
Abstract
Despite extensive study, the mechanisms of cell fate choice upon p53 activation remain poorly understood. Using genome-wide shRNA screening, we recently identified the ATM kinase as synthetic lethal with Nutlin-3, an MDM2 inhibitor that leads to non-genotoxic p53 activation. Here, we demonstrate that while this synthetic lethal interaction relies upon components of both the intrinsic and extrinsic apoptotic pathways (e.g., BAX and BID), it is not due to significant ATM effects on the expression of p53 target genes. Instead, loss of ATM activity results in increased mitochondria and reactive oxygen species that drive apoptosis. Finally, we provide evidence that pharmacologic inhibition of ATM blocks autophagy in direct opposition to p53, which activates this process, and that inhibition of autophagy is sufficient to elicit an apoptotic response when combined with Nutlin-3.
Introduction
The tumor suppressor p53 is a master regulator of the cellular response to stress. It is activated in response to a wide range of cellular insults, from oncogene activation to DNA damage, driving in turn diverse cellular outcomes such as cell cycle arrest and apoptosis.Citation1-3 This inherent pleiotropy has prevented a full understanding of the role of p53 in cell fate choice and hinders the development of effective p53-based therapeutics. Although TP53 is mutated in approximately 50% of human cancers, the remaining 50% inhibit p53 by other mechanisms such as MDM2 amplification, making wild type p53 an attractive target for therapeutic intervention.Citation4 Nutlin-3 is a small molecule inhibitor of MDM2, the cognate E3 ubiquitin ligase for p53, that effectively stabilizes and activates p53 without the genotoxic effects of traditional chemotherapeutics.Citation5 Unfortunately, owing to the pleiotropy associated with p53 activation, Nutlin-3 only triggers apoptosis in a small fraction of cancer cell types, and instead activates a reversible and therapeutically undesirable cell cycle arrest in the majority of cell lines tested.Citation6
We recently reported a genome-wide Synthetic Lethal with Nutlin-3 shRNA screen to identify genes whose activity protects cells from Nutlin-3-induced cell death.Citation7 We found that genetic or pharmacologic ablation of ATM kinase activity results in apoptosis in response to non-genotoxic p53 activation. Furthermore we demonstrated that this survival role of ATM is partially independent of its role in the DNA Damage Response (DDR), as knockdown of the damage sensor 53BP1 has no effect on the ability of the combination of Nutlin-3 and an ATM inhibitor (ATMi) to kill cancer cells. In fact, loss of 53BP1 increases relative viability in response to Nutlin-3.Citation7,8 In light of increasing evidence for roles of ATM in various cellular homeostatic processes, we examined how ATM protects cells from apoptosis in response to non-genotoxic p53 activation. In this report, we present evidence that Nutlin-3 and ATMi synthetic lethality utilizes a canonical apoptotic pathway with partial requirements for the p53 target gene BAX as well as Caspase 8, but is not due to significant changes in p53 transactivation of arrest versus apoptotic genes. We also show that loss of ATM activity leads to an increase in levels of mitochondria and mitochondrial reactive oxygen species (ROS) and that these ROS are required for apoptosis. Finally, we show that inhibition of ATM leads to a block in autophagy and that blocking autophagy is sufficient to kill cells in response to Nutlin-3 treatment. These findings broaden our understanding of the role of ATM with respect to the p53 network and highlight a functional interaction that may be exploited therapeutically.
Results and Discussion
Loss of ATM activity induces p53-dependent cell death in a manner independent of changes in expression of p53 target genes
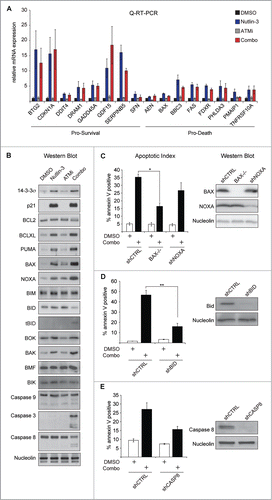
Nutlin-3 and ATMi (KU-55933) synergize to induce apoptosis in a number of cancer cell types, including colorectal cancer cell lines, but do not kill non-transformed cells (Fig. S1).Citation7 To explore the mechanism of Nutlin-3 and ATMi synthetic lethality, we began by examining the effects of each of these drugs individually or in combination on the expression of a panel of both pro-survival and pro-death p53 target genes in HCT116 cells. Although we previously demonstrated that the combination treatment did not affect transactivation of a few of the most prominent p53 target genes, we hypothesized that a number of additional targets may be responsible for the apoptotic phenotype.Citation7 Relative to Nutlin-3 treatment alone, combined treatment with Nutlin-3 and ATMi has no significant effects on the expression of a panel of 16 canonical direct p53 target genes with known pro-survival and pro-death functions (), suggesting that p53 transactivation is largely unaffected by ATM inhibition. Because ATM could be affecting expression of these genes at post-transcriptional steps, we next tested the expression levels of various pro-survival and pro-death proteins in response to the single or combination treatments. While many of these factors showed no change in their expression, levels of tBID and cleaved Caspases 3, 8, and 9 were increased in the combined treatment relative to either Nutlin-3 or ATMi alone (). We also observed greater increases in the expression of the pro-apoptotic proteins BAX and NOXA in response to combination treatment relative to either single agent despite the fact that there was no significant effect on the corresponding mRNA of these genes, BAX and PMAIP1 (). To test whether these alterations in gene expression are contributing to the synthetic lethal phenotype as opposed to indirect downstream effects, we compared BAX-/- cells to established HCT116 cell lines stably expressing a control shRNA or a NOXA-targeting shRNA.Citation9 While BAX-/- cells showed a partial rescue from apoptosis triggered by Nutlin-3 and ATMi combination treatment, NOXA knockdown had no significant effect (). HCT116 cell lines stably expressing shRNAs targeting BID and Caspase 8 were also tested to determine the roles of these proteins in Nutlin-3 and ATMi synthetic lethality.Citation9 Although both BID and Caspase 8 knockdowns partially blocked apoptosis in response to Nutlin-3 and ATMi, only BID knockdown had a statistically significant effect (). These data indicate that inhibition of ATMi in the context of non-genotoxic p53 activation by Nutlin-3 results in an apoptotic response involving components of the extrinsic pathway and that this apoptosis is partially dependent on the p53 target gene BAX. They also demonstrate that this synthetic lethal interaction is not due to widespread effects of ATM inhibition on the p53 gene expression program.
Figure 1. Synthetic Lethality of Nutlin-3 and ATMi is independent of changes to the p53 transcriptional program. (A) HCT116 cells were treated with DMSO, 20 μM Nutlin-3R, 10 μM ATMi or the combination for 24 hours and Q-RT-PCR (B) HCT116 cells were treated as in (A) and protein lysates prepared and western blots were performed using the indicated antibodies. (C) HCT116 cells stably expressing a non-targeting shRNA (shCTRL), an shRNA targeting NOXA or null for BAX, were treated as in (A) and apoptotic levels measured via Annexin-V staining (left). Western blots to confirm target gene knockdown (right). (D and E) HCT116 cell stably expressing the indicated shRNAs were treated and analyzed as in (C). Data presented are average of at least 3 independent replicates +/− SEM. *P < 0.05, **P < 0.01.
Loss of ATM activity increases reactive oxygen species and mitochondria levels
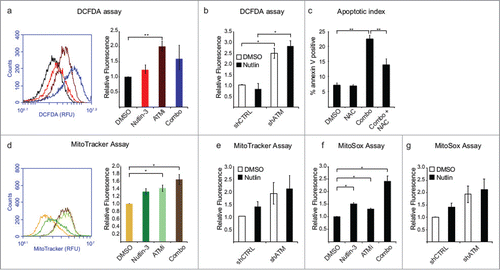
The known cellular roles for ATM have expanded beyond the DDR in recent years to include functions as diverse as NF-κB signaling and the oxidative stress response.Citation10,11 As we previously demonstrated that the synthetic lethality of ATM with non-genotoxic p53 activation by Nutlin-3 is partially independent of its role in the DDR; we next tested whether oxidative stress plays a role. To determine cellular levels of ROS upon treatment with Nutlin-3, ATMi, or the combination, cells were treated overnight and then probed with chloromethyl 2′, 7′-dichlorodihydrofluorescein diacetate (DCFDA), a cell-permeable indicator that fluoresces when oxidized. In fact, inhibition of ATM produces a marked increase in intracellular ROS levels, while the combination treatment exhibits a slightly smaller increase, possibly due to the loss from the population of cells with high ROS levels via apoptosis (). A recent report implicated mitochondrial p53 in ROS generation, consistent with the minor increase in ROS we saw in HCT116 cells treated with Nutlin-3 alone.Citation12 In RKO cells, both ATMi- and combination-treated cells display significant increases in ROS levels (Fig. S2A). To confirm that increased ROS levels were due to loss of ATM activity, we created HCT116 cells stably expressing an shRNA targeting ATM and compared their ROS levels to a non-targeting shRNA control cell line. Indeed, ATM knockdown cells display a substantial increase in ROS levels relative to the control cell line regardless of p53 activation status (). In order to more directly test the role of ROS in synthetic lethality, HCT116 cells were treated with the ROS scavenger N-acetyl cysteine (NAC) prior to Nutlin-3 and ATMi combination treatment and we observed that NAC prevented a complete apoptotic response indicating that ROS are required for this apoptotic response (). Interestingly, the MDM2 inhibitor RITA was recently shown to induce apoptosis via a mechanism that involves ROS generation, suggesting that this accumulation of ROS may be one of the key underlying differences in cell fate choice between RITA and Nutlin-3.Citation13 Next, we sought to determine the source of these ATMi-induced ROS by examining a major source of ROS production, the mitochondria. MitoTracker staining revealed that while Nutlin-3 treatment leads to a minor increase in mitochondria levels, both ATMi and combination treatments lead to significant increases in mitochondria (). ATM knockdown cells also display increased levels of mitochondria (). These data are consistent with reports that Ataxia Telangiectasia cells display defects in mitochondrial homeostasis.Citation14 Furthermore, treatment with Nutlin-3, ATMi, or the combination results in significantly increased levels of mitochondrial superoxide as measured with the MitoSox probe, which is also observed upon ATM knockdown (). Combination treatment produced increases in both mitochondria and mitochondrial ROS in RKO cells as well (Fig. S2B and C).
Figure 2. Loss of ATM activity promotes accumulation of ROS and mitochondria. (A) HCT116 cells were treated with DMSO, Nutlin-3, ATMi or the combination for 24 hours, prior to incubation for 30 min with 10 μM dichlorofluoresceine diacetate (DCFDA) at 37°C and analysis via flow cytometry. (Left) Representative histogram for DMSO (black), Nutlin-3 (red), ATMi (maroon) and combination (blue) treated cells. (Right) Histogram of DCFDA replicates. (B) HCT116 cells stably expressing an shRNA targeting ATM were treated as indicated for 24 hours prior to DCFDA analysis by flow cytometry. (C) HCT116 cells were treated with 5 mM N-acetyl cysteine (NAC) prior to addition of Nutlin-3 and ATMi for 24 hours followed by analysis of Annexin V levels by flow cytometry. (D) HCT116 cells were treated as in (A), prior to incubation for 30 min with 100 nM MitoTracker Green at 37°C and analysis via flow cytometry. (Left) Representative histogram for DMSO (yellow), Nutlin-3 (green), ATMi (light green) and combination (brown) treated cells. (Right) Histogram of DCFDA replicates. (E) HCT116 cells stably expressing an shRNA targeting ATM were treated as indicated for 24 hours prior to MitoTracker Green analysis by flow cytometry. (F) HCT116 cells were treated as in (A) prior to incubation with 5 μM MitoSox for 30 min at 37°C and analysis via flow cytometry. (G) HCT116 cells stably expressing an shRNA targeting ATM were treated as indicated for 24 hours prior to MitoTracker Green analysis by flow cytometry. Data presented are average of at least 3 independent replicates +/− SEM. *P < 0.05, **P < 0.01.
p53-inducible antioxidant genes protect cells from ATM synthetic lethality
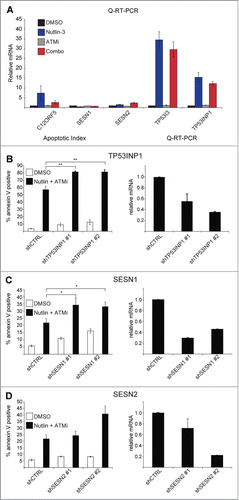
As a master regulator of the cellular response to stress, p53 controls a large transcriptional program often activating genes with seemingly opposite functions including both pro- and anti-oxidant genes, such as PIG3 (TP53I3) and TIGAR (C12ORF5), respectively. To determine what role, if any, several well documented p53 target genes play in the cellular response to Nutlin-3, ATMi or the combination treatment, we performed Q-RT-PCR to determine the effects of the various treatments on the expression levels of the antioxidant genes C12ORF5, SESN1, SESN2, and TP53INP1, and the pro-oxidant gene TP53I3. Although there were no significant changes in expression of any of these genes, there were very minor decreases in induction of C12ORF5, TP53I3 and TP53INP1, relative to Nutlin-3 single agent treatment (). Interestingly, there was no effect of ATMi on the induction of any of the genes tested, indicating that the increases in cellular ROS were not due to changes in expression of these genes. Next, we tested whether reducing the expression of the anti-oxidant p53 target genes TP53INP1, SESN1, or SESN2, could promote apoptosis. Indeed, knockdown of the p53 target gene TP53INP1, an antioxidant of poorly characterized function, leads to a greater increase in apoptosis in response to Nutlin-3 and ATMi combination treatment than a control cell line ().Citation15 We also examined the effect of Sestrin knockdown on combination-treated cells. The Sestrin genes function as antioxidants by regenerating peroxiredoxins, enzymes tasked with peroxide metabolism.Citation16-18 Although knockdown of SESN1 significantly increased levels of apoptosis in response to combination treatment, knockdown of SESN2 had a much more modest effect, indicating that these 2 genes may have non-redundant functions (). These data together with those presented previously demonstrate that ROS generated by the combination treatment are driving the apoptotic response and provide evidence that while p53 is required for this process, it may at the same time protect from a full apoptotic effect by increasing the expression of antioxidant genes.
Figure 3. Antioxidant p53 target genes protect from Nutlin-3 and ATMi synthetic lethality. (A) HCT116 cells were treated as indicated and Q-RT-PCR performed as in . (B–D) (Left) HCT116 cells stably expressing the indicated shRNAs were treated with DMSO or Nutlin-3 and ATMi for 24 hours prior to Annexin V staining and analysis by flow cytometry. (Right) Q-RT-PCR confirmation of shRNA knockdowns. Data presented are average of at least 3 independent replicates +/− SEM. *P < 0.05, **P < 0 .01.
Inhibition of ATM blocks autophagy
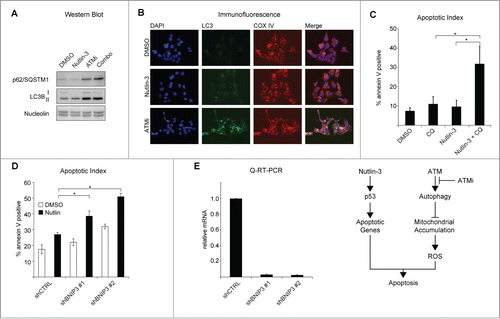
To further investigate the mechanism of ATMi and Nutlin-3 synthetic lethality, we focused on a cellular process that could contribute to accumulation of both mitochondria and ROS. Autophagy is the process by which the cell removes damaged organelles, a potential byproduct of high levels of intracellular ROS, and both p53 and ATM have been shown to regulate autophagy.Citation19-21 Interestingly, p53 and ATM both have paradoxical roles in the activation and repression of autophagy. Nuclear p53 transactivates a number of pro-autophagy genes such as DRAM1, while accumulation of cytoplasmic p53 blocks autophagy.Citation22,23 Additionally, p53 has been shown to inhibit mTOR to repress cellular senescence, and such inhibition of mTOR can also activate autophagy.Citation24-27 In response to ROS, ATM acts through TSC2 to induce autophagy, though there have been reports that inhibition of ATM activates autophagy as well.Citation28,29 We hypothesized that a blockage in autophagy could result in accumulation of mitochondria and ROS. To determine what effects non-genotoxic activation of p53 with Nutlin-3 and inhibition of ATM have on autophagy in HCT116 cells, we examined the levels of several proteins that are markers of autophagic flux.Citation30 Indeed, the levels of the autophagic adaptor protein, p62/SQSTM1, are increased when cells are treated with ATMi or the combination, consistent with a decrease in autophagic flux, and show a modest decrease upon Nutlin-3 treatment ().Citation31 Similarly, Nutlin-3 treatment results in an increase in the ratio of lipidated LC3B (LC3B-II) to full length LC3B (LC3B-I) indicating an increase in autophagic flux (; Fig. S3A).Citation30,32 Inhibition of ATM results in an increase in both LC3B-I and LC3B-II, however the ratio of the 2 is the same as for DMSO-treated cells, indicating a later stage blockage in autophagy (; Fig. S3A). Treatment of cells with Nutlin-3 and ATMi simultaneously results in an increase in LC3B-II and a ratio of LC3B-II: LC3BI that falls in between that of DMSO or Nutlin-3 single agent treatment (; Fig. S3A). Analysis of p62/SQSTM and LC3B levels in RKO colorectal carcinoma cells yielded similar results (Fig. S3B). To confirm the effects of p53 activation and ATM inhibition on autophagy, we analyzed LC3 localization by immunofluorescence after 24 hours of treatment. DMSO-treated HCT116 cells displayed very low levels of LC3 punctae, which are indicative of autophagosome formation, while Nutlin-3-treated cells showed numerous defined LC3 punctae indicating activation of autophagy ().Citation30 Inhibition of ATM, however, resulted in aggregation of LC3 and a partial colocalization with the mitochondrial marker COX IV, consistent with a blockage in autophagy (). Taken together, these data indicate that ATM activity promotes basal autophagy. To test whether autophagy protects from p53-dependent cell fate choice, we inhibited autolysosome turnover with chloroquine and found that this was sufficient to induce apoptosis in Nutlin-3-treated cells (). Finally, to define whether there is a role for mitophagy in Nutlin-3 and ATMi synthetic lethality, we knocked down the selective mitophagy receptor BNIP3 and tested the effects of Nutlin-3 on these cells.Citation33–35 Indeed, we found that depletion of BNIP3 was sufficient to induce apoptosis in response to Nutlin-3 treatment ().
Figure 4. Inhibition of ATM blocks autophagy. (A) HCT116 cells were treated as indicated for 24 hours prior to preparation of lysates and protein gel blotting with the antibodies shown. (B) Cells were treated as in (A) prior to methanol fixation and immunostaining with the indicated antibodies. Images were acquired using an Olympus IX71 inverted microscope with an Olympus DP73 digital camera. Images were analyzed using ImageJ. (C) HCT116 cells were treated with 25 μM chloroquine, Nutlin-3 or the combination for 24 hours prior to analysis of apoptotic levels by Annexin V staining. (D) HCT116 cells stably expressing shRNAs targeting BNIP3 were treated with DMSO or Nutlin-3 for 24 hours prior to analysis of Annexin V staining by flow cytometer. (E) Q-RT-PCR confirmation of shRNA knockdowns. Data presented are average of at least 3 independent replicates +/− SEM. *P < 0.05, **P < 0.01. (F) Model.
We previously reported that inhibition of ATM is synthetic lethal with non-genotoxic activation of p53, however, the mechanism of this interaction remained unclear. Here we report that loss of ATM activity results in an increase in mitochondria and ROS and that these ROS are drivers of the synthetic lethality. We also show that ATM and p53 have synergistic roles in the regulation of autophagy, with both proteins promoting an increase in mitochondrial flux. These data indicate a role for ATM in regulation of mitochondrial homeostasis and oxidative stress that prevent cells from undergoing apoptosis in response to non-genotoxic p53 activation. Of note, the ability of p53 to inhibit mTOR, which can block autophagy, thus affecting both mitochondria and ROS production, may also contribute to the synthetic lethality of combination of ATM inhibition and Nutlin-3.Citation24-27 While these findings shed new light on the functional interaction between p53 and ATM, they are also interesting in the light of combinatorial therapeutics. By studying mechanisms of synthetic lethality, we may uncover pharmacologically actionable targets not strictly identified by high throughput screening, thus highlighting novel therapeutic interventions.
Materials and Methods
Tissue culture and drug treatments
HEK293FT (Invitrogen, http://www.lifetechnologies.com/order/catalog/product/R70007) and RKO (ATCC, http://atcc.org/Products/All/CRL-2577.aspx), and IMR90 (ATCC, http://atcc.org/Products/All/CCL-247.aspx) cells were maintained in Dulbecco's Modified Eagle Medium DMEM, (Life Technologies, http://www.lifetechnologies.com/order/catalog/product/11995065) and HCT116 cells were maintained in McCoy's 5a Medium (Life Technologies, http://www.lifetechnologies.com/order/catalog/product/16600082). All media was (supplemented with 10% FBS (HyClone-SV30014.03) and 1X antibiotic/antimycotic (Life Technologies, http://www.lifetechnologies.com/order/catalog/product/15240062) and all cells were incubated at 37°C and 5% CO2. Nutlin-3 (Cayman Chemical, https://www.caymanchem.com/app/template/Product.vm/catalog/18585), ATMi (KU-55933, Cayman Chemical, https://www.caymanchem.com/app/template/Product.vm/catalog/16336), CM-H2DCFDA (Life Technologies, https://www.lifetechnologies.com/order/catalog/product/c6827), MitoTracker Green (Life Technologies, https://www.lifetechnologies.com/order/catalog/product/M7514) and MitoSox Red (Life Technologies, https://www.lifetechnologies.com/order/catalog/product/M36008) were resuspended in DMSO (Sigma-Aldrich, http://www.sigmaaldrich.com/catalog/product/sigma/d2650) and used at the indicated concentrations. Chloroquine (Sigma-Aldrich, http://www.sigmaaldrich.com/catalog/product/sigma/c6628) was resuspended in dH2O and used at the indicated concentration.
Lentiviral production
2 μg of pLKO target vector (Table S3) and 2 μg of a 9:1 ratio of pMISSIONgagpol (Sigma-Aldrich, http://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma-Aldrich/Vector/1/pmissiongagpol.pdf) and pMISSIONvsvg (Sigma-Aldrich, http://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma-Aldrich/General_Information/1/pmissionvsvg.pdf) were incubated with 12 μg of Polyethylenimine (Polysciences, Inc., http://www.polysciences.com/Catalog/Department/Product/98/categoryid–505/productid–1577/)for 15 minutes at RT prior to addition to HEK293FT cells. Media was changed 14 hours post-transfection. Target cells were transduced 48 hours later by filtering viral supernatant through a 0.45 μm cellulose acetate filter (VWR, https://us.vwr.com/store/catalog/product.jsp?product_id=4830563) directly onto them and incubating with 16 μg of polybrene for 16 hours (Sigma-Aldrich, http://www.sigmaaldrich.com/catalog/product/sigma/107689). Cells were then selected with 10 μg/μL puromycin (Sigma-Aldrich, http://www.sigmaaldrich.com/catalog/product/sigma/p8833).
Western blots, Q-RT-PCR, and flow cytometry
Were performed as previously described.Citation36 Oligonucleotide sequences used in this study are listed in Table S1. Antibodies used in this study are listed in Table S2. Western blots were imaged with an ImageQuant LAS4000 and ImageQuant software. Ratios of LC3B-II/LC3B-I were calculated using ImageJ software.
Microscopy
HCT116 cells were grown on coverslips and treated as described for 24 hours prior to fixation in ice-cold 100% methanol at −20 C for 15 minutes and wash 3X for 5 minutes in 1X PBS. After 60 minutes blocking, cells were incubated with the indicated antibody in Antibody Dilution Buffer (1X PBS/ 1% BSA/ 0.3% Triton-X 100) O/N at 4 C. Cells were washed 3X for 5 minutes in 1X PBS and incubated with fluorescently conjugated secondary antibodies for 2 hours at RT in Antibody Dilution Buffer. After a final 3X wash in 1X PBS cells were mounted on slides using Fluoromount (Sigma-Aldrich, http://www.sigmaaldrich.com/catalog/product/sigma/f4680). Images were acquired using an Olympus IX71 inverted microscope with an Olympus DP73 digital camera. Images were analyzed using ImageJ.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
973330_Supplementary_Materials.zip
Download Zip (3.4 MB)Acknowledgments
We are grateful to Dr. Hill and Dr. Caldwell for their inspirational work. JME is a Howard Hughes Medical Institute Early Career Scientist.
Funding
This work was supported by a grant from the National Institutes of Health (RO1CA117907) and a New Idea Award from the Leukemia and Lymphoma Society.
Supplemental Material
Supplemental data for this article is available online at the publisher's website.
References
- Sullivan KD, Gallant-Behm CL, Henry RE, Fraikin JL, Espinosa JM. The p53 circuit board. Biochim Biophys Acta 2012; 1825:229-44; PMID:22333261
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell 2009; 137:413-31; PMID:19410540; http://dx.doi.org/10.1016/j.cell.2009.04.037
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 2014; 14:359-70; PMID:24739573; http://dx.doi.org/10.1038/nrc3711
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307-10; PMID:11099028; http://dx.doi.org/10.1038/35042675
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844-8; PMID:14704432; http://dx.doi.org/10.1126/science.1092472
- Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A 2006; 103:1888-93; PMID:16443686; http://dx.doi.org/10.1073/pnas.0507493103
- Sullivan KD, Padilla-Just N, Henry RE, Porter CC, Kim J, Tentler JJ, Eckhardt SG, Tan AC, DeGregori J, Espinosa JM. ATM and MET kinases are synthetic lethal with nongenotoxic activation of p53. Nat Chem Biol 2012; 8:646-54; PMID:22660439; http://dx.doi.org/10.1038/nchembio.965
- Brummelkamp TR, Fabius AWM, Mullenders J, Madiredjo M, Velds A, Kerkhoven RM, Bernards R, Beijersbergen RL. An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat Chem Biol 2006; 2:202-6; PMID:16474381; http://dx.doi.org/10.1038/nchembio774
- Henry RE, Andrysik Z, París R, Galbraith MD, Espinosa JM. A DR4:tBID axis drives the p53 apoptotic response by promoting oligomerization of poised BAX. EMBO J 2012; 31:1266-78; PMID:22246181; http://dx.doi.org/10.1038/emboj.2011.498
- Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, Tergaonkar V. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-Mediated IKK activation in response to genotoxic stress. Mol Cell 2010; 40:75-86; PMID:20932476; http://dx.doi.org/10.1016/j.molcel.2010.09.010
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science 2010; 330:517-21; PMID:20966255; http://dx.doi.org/10.1126/science.1192912
- Lee S-Y, Shin SJ, Kim H-S. ERK12 activation mediated by the nutlin‑3‑induced mitochondrial translocation of p53. Int J Oncol 2013; 42:1027-35; PMID:23314357
- Shi Y, Nikulenkov F, Zawacka-Pankau J, Li H, Gabdoulline R, Xu J, Eriksson S, Hedström E, Issaeva N, Kel A, et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ 2014; 21:612-23; PMID:24413150; http://dx.doi.org/10.1038/cdd.2013.186
- Valentin-Vega YA, Maclean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, Cleveland JL, Green DR, Kastan MB. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012; 119:1490-500; PMID:22144182; http://dx.doi.org/10.1182/blood-2011-08-373639
- N’guessan P, Pouyet L, Gosset G, Hamlaoui S, Seillier M, Cano CE, Seux M, Stocker P, Culcasi M, Iovanna JL, et al. Absence of tumor suppressor tumor protein 53-induced nuclear protein 1 (TP53INP1) sensitizes mouse thymocytes and embryonic fibroblasts to redox-driven apoptosis. Antioxid Redox Signal 2011; 15:1639-53; PMID:21235351; http://dx.doi.org/10.1089/ars.2010.3553
- Budanov A V, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 2002; 21:6017-31; PMID:12203114; http://dx.doi.org/10.1038/sj.onc.1205877
- Budanov A V, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004; 304:596-600; PMID:15105503; http://dx.doi.org/10.1126/science.1095569
- Velasco-Miguel S, Buckbinder L, Jean P, Gelbert L, Talbott R, Laidlaw J, Seizinger B, Kley N. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene 1999; 18:127-37; PMID:9926927; http://dx.doi.org/10.1038/sj.onc.1202274
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 2006; 160:1-40; PMID:16430879; http://dx.doi.org/10.1016/j.cbi.2005.12.009
- Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC, Kroemer G. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol 2013; 23:310-22; PMID:23726895; http://dx.doi.org/10.1016/j.semcancer.2013.05.008
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14:197-210; http://dx.doi.org/10.1038/nrm3546
- Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, Is critical for apoptosis. Cell 2006; 126:121-34; PMID:16839881; http://dx.doi.org/10.1016/j.cell.2006.05.034
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10:676-87; PMID:18454141; http://dx.doi.org/10.1038/ncb1730
- Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A 2010; 107:9660-4; PMID:20457898; http://dx.doi.org/10.1073/pnas.1002298107
- Leontieva O V, Gudkov A V, Blagosklonny M V. Weak p53 permits senescence during cell cycle arrest. Cell Cycle 2010; 9:4323-7; PMID:21051933; http://dx.doi.org/10.4161/cc.9.21.13584
- McCubrey JA, Demidenko ZN. Recent discoveries in the cycling, growing and aging of the p53 field. Aging (Albany. NY) 2012; 4:887-93; PMID:23425920
- Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ 2008; 15:1572-81; PMID:18551130; http://dx.doi.org/10.1038/cdd.2008.84
- Alexander A, Cai S-L, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A 2010; 107:4153-8; PMID:20160076; http://dx.doi.org/10.1073/pnas.0913860107
- Lin C-S, Wang Y-C, Huang J-L, Hung CC, Chen JY. Autophagy and reactive oxygen species modulate cytotoxicity induced by suppression of ATM kinase activity in head and neck cancer cells. Oral Oncol 2012; 48:1152-8; PMID:22763242; http://dx.doi.org/10.1016/j.oraloncology.2012.05.020
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Ichimura Y, Kominami E, Tanaka K, Komatsu M. Selective turnover of p62A170SQSTM1 by autophagy. Autophagy 2008; 4:1063-6; PMID:18776737; http://dx.doi.org/10.4161/auto.6826
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 3:542-5; PMID:17611390; http://dx.doi.org/10.4161/auto.4600
- Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, Gustafsson ÅB. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ 2011; 18:721-31; PMID:21278801; http://dx.doi.org/10.1038/cdd.2010.146
- Thomas RL, Kubli DA, Gustafsson AB. Bnip3-mediated defects in oxidative phosphorylation promote mitophagy. Autophagy 2011; 7:775-7; PMID:21460627; http://dx.doi.org/10.4161/auto.7.7.15536]Z
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 2012; 287:19094-104; PMID:22505714; http://dx.doi.org/10.1074/jbc.M111.322933
- Gomes NP, Bjerke G, Llorente B, Szostek SA, Emerson BM, Espinosa JM. Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program. Genes Dev 2006; 20:601-12; PMID:16510875; http://dx.doi.org/10.1101/gad.1398206