden, and Chunling Yi for sharing reagents, helpful discussions and intellectual insights, and/or critical reading of the manuscript.
Funding
This research was generously supported by 2012–2013 seed funding from Partners in Research (partnersinresearch.gumc.georgetown.edu) and R21CA191444 awarded to RBR, and by start-up funds from the Lombardi Comprehensive Cancer Center (LCCC) Cancer Center Support Grant (P30-CA-51008; PI Dr. Louis M. Weiner). Fellowship funding for MMH was provided by the LCCC Tumor Biology Training Grant (T32-CA-009686; PI Dr. Anna T. Riegel), and Post Baccalaureate Training in Cancer Health Disparities Research (PBTDR12228366; PI Dr. Lucile Adams-Campbell). Technical services were provided by the Flow Cytometry, Genomics & Epigenomics, Microscopy & Imaging, and Tissue Culture Shared Resources, which are also supported by P30-CA-51008. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute, the National Institutes of Health, or Susan G. Komen for the Cure.
References
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008; 40:1413-5; PMID:18978789; http://dx.doi.org/10.1038/ng.259
- Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol 2013; 14:153-65; PMID:23385723; http://dx.doi.org/10.1038/nrm3525
- Busslinger M, Moschonas N, Flavell RA. Beta + thalassemia: aberrant splicing results from a single point mutation in an intron. Cell 1981; 27:289–98; PMID:6895866; http://dx.doi.org/10.1016/0092-8674(81)90412-8
- Spritz RA, Jagadeeswaran P, Choudary PV, Biro PA, Elder JT, deRiel JK, Manley JL, Gefter ML, Forget BG, Weissman SM. Base substitution in an intervening sequence of a beta+-thalassemic human globin gene. Proc Natl Acad Sci U S A 1981; 78:2455–9; PMID:6264477; http://dx.doi.org/10.1073/pnas.78.4.2455
- Helmken C, Hofmann Y, Schoenen F, Oprea G, Raschke H, Rudnik-Schöneborn S, Zerres K, Wirth B. Evidence for a modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet 2003; 114:11–21; PMID:14520560; http://dx.doi.org/10.1007/s00439-003-1025-2
- Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993; 363:640–4; PMID:8510758; http://dx.doi.org/10.1038/363640a0
- Imai H, Chan EK, Kiyosawa K, Fu XD, Tan EM. Novel nuclear autoantigen with splicing factor motifs identified with antibody from hepatocellular carcinoma. J Clin Invest 1993; 92:2419–26; PMID:8227358; http://dx.doi.org/10.1172/JCI116848
- Golan-Gerstl R, Cohen M, Shilo A, Suh SS, Bakàcs A, Coppola L, Karni R. Splicing factor hnRNP A2B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res 2011; 71:4464-72; PMID:21586613; http://dx.doi.org/10.1158/0008-5472.CAN-10-4410
- Choi EJ, Jang YJ, Cha EY, Shin JG, Lee SS. Identification and characterization of novel alternative splice variants of human constitutive androstane receptor in liver samples of Koreans and Caucasians. Drug Metab Dispos 2013; 41:888-96; PMID:23378627; http://dx.doi.org/10.1124/dmd.112.049791
- Matic M, Corradin AP, Tsoli M, Clarke SJ, Polly P, Robertson GR. The alternatively spliced murine pregnane X receptor isoform, mPXR(delta171-211) exhibits a repressive action. Int J Biochem Cell Biol 2010; 42:672-82; PMID:20060928; http://dx.doi.org/10.1016/j.biocel.2010.01.001
- Thomas-Chollier M, Watson LC, Cooper SB, Pufall MA, Liu JS, Borzym K, Vingron M, Yamamoto KR, Meijsing SH. A naturally occurring insertion of a single amino acid rewires transcriptional regulation by glucocorticoid receptor isoforms. Proc Natl Acad Sci U S A 2013; 110:17826-31; PMID:24127590; http://dx.doi.org/10.1073/pnas.1316235110
- Benoit G, Cooney A, Giguere V, Ingraham H, Lazar M, Muscat G, Perlmann T, Renaud JP, Schwabe J, Sladek F, et al. International union of pharmacology. LXVI. Orphan nuclear receptors. Pharmacol Rev 2006; 58:798-836; PMID:17132856; http://dx.doi.org/10.1124/pr.58.4.10
- Eichner LJ, Giguère V. Estrogen related receptors (ERRs): a new dawn in transcriptional control of mitochondrial gene networks. Mitochondrion 2011; 11:544-52; PMID:21497207; http://dx.doi.org/10.1016/j.mito.2011.03.121
- Mitsunaga K, Araki K, Mizusaki H, Morohashi K, Haruna K, Nakagata N, Giguère V, Yamamura K, Abe K. Loss of PGC-specific expression of the orphan nuclear receptor ERR-beta results in reduction of germ cell number in mouse embryos. Mech Dev 2004; 121:237-46; PMID:15003627; http://dx.doi.org/10.1016/j.mod.2004.01.006
- Luo J, Sladek R, Bader JA, Matthyssen A, Rossant J, Giguère V. Placental abnormalities in mouse embryos lacking the orphan nuclear receptor ERR-beta. Nature 1997; 388:778-82; PMID:9285590; http://dx.doi.org/10.1038/42022
- Byerly MS, Al Salayta M, Swanson RD, Kwon K, Peterson JM, Wei Z, Aja S, Moran TH, Blackshaw S, Wong GW. Estrogen-related receptor β deletion modulates whole-body energy balance via estrogen-related receptor γ and attenuates neuropeptide Y gene expression. Eur J Neurosci 2013; 37:1033-47; PMID:23360481
- Byerly MS, Swanson RD, Wong GW, Blackshaw S. Estrogen-related receptor β deficiency alters body composition and response to restraint stress. BMC Physiol 2013; 13:10; PMID:24053666; http://dx.doi.org/10.1186/1472-6793-13-10
- Collin RW, Kalay E, Tariq M, Peters T, van der Zwaag B, Venselaar H, Oostrik J, Lee K, Ahmed ZM, Caylan R, et al. Mutations of ESRRB encoding estrogen-related receptor beta cause autosomal-recessive nonsyndromic hearing impairment DFNB35. Am J Hum Genet 2008; 82:125-38; PMID:18179891; http://dx.doi.org/10.1016/j.ajhg.2007.09.008
- Zhou W, Liu Z, Wu J, Liu JH, Hyder SM, Antoniou E, Lubahn DB.. Identification and characterization of two novel splicing isoforms of human estrogen-related receptor beta. J Clin Endocrinol Metab 2006; 91:569-79; PMID:16332939; http://dx.doi.org/10.1210/jc.2004-1957
- Bombail V, Collins F, Brown P, Saunders PT. Modulation of ER alpha transcriptional activity by the orphan nuclear receptor ERR beta and evidence for differential effects of long- and short-form splice variants. Mol Cell Endocrinol 2010; 314:53-61; PMID:19755138; http://dx.doi.org/10.1016/j.mce.2009.09.007
- Sengupta D, Bhargava DK, Dixit A, Sahoo BS, Biswas S, Biswas G, Mishra SK. ERRβ signalling through FST and BCAS2 inhibits cellular proliferation in breast cancer cells. Br J Cancer 2014; 110:2144-58; PMID:24667650; http://dx.doi.org/10.1038/bjc.2014.53
- Yu S, Wong YC, Wang XH, Ling MT, Ng CF, Chen S, Chan FL. Orphan nuclear receptor estrogen-related receptor-beta suppresses in vitro and in vivo growth of prostate cancer cells via p21(WAF1CIP1) induction and as a potential therapeutic target in prostate cancer. Oncogene 2008; 27:3313-28; PMID:18071305; http://dx.doi.org/10.1038/sj.onc.1210986
- Yu DD, Forman BM. Identification of an agonist ligandfor estrogen-related receptors ERRbetagamma 1. BioorgMedChemLett 2005; 15:1311-3.
- Zuercher WJ, Gaillard S, Orband-Miller LA, Chao EY, Shearer BG, Jones DG, Miller AB, Collins JL, McDonnell DP, Willson TM. Identification and structure-activity relationship of phenolic acyl hydrazones as selective agonists for the estrogen-related orphan nuclear receptors ERRbeta and ERRgamma. J Med Chem 2005; 48:3107-9; PMID:15857113; http://dx.doi.org/10.1021/jm050161j
- Yu S, Wang X, Ng CF, Chen S, Chan FL. ERRgamma suppresses cell proliferation and tumor growth of androgen-sensitive and androgen-insensitive prostate cancer cells and its implication as a therapeutic target for prostate cancer. Cancer Res 2007; 67:4904-14; PMID:17510420; http://dx.doi.org/10.1158/0008-5472.CAN-06-3855
- Wang L, Zuercher WJ, Consler TG, Lambert MH, Miller AB, Orband-Miller LA, McKee DD, Willson TM, Nolte RT. X-ray crystal structures of the estrogen-related receptor-gamma ligand binding domain in three functional states reveal the molecular basis of small molecule regulation. J Biol Chem 2006; 281:37773-81; PMID:16990259; http://dx.doi.org/10.1074/jbc.M608410200
- Wang Y, Arvanites AC, Davidow L, Blanchard J, Lam K, Yoo JW, Coy S, Rubin LL, McMahon AP. Selective identification of hedgehog pathway antagonists by direct analysis of smoothened ciliary translocation. ACS Chem Biol 2012; 7:1040-8; PMID:22554036; http://dx.doi.org/10.1021/cb300028a
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000; 406:1005-9; PMID:10984056; http://dx.doi.org/10.1038/35023008
- Robarge KD, Brunton SA, Castanedo GM, Cui Y, Dina MS, Goldsmith R, Gould SE, Guichert O, Gunzner JL, Halladay J, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett 2009; 19:5576-81; PMID:19716296; http://dx.doi.org/10.1016/j.bmcl.2009.08.049
- Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 1997; 106:348-60; PMID:9362543; http://dx.doi.org/10.1007/s004120050256
- Guo XW, Th’ng JP, Swank RA, Anderson HJ, Tudan C, Bradbury EM, Roberge M. Chromosome condensation induced by fostriecin does not require p34cdc2 kinase activity and histone H1 hyperphosphorylation, but is associated with enhanced histone H2A and H3 phosphorylation. EMBO J 1995; 14:976-85; PMID:7889943
- Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol 2007; 27:662-77; PMID:17060456; http://dx.doi.org/10.1128/MCB.00537-06
- Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Kinzler KW, Vogelstein B, Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A 2009; 106:3964-9; PMID:19225112; http://dx.doi.org/10.1073/pnas.0813333106
- Zeng YX, Somasundaram K, el-Deiry WS. AP2 inhibits cancer cell growth and activates p21WAF1CIP1 expression. Nat Genet 1997; 15:78-82; PMID:8988173; http://dx.doi.org/10.1038/ng0197-78
- Chan CH, Gao Y, Moten A, Lin HK. Novel ARFp53-independent senescence pathways in cancer repression. J Mol Med (Berl) 2011; 89:857-67; PMID:21594579; http://dx.doi.org/10.1007/s00109-011-0766-y
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31:3406-15; PMID:12824337; http://dx.doi.org/10.1093/nar/gkg595
- Gao L, Zhang L, Hu J, Li F, Shao Y, Zhao D, Kalvakolanu DV, Kopecko DJ, Zhao X, Xu DQ. Down-regulation of signal transducer and activator of transcription 3 expression using vector-based small interfering RNAs suppresses growth of human prostate tumor in vivo. Clin Cancer Res 2005; 11:6333-41; PMID:16144938; http://dx.doi.org/10.1158/1078-0432.CCR-05-0148
- Luo KQ, Chang DC. The gene-silencing efficiency of siRNA is strongly dependent on the local structure of mRNA at the targeted region. Biochem Biophys Res Commun 2004; 318:303-10; PMID:15110788; http://dx.doi.org/10.1016/j.bbrc.2004.04.027
- Rudnick SI, Swaminathan J, Sumaroka M, Liebhaber S, Gewirtz AM. Effects of local mRNA structure on posttranscriptional gene silencing. Proc Natl Acad Sci U S A 2008; 105:13787-92; PMID:18784366; http://dx.doi.org/10.1073/pnas.0805781105
- Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nat Rev Clin Oncol 2011; 8:25-37; PMID:20975744; http://dx.doi.org/10.1038/nrclinonc.2010.174
- Mahyar-Roemer M, Roemer K. p21 Waf1Cip1 can protect human colon carcinoma cells against p53-dependent and p53-independent apoptosis induced by natural chemopreventive and therapeutic agents. Oncogene 2001; 20:3387-98; PMID:11423989; http://dx.doi.org/10.1038/sj.onc.1204440
- Shaulian E, Schreiber M, Piu F, Beeche M, Wagner EF, Karin M. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell 2000; 103:897-907; PMID:11136975; http://dx.doi.org/10.1016/S0092-8674(00)00193-8
- Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, Ozawa T, James CD, Waldman T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res 2010; 70:3228-38; PMID:20354191; http://dx.doi.org/10.1158/0008-5472.CAN-09-4559
- Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A 2007; 104:15472-7; PMID:17875987; http://dx.doi.org/10.1073/pnas.0707351104
- Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nuñez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993; 74:597-608; PMID:8358789; http://dx.doi.org/10.1016/0092-8674(93)90508-N
- Laitem C, Leprivier G, Choul-Li S, Begue A, Monte D, Larsimont D, Dumont P, Duterque-Coquillaud M, Aumercier M. Ets-1 p27: a novel Ets-1 isoform with dominant-negative effects on the transcriptional properties and the subcellular localization of Ets-1 p51. Oncogene 2009; 28:2087-99; PMID:19377509; http://dx.doi.org/10.1038/onc.2009.72
- Sierens JE, Scobie GA, Wilson J, Saunders PT. Cloning of oestrogen receptor beta from Old and New World primates: identification of splice variants and functional analysis. J Mol Endocrinol 2004; 32:703-18; PMID:15171710; http://dx.doi.org/10.1677/jme.0.0320703
- Bonnal S, Vigevani L, Valcárcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov 2012; 11:847-59; PMID:23123942; http://dx.doi.org/10.1038/nrd3823
- Bouker KB, Skaar TC, Fernandez DR, O’Brien KA, Clarke R. Interferon regulatory factor-1 mediates the proapoptotic but not cell cycle arrest effects of the steroidal antiestrogen ICI 182,780 (Faslodex, Fulvestrant). Cancer Res 2004; 64:4030-9; PMID:15173018; http://dx.doi.org/10.1158/0008-5472.CAN-03-3602
- Riggins RB, Lan JP, Zhu Y, Klimach U, Zwart A, Cavalli LR, Haddad BR, Chen L, Gong T, Xuan J, et al. ERR{gamma} mediates tamoxifen resistance in novel models of invasive lobular breast cancer. Cancer Res 2008; 68:8908-17; PMID:18974135; http://dx.doi.org/10.1158/0008-5472.CAN-08-2669
- Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 2012; 150:1209-22; PMID:22980981; http://dx.doi.org/10.1016/j.cell.2012.08.023