
Abstract
Xeroderma pigmentosum group G (XPG) protein is a structure-specific repair endonuclease, which cleaves DNA strands on the 3′ side of the DNA damage during nucleotide excision repair (NER). XPG also plays a crucial role in initiating DNA repair synthesis through recruitment of PCNA to the repair sites. However, the fate of XPG protein subsequent to the excision of DNA damage has remained unresolved. Here, we show that XPG, following its action on bulky lesions resulting from exposures to UV irradiation and cisplatin, is subjected to proteasome-mediated proteolytic degradation. Productive NER processing is required for XPG degradation as both UV and cisplatin treatment-induced XPG degradation is compromised in NER-deficient XP-A, XP-B, XP-C, and XP-F cells. In addition, the NER-related XPG degradation requires Cdt2, a component of an E3 ubiquitin ligase, CRL4Cdt2. Micropore local UV irradiation and in situ Proximity Ligation assays demonstrated that Cdt2 is recruited to the UV-damage sites and interacts with XPG in the presence of PCNA. Importantly, Cdt2-mediated XPG degradation is crucial to the subsequent recruitment of DNA polymerase δ and DNA repair synthesis. Collectively, our data support the idea of PCNA recruitment to damage sites which occurs in conjunction with XPG, recognition of the PCNA-bound XPG by CRL4Cdt2 for specific ubiquitylation and finally the protein degradation. In essence, XPG elimination from DNA damage sites clears the chromatin space needed for the subsequent recruitment of DNA polymerase δ to the damage site and completion of gap-filling DNA synthesis during the final stage of NER.
Introduction
Genomic instability is one of the main forces driving the onset and progression of carcinogenesis. Eukaryotic cells have developed a wide range of DNA repair pathways in order to maintain genomic stability and cell viability when exposed to environmental and endogenous metabolic DNA damaging agents. Nucleotide excision repair (NER) is a versatile DNA repair pathway that eliminates a wide variety of DNA helix-distorting lesions, such as UV-induced cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6–4) pyrimidone photoproducts (6-4PPs),Citation1 and bulky adducts induced by exposure to numerous chemical compounds, such as 7,8-dihydroxy-anti-9,10-epoxy-7,8,9,10-tetrahydro-benzo[a]pyrene [(±)-anti-BPDE], a carcinogenic metabolite of environmentally ubiquitous benzo[a]pyrene, and the widely used chemotherapeutic agent cisplatin. NER entails a multistep reaction and requires the coordinated action of ∼30 proteins that are involved in damage detection, helix opening, lesion verification, dual incision of the damaged strand, removal of the lesion-containing DNA strand, gap-filling DNA synthesis, and ligation.Citation2 In human cells, the minimal set of NER components involved in performing repair reaction comprises XPA, XPC-RAD23B, XPG, RPA, ERCC1-XPF, TFIIH, PCNA, DNA polymerase δ or ϵ, and DNA ligase I.Citation3,4 It is now well resolved that repair proteins are sequentially assembled at the site of DNA lesions in chromatin.Citation5-7 The XPC–RAD23B complex is most likely the initial damage recognition factor when the lesions are situated in a non-transcribed strand.Citation8,9 Following damage recognition, the DNA around the site of the lesion is opened asymmetrically in an ATP-dependent manner, employing the TFIIH complex with its 2 DNA helicases, XPB and XPD.Citation10 XPA, RPA and XPG subsequently join the complex, and the recruitment of ERCC1-XPF by XPA initiates the dual incision of the DNA 5′ and 3′ to the lesion by ERCC1-XPF and XPG, respectively.Citation11,12 Once the incisions have been placed, a damage-containing oligonucleotide of approximately 24–32 nucleotides is released and the DNA structure is restored by subsequent DNA synthesis with the assistance of proliferating cell nuclear antigen (PCNA).Citation13
XPG is an indispensable factor in the NER pathway, functioning as a structure-specific endonuclease to cleave DNA lesion from 3′ side and serving to recruit PCNA to the damage sites for the subsequent gap-filling DNA synthesis.Citation14 Protein-protein interactions are essential in the recruitment of XPG to the damage sites and for XPG to recruit PCNA.Citation14 XPG is recruited to the damage sites by interacting with TFIIH,Citation15,16 while the interaction between XPG and PCNA brings PCNA to the DNA damage sites and facilitates the initiation of the resynthesis step.Citation17,13 Immediately following induction of UV-induced DNA damage in cells, XPG is incorporated into the NER complex with a half-life of 200 seconds and remains intact and associated for 3 to 4 min before disappearing from the damage sites.Citation18 However, the fate of XPG following successful DNA repair has remained unresolved. It might dissociate from the repair complex and recycled for fresh assembly on another lesion or could be simply destroyed following its initial action.
Protein degradation has been shown to play a critical role in DNA repair, probably by facilitating the disassembly of the repair complex in a timely manner.Citation19 Our studies and others have demonstrated that both DDB2 and XPC are degraded in response to UV irradiation, and their degradation is a prerequisite for the subsequent recruitment of downstream NER pathway factors.Citation20-23 For instance, the Cullin4 RING E3 ligases (CRL4) play an important role in the NER pathway.Citation20 CRL4 complex consists of a Cullin 4 (Cul4) scaffold, an adaptor protein (DDB1), and a substrate receptor that binds directly to DDB1. DDB2 and Cdt2 are 2 such substrate receptors. The substrates of CRL4DDB2 E3 ligase include DDB2,Citation24,21 XPC,Citation23 and histone H2A.Citation25 There are more than 6 confirmed mammalian substrates of CRL4Cdt2 ligase, including Cdt1, p21, Set8,Citation26 and DNA Pol δ.Citation27 All these confirmed substrates of CRL4Cdt2 ligase contain a PCNA interaction protein motif (PIP box), and CRL4Cdt2 itself also binds to chromatin after DNA damage, which is dependent on prior interaction of a CRL4Cdt2 substrate with PCNA on chromatin.Citation26 Here, we report a newly identified substrate of CRL4Cdt2, XPG. XPG is degraded in response to both UV and cisplatin treatments. Cdt2 is able to interact with XPG in the presence of PCNA and facilities the XPG degradation. Subsequently, Cdt2-mediated XPG degradation promotes the gap-filling DNA synthesis in NER.
Results
XPG is degraded during NER
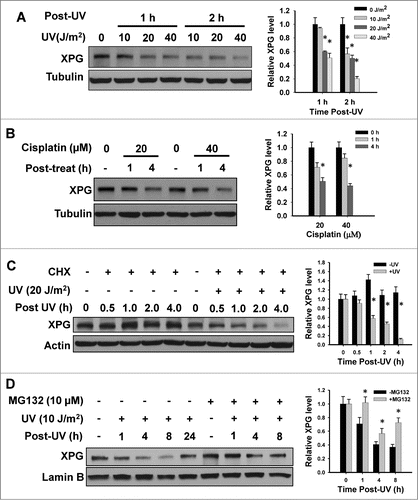
The DNA repair process is controlled not only by the timely orchestrated expression of distinct proteins, but also by the recruitment and assembly of these proteins into the functionally productive repair machinery. We have previously reported that NER factors DDB2 and XPC are degraded in response to UV irradiation, most probably for making necessary space for the subsequent recruitment and accommodation of other NER factors.Citation20,22 To understand the fate of downstream NER factor XPG during NER, we first determined its protein expression level in response to UV and cisplatin treatments, both of which produce NER-specific bulky lesions. As shown in , XPG protein levels exhibited a robust time and dose dependent decrease following UV and cisplatin treatments. Additionally, continuous treatment of HeLa cells with 20 or 40 μM of cisplatin induces XPG degradation at 4 h (). To confirm this phenomenon, we compared the decay rates of XPG in UV- or mock-irradiated HeLa cells in the presence of cycloheximide (CHX), an inhibitor of de novo protein synthesis. shows that in the absence of new XPG synthesis, XPG protein exhibits a high decay rate following UV irradiation. This result indicates that the decrease of XPG observed after UV irradiation is most likely due to the protein degradation. Moreover, we treated the cells with proteasome inhibitor MG132 prior to UV irradiation to test the involvement of 26S proteasome in this UV-induced XPG degradation. We found that the UV-induced reduction of XPG level is promptly inhibited in the presence of MG132 (), albeit the incomplete inhibition by 10 μM MG132 could be due to the drug resistance of HeLa cells to MG132. Given that both UV and cisplatin-induced DNA lesions are repaired through NER pathway, these combined data indicate that XPG protein is degraded via proteasome-mediated proteolysis during NER.
Figure 1. (See previous page). XPG is degraded upon DNA damaging agent treatment. (A and B) UV and cisplatin treatment causes a reduction of XPG protein level. HeLa cells were treated with UV (A) or cisplatin (B) for the indicated time periods. Whole cell lysates were prepared and subjected to immunoblotting to detect the expression of XPG. Tubulin was also detected as a loading control. (C) XPG protein exhibits a high decay rate following UV irradiation in the absence of new XPG synthesis. HeLa cells were pretreated with CHX for 0.5 h, UV irradiated and further cultured in the presence of CHX for various time periods. Whole cell lysates were prepared and subjected to immunoblotting to detect the expression level of XPG. Tubulin was also detected as a loading control. (D) XPG is degraded by proteasome upon UV irradiation. HeLa cells were pretreated with MG132 for 0.5 h, UV irradiated and further cultured in the presence of MG132 for various time periods. Whole cell lysates were prepared and subjected to immunoblotting to detect the expression level of XPG. Lamin B was used to serve as a loading control. The levels of total XPG in each lane were quantified and normalized to the loading control and then to the initial amount of XPG. Data from 3 independent experiments were plotted on the right of each figure.
Efficient assembly of incision complex is required for UV-induced XPG degradation
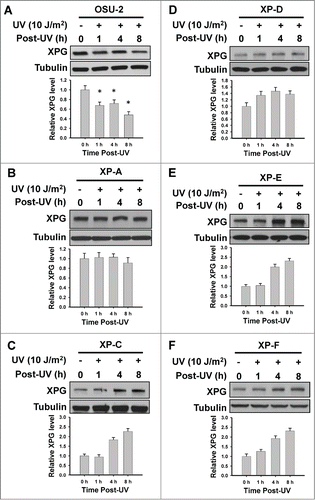
To further investigate the relationship between XPG degradation and the NER process, we determined the kinetics of XPG protein level changes upon UV irradiation in normal human fibroblasts as well as human cell lines belonging to different XP complementation groups. As shown in , normal human fibroblast OSU-2 cells exhibited a typical XPG degradation in response to UV, while all NER-deficient XP cells failed to show UV-induced XPG degradation (). XPC, DDB2 and XPD are believed to be recruited to DNA damage sites prior to XPG in the sequential assembly of NER machinery, and their absence blocks the recruitment of XPG.Citation5,28 Thus, it appears that XPG must be recruited to the damage sites for degradation. In fact, the failure to utilize XPG for repair assembly in the absence of upstream factors DDB2, XPC, and XPD leads to an accumulation of this factor following irradiation (). In addition, XPA and XPF are not required for the recruitment of XPG to UV damage sites,Citation5 but are indispensable for successful excision of damage-containing DNA fragment. Therefore, the finding that XPG is unable to degrade in the absence of XPA or XPF indicates the requirement of the fully formed incision complex for XPG degradation in response to UV irradiation.
Figure 2. UV-induced XPG degradation is dependent on efficient NER. NER-proficient human fibroblast OSU-2 cells (A) and various NER-deficient human fibroblasts (B–F) were UV irradiated at 10 J/m2 and further cultured for the indicated time periods. Whole cell lysates were prepared and subjected to immunoblotting to detect the expression level of XPG. Tubulin was used to serve as a loading control. XPG bands were scanned, quantified, normalized to the tubulin and then to the initial amount of XPG. Data from 3 independent experiments were plotted on the bottom of reach figure.
DDB2 participates in the DNA damage-induced XPG degradation but not through its function in CRL4DDB2 ubiquitin ligase
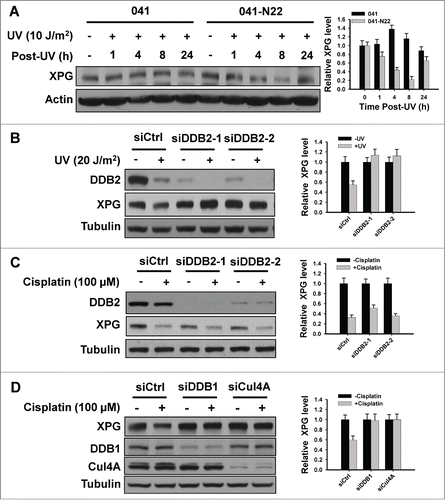
Besides its DNA damage binding function, DDB2 also serves as a substrate-recognition protein via the E3 ubiquitin ligase complex containing Cul4A and DDB1 (CRL4DDB2).Citation29 shows DDB2-deficient XP-E cells failed to show XPG degradation upon UV irradiation, indicating a possibility that DDB2 may participate in DNA damage-induced XPG degradation through either recognizing DNA damage, or its role in CRL4DDB2. We further confirmed the requirement of DDB2 in UV-induced XPG degradation in a DDB2-deficient Li-Fraumani Syndrome 041 cells and the DDB2-restored 041-N22 cells (), as well as HeLa cells with quantitatively downregulated DDB2 expression upon UV irradiation (). 20 J/m2 UV irradiation was able to induce XPG degradation at 2 h post treatment of HeLa cells, while DDB2 downregulation inhibited this UV-induced XPG degradation (). To induce significant XPG degradation in HeLa cells by cisplatin, we treated DDB2-proficient and -downregulated HeLa cells with higher dose of cisplatin (100 μM) for 1 h and further cultured for additional 2 h. As shown in , we were unable to detect the blockage of cisplatin-induced XPG degradation in DDB2-downregulated HeLa cells (). Both UV-induced dimers and cisplatin-induced DNA crosslinks are substrates of NER pathway, and XPG participates in this process through cleaving 3′ of the lesion.Citation11 However, DDB2 is required for the removal of UV-induced CPD but is not required for the repair of cisplatin-induced intra-strand crosslinks.Citation28,30–32 Thus, it appears that the requirement of DDB2 in UV-induced, but not cisplatin-induced, XPG degradation is attributable to the ability of DDB2 to recognize DNA damage.
Figure 3. (See previous page). DDB2 indirectly participates in XPG degradation upon DNA damage. (A–C) DDB2 is required for UV-induced, but not cisplatin-induced XPG degradation. DDB2-deficient 041 cells and DDB2 stably transfected 041-N22 cells were UV irradiated at 10 J/m2, further cultured for the indicated time periods. Whole cell lysates were prepared and subjected to immunoblotting to detect the expression level of XPG. Actin was used to serve as a loading control. (A) HeLa cells were transfected with either control or 2 different DDB2 siRNA for 48 h, UV irradiated at 20 J/m2 (B) or treated with cisplatin at 100 μM for 1 h. (C) Cells were further cultured for 2 h, whole cell lysates were prepared and subjected to immunoblotting to detect the expression level of XPG and DDB2. Tubulin was used to serve as a loading control. (D) Both DDB1 and Cul4A are required for cisplatin-induced XPG degradation. HeLa cells were transfected with either control siRNA or siRNA specific to DDB1, or Cul4A for 48 h, then treated with cisplatin at 100 μM for 1 h. Cells were further cultured for 2 h, whole cell lysates were prepared and subjected to immunoblotting to detect the expression level of XPG, DDB1, and Cul4A. Tubulin was used to serve as a loading control. The levels of total XPG in each lane were quantified and normalized to the loading control and then to the initial amount of XPG. Data from 3 independent experiments were plotted on the right.
DNA damage-induced XPG degradation is mediated by CRL4Cdt2 ubiquitin ligase
In order to determine the participation of CRL4 E3 ligase in DNA damage-induced XPG degradation, we downregulated either DDB1 or Cul4A in HeLa cells and detected the XPG degradation in response to cisplatin treatment. As shown in , downregulation of either one of the crucial subunits of CRL4 complex is able to block cisplatin-induced XPG degradation, indicating that CRL4 ligase complex is required for this process.
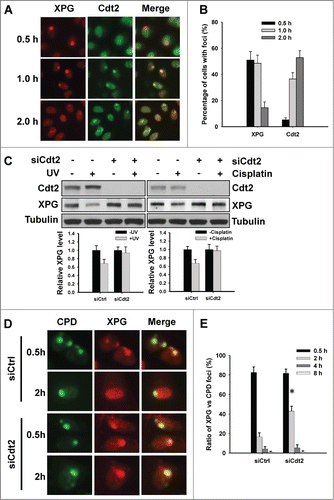
Cdt2 is another substrate-recognition component protein in CRL4 ligase complex. It has been reported that Cdt2 is recruited to UV-induced DNA damage.Citation33 So, we compared the dynamics of Cdt2 and XPG recruitment to DNA damage sites upon micropore local UV irradiation. As shown in , XPG foci can be readily detected at 0.5 h post-UV, but majority of these foci disappeared after 2 h. In contrast, we were unable to detect Cdt2 foci at 0.5 h post-UV, but could detect them at later 1 and 2 h time points. These data indicate that XPG degradation at damage sites is accompanied by the accumulation of Cdt2. We then knocked down Cdt2 expression in HeLa cells and determined XPG degradation in response to either UV or cisplatin treatment. As shown in , both UV and cisplatin-induced XPG degradations were blocked by downregulation of Cdt2. Furthermore, downregulation of Cdt2 also delayed the disappearance of XPG from UV-induced CPD sites (). These results suggest that Cdt2 is the substrate-recognition protein in CRL4 complex that is responsible for the observed XPG degradation upon DNA damage.
Figure 4. Cdt2 is responsible for XPG degradation upon DNA damage. (A and B) Cdt2 is recruited to the UV-induced DNA damage sites. HeLa cells were UV irradiated at 100 J/m2 through a 5 μm isopore filter and maintained in medium for the indicated time periods. The cells were fixed and double immunostained with anti-XPG (red) and anti-Cdt2 (green) antibodies (A), as described in Materials and Methods. The total number of cells with foci was counted from at least 5 separate fields, and the percentage of cells with foci was calculated and plotted. (B) Bar: SD. (C–E) Cdt2 is required for UV and cisplatin-induced XPG degradation. HeLa cells were transfected with either control or Cdt2 siRNA for 48 h, UV irradiated at 40 J/m2 or treated with cisplatin at 100 μM for 1 h. Cells were further cultured for 2 h, whole cell lysates were prepared and subjected to immunoblotting to detect the expression levels of XPG and Cdt2. Tubulin was used to serve as a loading control. The levels of total XPG in each lane were quantified and normalized to the tubulin and then to the initial amount of XPG. Data from 3 independent experiments were plotted at the bottom. (C) HeLa cells growing on coverslips were transfected with either control or Cdt2 siRNA for 48 h, UV irradiated at 100 J/m2 through a 5 μm isopore filter and maintained in medium for 0.5, 2, 4 and 8 h. The cells were fixed and double immunostained with anti-CPD (green) and anti-XPG (red) antibodies. (D) The total numbers of XPG and CPD foci were counted from at least 5 separate fields, the ratios of XPG to CPD foci were calculated and plotted. (E) Bar: SD, *: P < 0.05 compared with siCtrl 2 h.
PCNA mediates XPG degradation by CRL4Cdt2 ubiquitin ligase in response to NER-specific DNA damage
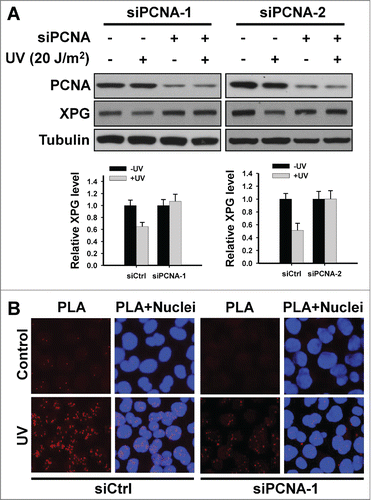
Most of CRL4Cdt2 substrates contain a PCNA interaction protein motif (PIP box).Citation26 XPG protein has been reported to contain this PIP-box motif that mediates an interaction with PCNA as well.Citation17 Thus, we investigated the requirement of PCNA in UV-induced XPG degradation and the interaction between XPG and Cdt2. As shown in , downregulation of PCNA inhibited UV-induced XPG degradation in HeLa cells. By performing in situ proximity ligation assay,Citation34 we clearly visualized the interaction of endogenous XPG and Cdt2 following UV irradiation, as demonstrated by red discrete fluorescent spots (). The requirement of PCNA in this interaction is also confirmed because knockdown of PCNA dramatically reduced the PLA signals. These data indicate that PCNA plays a critical role in CRL4Cdt2-mediated XPG degradation during NER through facilitating the interaction between XPG and Cdt2.
Figure 5. PCNA is required for Cdt2-mediated XPG degradation by mediating the interaction between XPG and Cdt2 upon UV irradiation. (A) PCNA is required for UV-induced XPG degradation. HeLa cells were transfected with either control or 2 different PCNA siRNA for 48 h, UV irradiated at 20 J/m2. Cells were further cultured for 2 h. Whole cell lysates were prepared and subjected to immunoblotting to detect the expression levels of XPG and PCNA. Tubulin was used to serve as a loading control. The levels of total XPG in each lane were quantified and normalized to the loading control and then to the initial amount of XPG. Data from 3 independent experiments were plotted at the bottom. (B) PCNA is required for the interaction between XPG and Cdt2 upon UV irradiation. HeLa cells growing on coverslips were transfected with either siCtrl or siPCNA-1 for 48 h, UV irradiated at 20 J/m2, and further cultured for 2 h. Cells were fixed and subjected to in situ PLA analysis. Primary mouse anti-XPG and rabbit anti-Cdt2 antibodies were combined with secondary PLA probes as described in the Materials and Methods. The interaction events are visible as red dots (nuclear staining in blue).
Cdt2-mediated XPG degradation facilities the recruitment of DNA Pol δ and subsequent gap-filling DNA synthesis during NER
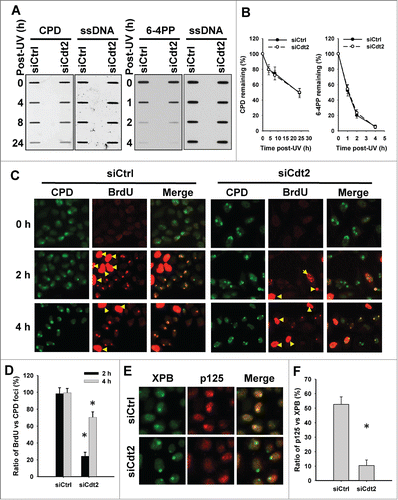
In order to understand the functional significance of Cdt2-mediated XPG degradation in the process of NER, we first determined the effect of inhibiting XPG degradation by Cdt2 knockdown on the removal of CPD upon UV irradiation. As shown in , downregulation of Cdt2 did not affect the removal rate of UV-induced CPD and 6-4PP, indicating that XPG degradation is not for the purpose of lesion excision following UV irradiation. We further analyzed the gap-filling DNA synthesis in HeLa cells with or without Cdt2 knockdown by using BrdU incorporation. As shown in , strong signals from scheduled DNA synthesis in S-phase cells can be distinguished from much weaker cell-cycle independent unscheduled gap-filling BrdU incorporation, resulting from repair synthesis. In these non S-phase cells, gap-filling DNA synthesis occurs efficiently at the UV-induced DNA damage sites in the presence of Cdt2, evidenced by co-localization of CPD and incorporated BrdU. It should be pointed out that the co-localization of CPD and BrdU foci does not mean that the damaged oligonucleotides resulting from dual incision remain bound to genome during the gap-filling DNA synthesis. Each damage focus contains numerous DNA lesions that follow diverse repair dynamics. Thus, the co-localization of CPD and BrdU signals indicate the existence of un-incised DNA lesions mixed with gap-filling DNA synthesis following oligonucleotide excision within the same UV-irradiated focus. In contrast, the incorporation of BrdU into UV-induced DNA damage sites is severely compromised when Cdt2 was downregulated, especially at 2 h time point post UV. This result indicates that Cdt2 is required for the gap-filling DNA synthesis during NER, probably through facilitating XPG degradation. It has been reported that XPG and RPA are released as soon as Pol δ is recruited by the RF-C/PCNA complex.Citation13 Thus, we hypothesized that the purpose for XPG degradation is to clear the necessary space for the subsequent recruitment of DNA Pol δ. To test this, we analyzed the recruitment of DNA Pol δ catalytic subunit p125 to UV-induced DNA damage sites in the presence and absence of Cdt2. As shown in , p125 can be clearly visualized at the damage sites represented by XPB foci at 1 h post UV irradiation. However, Cdt2 knockdown significantly reduced the co-localization kinetics of p125 and XPB, indicating that Cdt2 is required for the recruitment of Pol δ to UV-induced DNA damage sites, likely through degradation and eviction of XPG.
Figure 6. (See previous page). Cdt2-mediated XPG degradation facilitates the recruitment of DNA Pol δ to UV-damaged sites and subsequent gap-filling DNA synthesis. (A and B) Cdt2 is not required for the removal of UV-induced DNA lesions. HeLa cells were transfected with either control siRNA or siCdt2 for 48 h. Cells were UV irradiated at 10 J/m2, and further cultured for the indicated time periods. Genomic DNA was isolated and the same amount of DNA was loaded for ISB with anti-CPD or anti-6-4PP antibody. Single strand DNA (ssDNA) was detected to serve as a loading control. (A) The intensity of each band was scanned and plotted. (B) (C and D) Cdt2 is required for the gap-filling DNA synthesis upon UV irradiation. HeLa cells growing on coverslips were transfected with either control or Cdt2 siRNA for 48 h, UV irradiated at 100 J/m2 through a 5 μm isopore filter and maintained in medium containing BrdU for 2 and 4 h. The cells were fixed and double immunostained with anti-CPD (green) and anti-BrdU (red) antibodies. (C) The total numbers of CPD and BrdU foci in non-S phase cells were counted from at least 5 separate fields, the ratios of BrdU to CPD foci were calculated and plotted. (D) Yellow arrow: S phase cells. Bar: SD, *: P < 0.01 compared with their corresponding siCtrl. (E and F) Cdt2 is required for the recruitment of DNA Pol δ to UV damaged sites. siCtrl and siCdt2 transfected HeLa cells were UV irradiated at 100 J/m2 through a 5 μm isopore filter and maintained in medium for 1 h. The cells were fixed and double immunostained with anti-XPB (green) and anti-p125 (red) antibodies. (E) The total numbers of XPB and p125 foci were counted and the ratios of p125 to XPB foci were calculated and plotted. (F) n = 5, Bar: SD, *: P < 0.01 compared with siCtrl.
Discussion
The structure-specific endonuclease XPG plays an indispensable role in the removal of DNA damage through NER pathway. Besides its role in the incision of DNA lesions, the subsequent degradation of XPG is also critical for the recruitment of gap-filling DNA synthesis complex to the sites of damage, and facilitates the completion of NER.
In the NER mediated damage processing, many proteins are sequentially recruited to the damage site, perform their inherent actions and are released. Two mechanisms might be involved in the release of repair proteins from the damage site — the proteins either just dissociate and are then reused in the repair of other DNA lesions, such as TFIIH,Citation35 or are degraded, such as DDB2 and XPC.Citation20-22 In the present study, we have demonstrated that XPG is another NER pathway protein that is degraded during NER. A recent report has indicated that the XPG protein is released from DNA damage sites in a complex with excised oligonucleotides containing DNA damage, because XPG was present in the dual incision product along with TFIIH following UV irradiation.Citation36 Given that XPG is required for the subsequent recruitment of PCNA to the gapped DNA and plays a key role in DNA resynthesis,Citation13 XPG is unlikely to release before the recruitment of PCNA. We reasoned that XPG recruits PCNA to the damage site subsequent to the dual nucleolytic incision. PCNA then brings Cdt2 to the damage site and mediates the ubiquitylation and degradation of XPG by CRL4Cdt2 in situ.
Most of Cdt2-dependent protein degradation is dependent on a degron sequence that includes a PIP box, a TD motif at position 5 and 6 of the PIP box, and a basic residue (R/K) 4 amino acids downstream from the PIP box (B+4).Citation26 The TD motif promotes high affinity binding to PCNA, whereas the B+4 residue is required for docking of CRL4Cdt2 onto the PCNA-PIP degron complex. However, although we did recognize a PIP box in XPG protein, we failed to find a TD motif or B+4 residue. Our findings further indicate that TD motif and B+4 residues are not essential for a protein to be a CRL4Cdt2 substrate.Citation26
The role of Cdt2 in NER is not fully clear. Although several reports have demonstrated that Cdt2 is able to recruit to the UV-induced DNA damage site,Citation33,37 there is an inconsistency regarding the dynamics of Cdt2 recruitment. It was reported that Cdt2 accumulates at sites of DNA damage soon after UV irradiation in mammalian cells, and Cdt2 spots were only detected in G1 phase cells because Ctd1 is present only in G1 phase.Citation33 However, another report indicated that Cdt2 could be recruited to the site of damage in the absence of Cdt1, and Cdt2 continues to accumulate on chromatin after Cdt1 reaches saturation, indicating that Cdt2 might be recruited to sites of damage by multiple pathways.Citation37 Indeed, at least one more CRL4Cdt2 substrate p21,Citation38 accumulates at the site of damage.Citation39 In the present study, we found that Cdt2 was recruited to UV-induced DNA damage sites in a delayed manner and was continuously retained at the damage sites for a longer time than NER factor XPG. In addition, we also demonstrated that XPG is another CRL4Cdt2 substrate. The delayed accumulation of Cdt2 to UV damage sites might provide sufficient time for XPG to cleave damage-containing DNA oligonucleotides before being proteolytically degraded. Meanwhile, the Cdt2-mediated XPG degradation creates the space for subsequent recruitment of DNA Pol δ for facilitating the gap-filling DNA synthesis.
UV irradiation is known to inhibit transcription, and this was attributed to the blocked elongation at sites of lesions, and repression of transcription initiation by phosphorylation of RNA polymerase II.Citation40 Transcription initiation is also likely to be inhibited as a result of sequestration of TFIIH in repair complexes at damage sites and the post-excision complex.Citation35 The discovery that XPG is also involved in transcription Citation41–43 has led to the realization that transcription initiation would also be inhibited as a result of degradation of XPG in repair complexes at damage sites.
The recruitment of XPG to the damage site is a prerequisite for the subsequent XPG degradation, because the deficiency of any NER factor required for XPG recruitment inhibited the UV-induced XPG degradation. However, XPG recruitment alone to the damage site is not sufficient for XPG degradation. XPA and XPF are not required for the XPG recruitment, but their deficiency also inhibited the UV-induced XPG degradation. Our data has indicated that XPG degradation requires PCNA-mediated recruitment of Cdt2 and, thus, the recruitment of PCNA to the damage sites is another prerequisite for the XPG degradation. The recruitment of PCNA to UV-induced DNA damage site is dependent not only on the presence of XPG on the damaged DNA, but also on the 5′ incision by XPF.Citation13 Given that the recruitment and incision of XPF depend on XPA, we believe that XPA or XPF deficiency can impair the recruitment of PCNA to the damage site. As a result, the XPG protein accumulated at the damage site is unable to get ubiquitylated by Cdt2, and XPG fails to get degraded.
It is interesting that XPG protein level increased upon UV irradiation in XP-C, XP-D, XP-E and XP-F cells, in comparison to the NER-proficient cells. It has been reported that XPG expression can be increased by UV irradiation, probably through the induction of c-fos.Citation44,45 Given that XPG is degraded during the repair of DNA lesions, cells must have developed a mechanism to increase XPG protein level. Thus, the re-synthesized XPG could compensate the destructed XPG, and maintain a certain level of cellular XPG protein for the subsequent repair of DNA damage. However, XPG protein is unable to degrade in NER-deficient XP cells, but the induction of XPG expression is still invoked by UV or cisplatin treatments, leading to an increased XPG protein level in these cells.
CRL4Cdt2 E3 ubiquitin ligase is critical for the cell's response to DNA damage, because it not only suppresses replication licensing by targeting Cdt1 for degradation,Citation46,47 but also promotes DNA repair by promoting the degradation of p21.Citation38 The degradation of Cdt1 after DNA damage has been postulated to constitute a novel checkpoint,Citation37 while the degradation of p21 in response to UV is critical for optimal DNA repair by alleviating the repression of PCNA and promoting translesion DNA synthesis.Citation48 However, there is no direct evidence to show the relationship between Cdt2 and NER. Here, we have demonstrated that Cdt2 does not affect the recognition and dual incision of UV-induced DNA lesions, but is required for the gap-filling DNA synthesis. Cdt2 is recruited to the DNA damage site through PCNA and ubiquitylates XPG for degradation, and more importantly, the destruction of XPG facilitates the recruitment of DNA Pol δ, which catalyzes the gap-filling DNA repair synthesis at the damage site.
Materials and Methods
Cell culture and treatment
HeLa cells were purchased from ATCC (Manassas, VA). Normal human fibroblasts OSU-2 cells were established and maintained in culture in our laboratory, DDB2-deficient Li-Fraumeni Syndrome fibroblast strain, designated 041 fibroblasts, were kindly provided by Dr. Michael Tainsky (MD Anderson Cancer center, Houston, TX), DDB2-expressing 041 cell line (041-N22) was established in our laboratory by stably transfecting pcDNA3.1-His-DDB2 into 041 cells.Citation32 These cells were grown in DMEM supplemented with 10% fetal calf serum (FCS) and antibiotics. XP-A (GM05509), XP-C (GM02096), XP-D (GM03615), XP-E (GM01389), and XP-F (GM04313) fibroblasts were purchased from NIGMS Human Genetic Cell Repository (Camden, NJ), and were grown in MEM supplemented with 10% FCS and antibiotics. For overall UV exposure, cells were washed with PBS, irradiated with varying UV doses and incubated in suitable medium for the desired time period. The irradiation was performed with a germicidal lamp at a dose rate of 0.8 J/m2/s as measured by a Kettering model 65 radiometer (Cole Palmer Instrument Co., Vernon Hill, IL). For cisplatin treatment, cells were maintained in medium with the desired doses of cisplatin (Sigma, St. Louis, MO) for 1 h and then washed with PBS and followed by incubation in fresh drug-free medium for varying times post-treatment.
RNA interference
All small interfering RNA (siRNA) oligonucleotides were synthesized by Dharmacon (Lafayette, CO) in a purified and annealed duplex form. The targeting sequences are siCul4A: 5′-GAA CAG CGA UCG UAA UCA AUU-3′; siDDB1: 5′-UAA CAU GAG AAC UCU UGU C-3′; siDDB2–1: 5′-CAA CUA GGC UGC AAG ACU U-3′; siDDB2–2: 5′-GAU AUC AUG CUC UGG AAU U-3′; siCdt2: 5′-GAA UUA UAC UGC UUA UCG AdTdT-3′; siPCNA-1: 5′-GCC GAG AUC UCA GCC AUA UdTdT-3′; siPCNA-2: 5′-GAU CGA GGA UGA AGA AGG AdTdT-3′; and siCtrl: 5′-UUC UCC GAA CGU GUC ACG UdTdT-3′. siRNA transfections were performed with Lipofectamine 2000 (Life Technologies, Grand Island, NY) according to manufacturer's instruction.
Immunoblotting
Whole cell lysates were prepared by boiling cell pellets for 10 min in lysis buffer (2% SDS, 10% glycerol, 62 mM Tris-HCl, pH 6.8 and a complete mini-protease inhibitor cocktail [Roche Applied Science]). After protein quantification with Bio-Rad Dc Protein Assay (Bio-Rad Laboratories, Hercules, CA), equal amounts of proteins were loaded and separated on a polyacrylamide gel and transferred to a nitrocellulose membrane. Protein bands were immunodetected with appropriate antibodies, e.g., rabbit anti-XPG (A301-484A, 1:3000), rabbit anti-DDB1 (A300-462A, 1:1000), and rabbit anti-Cdt2 (A300-948A, 1:1000) purchased from Bethyl Laboratories, Inc. (Montgomery, TX); goat anti-DDB2 (AF3297, 1:2000) purchased from R&D Systems (Minneapolis, MN); mouse anti-Tubulin, mouse anti-Actin, and goat anti-Lamin B purchased from Santa Cruz Biotechnology (Dallas, TX); and rabbit anti-Cul4A (a gift from Dr. Yue Xiong, University of North Carolina).
Micropore local UV irradiation and Immunofluorescence
HeLa cells growing on glass coverslips were UV irradiated at 100 J/m2 through an isopore polycarbonate filter (Millipore, Bedford, MA), containing pores of 5 μm diameter, as described previously.Citation49 The cells were fixed and permeabilized with 2% paraformaldehyde and 0.5% Triton X-100 after further cultured for the desired time periods. For double staining of XPG and Cdt2, as well as XPB and p125, the coverslips were incubated with mouse anti-XPG (8H7, 1:50, Santa Cruz Biotechnology) and rabbit anti-Cdt2 (A300-948A, 1:200, Bethyl Laboratories) antibodies simultaneously, or mouse anti-p125 (A-9, 1:25, Santa Cruz Biotechnology) and rabbit anti-XPB (S-19, 1:200, Santa Cruz Biotechnology) antibodies simultaneously. After washing 4 times with PBST, coverslips were incubated with Alexa Fluor 488-conjugated goat anti-rabbit and Alexa Fluor 594-conjugated goat anti-mouse (1:200, Life Technologies). For double staining of CPD and XPG, cells were first incubated with mouse anti-XPG (8H7), stained with goat anti-mouse antibody conjugated with Alexa Fluor 594. The cells were then denatured with 2M HCl at 37°C for 10 min, incubated with rabbit anti-CPD (UV-2, 1:1000),Citation50 and stained with goat anti-rabbit antibody conjugated with Alexa Fluor 488. Fluorescence images were obtained with a Nikon Fluorescence Microscope E80i (Nikon, Tokyo, Japan) fitted with appropriate filters for FITC and Texas Red. The digital images were then captured with a cooled CCD camera and processed with the help of its SPOT software (Diagnostic Instruments, Sterling Heights, MI, USA).
Proximate ligation assay (PLA)
HeLa cells were cultured on glass coverslips and UV irradiated at 20 J/m2. The cells were fixed and permeabilized with 2% paraformaldehyde and 0.5% Triton X-100 after further cultured for 2 h. PLA were then performed according to the manufacturer's instruction using mouse anti-XPG (8H7, 1:50, Santa Cruz Biotechnology) and rabbit anti-Cdt2 (A300-948A, 1:200, Bethyl Laboratories) antibodies. Briefly, the slides were blocked with 20% goat serum overnight at 4°C, then incubated with mouse anti-XPG and rabbit anti-Cdt2 antibodies for 1 h at room temperature. After washing, the slides were incubated with Dulink PLA Rabbit MINUS and PLA Mouse PLUS proximity probes (Sigma) and proximity ligation was performed using the Duolink detection reagent kit (Sigma) according to the manufactures protocol. Fluorescence was detected using a Nikon E80i microscope. Images were acquired with a cooled CCD camera and processed with the help of its SPOT software.
Immuno-slot blot (ISB) analysis
HeLa cells growing to confluent were cultured with serum-free medium for 24 h to exclude cells from S phase, then UV irradiated at 10 J/m2, and further cultured in serum-free medium for the desired time periods. The nuclei were isolated and treated with RNase for 1 h. The genomic DNA was then isolated with phenol/chloroform/isoamyl alcohol (25:24:1), precipitated with ethanol, and quantified using PicoGreen kit assay (Life Technologies). The same amounts of denatured DNA were applied to nitrocellulose membranes through a slot blot apparatus. UV-induced CPD were detected with anti-CPD antibody (UV-2, 1:5000) and visualized by Li-Cor Odyssey Imager (Li-COR Biosciences, Lincoln, NE). The intensity of each band was quantified, and the lesion concentrations were determined from a reference standards run in parallel to calculate the relative amounts of CPD remaining at each time point.
Detection of unscheduled DNA synthesis using Bromodeoxyuridine (BrdU) incorporation
HeLa cells were cultured on glass coverslips to grow to confluent. Cells were UV irradiated at 100 J/m2 through an isopore polycarbonate filter containing pores of 5 μm diameter, and further cultured in the medium containing 5 μM BrdU for 2 and 4 h. The cells were fixed and permeabilized with 2% paraformaldehyde and 0.5% Triton X-100, denatured by incubating coverslips in 2M HCl for 10 min at 37°C. CPD and incorporated BrdU moieties were then detected by 1-h incubation with mouse-anti-BrdU (BD, 1:200) and rabbit anti-CPD (UV-2, 1:1000) antibodies, followed by 45-min incubation with Alexa Fluor 488-conjugated goat anti-rabbit and Alex Fluor 594-conjugated goat anti-mouse IgG. Images were captured by using a Nikon Fluorescence Microscope. At least 50 non-S-phase cells were randomly selected for foci counting.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Acknowledgments
We would like to thank Dr. Michael Tainsky (MD Anderson Cancer Center) for providing LFS 041 cells, and Dr. Yue Xiong (University of North Carolina) for providing anti-Cul4A antibody.
Funding
This work was supported by NIH grants ES002388 (AAW) and CA151248 (QEW).
References
- Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev 2006; 106:253-76; PMID:16464005; http://dx.doi.org/10.1021/cr040483f
- Kamileri I, Karakasilioti I, Garinis GA. Nucleotide excision repair: new tricks with old bricks. Trends Genet 2012; 28:566-73; PMID:22824526; http://dx.doi.org/10.1016/j.tig.2012.06.004
- Mu D, Park C-H, Matsunaga T, Hsu DS, Reardon JT, Sancar A. Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem 1995; 270:2415-8; PMID:7852297; http://dx.doi.org/10.1074/jbc.270.6.2415
- Araujo SJ, Tirode F, Coin F, Pospiech H, Syvaoja JE, Stucki M, Hubscher U, Egly JM, Wood RD. Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev 2000; 14:349-59; PMID:10673506
- Volker M, Mone MJ, Karmakar P, Van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JH, van Driel R, Van Zeeland AA, Mullenders LH. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell 2001; 8:213-24; PMID:11511374; http://dx.doi.org/10.1016/S1097-2765(01)00281-7
- Riedl T, Hanaoka F, Egly JM. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J 2003; 22:5293-303; PMID:14517266; http://dx.doi.org/10.1093/emboj/cdg489
- Luijsterburg MS, von BG, Gourdin AM, Politi AZ, Mone MJ, Warmerdam DO, Goedhart J, Vermeulen W, van DR, Hofer T. Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J Cell Biol 2010; 189:445-63; PMID:20439997; http://dx.doi.org/10.1083/jcb.200909175
- Batty D, Rapic’-Otrin V, Levine AS, Wood RD. Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J Mol Biol 2000; 300:275-90; PMID:10873465; http://dx.doi.org/10.1006/jmbi.2000.3857
- Sugasawa K, Ng JMY, Masutani C, Iwai S, Van der Spek P, Eker A, Hanoaka F, Bootsma D, Hoeijmakers JHJ. Xeroderma pigmentosum group C complex is the initiator of global genome nucleotide excision repair. Mol Cell 1998; 2:223-32; PMID:9734359; http://dx.doi.org/10.1016/S1097-2765(00)80132-X
- Sugasawa K, Akagi J, Nishi R, Iwai S, Hanaoka F. Two-step recognition of DNA damage for mammalian nucleotide excision repair: Directional binding of the XPC complex and DNA strand scanning. Mol Cell 2009; 36:642-53; PMID:19941824; http://dx.doi.org/10.1016/j.molcel.2009.09.035
- Evans E, Fellows J, Coffer A, Wood RD. Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. EMBO J 1997; 16:625-38; PMID:9034344; http://dx.doi.org/10.1093/emboj/16.3.625
- Matsunaga T, Mu D, Park CH, Reardon JT, Sancar A. Human DNA repair excision nuclease - analysis of the roles of the subunits involved in dual incisions by using anti- XPG and anti-ERCC1 antibodies. J Biol Chem 1995; 270:20862-9; PMID:7657672; http://dx.doi.org/10.1074/jbc.270.35.20862
- Mocquet V, Laine JP, Riedl T, Yajin Z, Lee MY, Egly JM. Sequential recruitment of the repair factors during NER: the role of XPG in initiating the resynthesis step. EMBO J 2008; 27:155-67; PMID:18079701; http://dx.doi.org/10.1038/sj.emboj.7601948
- Scharer OD. XPG: its products and biological roles. Adv Exp Med Biol 2008; 637:83-92; PMID:19181113; http://dx.doi.org/10.1007/978-0-387-09599-8_9
- Iyer N, Reagan MS, Wu KJ, Canagarajah B, Friedberg EC. Interactions involving the human RNA polymerase II transcriptionnucleotide excision repair complex TFIIH, the nucleotide excision repair protein XPG, and Cockayne syndrome group B (CSB) protein. Biochemistry 1996; 35:2157-67; PMID:8652557; http://dx.doi.org/10.1021/bi9524124
- Thorel F, Constantinou A, Dunand-Sauthier I, Nouspikel T, Lalle P, Raams A, Jaspers NG, Vermeulen W, Shivji MK, Wood RD, et al. Definition of a short region of XPG necessary for TFIIH interaction and stable recruitment to sites of UV damage. Mol Cell Biol 2004; 24:10670-80; PMID:15572672; http://dx.doi.org/10.1128/MCB.24.24.10670-10680.2004
- Gary R, Ludwig DL, Cornelius HL, MacInnes MA, Park MS. The DNA repair endonuclease XPG binds to proliferating cell nuclear antigen (PCNA) and shares sequence elements with the PCNA-binding regions of FEN-1 and cyclin-dependent kinase inhibitor p21. J Biol Chem 1997; 272:24522-9; PMID:9305916; http://dx.doi.org/10.1074/jbc.272.39.24522
- Zotter A, Luijsterburg MS, Warmerdam DO, Ibrahim S, Nigg A, van Cappellen WA, Hoeijmakers JH, van DR, Vermeulen W, Houtsmuller AB. Recruitment of the nucleotide excision repair endonuclease XPG to sites of UV-induced dna damage depends on functional TFIIH. Mol Cell Biol 2006; 26:8868-79; PMID:17000769; http://dx.doi.org/10.1128/MCB.00695-06
- Kouranti I, Peyroche A. Protein degradation in DNA damage response. Semin Cell Dev Biol 2012; 23:538-45; PMID:22353182; http://dx.doi.org/10.1016/j.semcdb.2012.02.004
- El-Mahdy MA, Zhu Q, Wang QE, Wani G, Praetorius-Ibba M, Wani AA. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J Biol Chem 2006; 281:13404-11; PMID:16527807; http://dx.doi.org/10.1074/jbc.M511834200
- Matsuda N, Azuma K, Saijo M, Iemura S, Hioki Y, Natsume T, Chiba T, Tanaka K, Tanaka K. DDB2, the xeroderma pigmentosum group E gene product, is directly ubiquitylated by Cullin 4A-based ubiquitin ligase complex. DNA Repair (Amst) 2005; 4:537-45; PMID:15811626; http://dx.doi.org/10.1016/j.dnarep.2004.12.012
- Wang QE, Praetorius-Ibba M, Zhu Q, El-Mahdy MA, Wani G, Zhao Q, Qin S, Patnaik S, Wani AA. Ubiquitylation-independent degradation of Xeroderma pigmentosum group C protein is required for efficient nucleotide excision repair. Nucleic Acids Res 2007; 35:5338-50; PMID:17693435; http://dx.doi.org/10.1093/nar/gkm550
- Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Tanaka K, et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 2005; 121:387-400; PMID:15882621; http://dx.doi.org/10.1016/j.cell.2005.02.035
- Nag A, Bondar T, Shiv S, Raychaudhuri P. The xeroderma pigmentosum group E gene product DDB2 is a specific target of cullin 4A in mammalian cells. Mol Cell Biol 2001; 21:6738-47; PMID:11564859; http://dx.doi.org/10.1128/MCB.21.20.6738-6747.2001
- Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapic-Otrin V, Levine AS. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci U S A 2006; 103:2588-93; PMID:16473935; http://dx.doi.org/10.1073/pnas.0511160103
- Havens CG, Walter JC. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev 2011; 25:1568-82; PMID:21828267; http://dx.doi.org/10.1101/gad.2068611
- Zhang S, Zhao H, Darzynkiewicz Z, Zhou P, Zhang Z, Lee EY, Lee MY. A novel function of CRL4(Cdt2): regulation of the subunit structure of DNA polymerase delta in response to DNA damage and during the S phase. J Biol Chem 2013; 288:29550-61; PMID:23913683; http://dx.doi.org/10.1074/jbc.M113.490466
- Wang QE, Zhu Q, Wani G, Chen J, Wani AA. UV radiation-induced XPC translocation within chromatin is mediated by damaged-DNA binding protein, DDB2. Carcinogenesis 2004; 25:1033-43; PMID:14742321; http://dx.doi.org/10.1093/carcin/bgh085
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 2003; 113:357-67; PMID:12732143; http://dx.doi.org/10.1016/S0092-8674(03)00316-7
- Fitch ME, Nakajima S, Yasui A, Ford JM. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J Biol Chem 2003; 278:46906-10; PMID:12944386; http://dx.doi.org/10.1074/jbc.M307254200
- Moser J, Volker M, Kool H, Alekseev S, Vrieling H, Yasui A, Van Zeeland AA, Mullenders LH. The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions. DNA Repair (Amst) 2005; 4:571-82; PMID:15811629; http://dx.doi.org/10.1016/j.dnarep.2005.01.001
- Barakat BM, Wang QE, Han C, Milum K, Yin DT, Zhao Q, Wani G, Arafa ES, El-Mahdy MA, Wani AA. Overexpression of DDB2 enhances the sensitivity of human ovarian cancer cells to cisplatin by augmenting cellular apoptosis. Int J Cancer 2009; 127:977-88; PMID:20013802; http://dx.doi.org/10.1002/ijc.25112
- Ishii T, Shiomi Y, Takami T, Murakami Y, Ohnishi N, Nishitani H. Proliferating cell nuclear antigen-dependent rapid recruitment of Cdt1 and CRL4Cdt2 at DNA-damaged sites after UV irradiation in HeLa cells. J Biol Chem 2010; 285:41993-2000; PMID:20929861; http://dx.doi.org/10.1074/jbc.M110.161661
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 2006; 3:995-1000; PMID:17072308; http://dx.doi.org/10.1038/nmeth947
- Kemp MG, Reardon JT, Lindsey-Boltz LA, Sancar A. Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J Biol Chem 2012; 287:22889-99; PMID:22573372; http://dx.doi.org/10.1074/jbc.M112.374447
- Hu J, Choi JH, Gaddameedhi S, Kemp MG, Reardon JT, Sancar A. Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J Biol Chem 2013; 288:20918-26; PMID:23749995; http://dx.doi.org/10.1074/jbc.M113.482257
- Roukos V, Kinkhabwala A, Colombelli J, Kotsantis P, Taraviras S, Nishitani H, Stelzer E, Bastiaens P, Lygerou Z. Dynamic recruitment of licensing factor Cdt1 to sites of DNA damage. J Cell Sci 2011; 124:422-34; PMID:21224399; http://dx.doi.org/10.1242/jcs.074229
- Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev 2008; 22:2496-506; PMID:18794347; http://dx.doi.org/10.1101/gad.1676108
- Perucca P, Cazzalini O, Mortusewicz O, Necchi D, Savio M, Nardo T, Stivala LA, Leonhardt H, Cardoso MC, Prosperi E. Spatiotemporal dynamics of p21CDKN1A protein recruitment to DNA-damage sites and interaction with proliferating cell nuclear antigen. J Cell Sci 2006; 119:1517-27; PMID:16551699; http://dx.doi.org/10.1242/jcs.02868
- Rockx DA, Mason R, van HA, Barton MC, Citterio E, Bregman DB, Van Zeeland AA, Vrieling H, Mullenders LH. UV-induced inhibition of transcription involves repression of transcription initiation and phosphorylation of RNA polymerase II. Proc Natl Acad Sci U S A 2000; 97:10503-8; PMID:10973477; http://dx.doi.org/10.1073/pnas.180169797
- Lee SK, Yu SL, Prakash L, Prakash S. Requirement of yeast RAD2, a homolog of human XPG gene, for efficient RNA polymerase II transcription. implications for Cockayne syndrome. Cell 2002; 109:823-34; PMID:12110180; http://dx.doi.org/10.1016/S0092-8674(02)00795-X
- Le MN, Fradin D, Iltis I, Bougneres P, Egly JM. XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol Cell 2012; 47:622-32; PMID:22771116; http://dx.doi.org/10.1016/j.molcel.2012.05.050
- Schmitz KM, Schmitt N, Hoffmann-Rohrer U, Schafer A, Grummt I, Mayer C. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol Cell 2009; 33:344-53; PMID:19217408; http://dx.doi.org/10.1016/j.molcel.2009.01.015
- Christmann M, Tomicic MT, Origer J, Aasland D, Kaina B. c-Fos is required for excision repair of UV-light induced DNA lesions by triggering the re-synthesis of XPF. Nucleic Acids Res 2006; 34:6530-39; PMID:17130154; http://dx.doi.org/10.1093/nar/gkl895
- Tomicic MT, Reischmann P, Rasenberger B, Meise R, Kaina B, Christmann M. Delayed c-Fos activation in human cells triggers XPF induction and an adaptive response to UVC-induced DNA damage and cytotoxicity. Cell Mol Life Sci 2011; 68:1785-98; PMID:20976523; http://dx.doi.org/10.1007/s00018-010-0546-9
- Arias EE, Walter JC. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat Cell Biol 2006; 8:84-90; PMID:16362051; http://dx.doi.org/10.1038/ncb1346
- Sansam CL, Shepard JL, Lai K, Ianari A, Danielian PS, Amsterdam A, Hopkins N, Lees JA. DTLCDT2 is essential for both CDT1 regulation and the early G2M checkpoint. Genes Dev 2006; 20:3117-29; PMID:17085480; http://dx.doi.org/10.1101/gad.1482106
- Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, Fotedar A, Fotedar R. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 2003; 114:599-610; PMID:13678583; http://dx.doi.org/10.1016/j.cell.2003.08.001
- Wang QE, Zhu Q, Wani MA, Wani G, Chen J, Wani AA. Tumor supressor p53 dependent recruitment of nucleotide excision repair ractors XPC and TFIIH to DNA damage. DNA Repair 2003; 2:483-99; PMID:12713809; http://dx.doi.org/10.1016/S1568-7864(03)00002-8
- Wani AA, Gibson-D’Ambrosio RE, D’Ambrosio SM. Antibodies to UV irradiated DNA: The monitoring of DNA damage by ELISA and indirect immunofluorescence. Photochem Photobiol 1984; 40:465-71; PMID:6390471; http://dx.doi.org/10.1111/j.1751-1097.1984.tb04619.x