
Abstract
Cancer stem cells (CSCs) represent a subpopulation of tumor cells endowed with self-renewal capacity and are considered as an underlying cause of tumor recurrence and metastasis. The metabolic signatures of CSCs and the mechanisms involved in the regulation of their stem cell-like properties still remain elusive. We utilized nasopharyngeal carcinoma (NPC) CSCs as a model to dissect their metabolic signatures and found that CSCs underwent metabolic shift and mitochondrial resetting distinguished from their differentiated counterparts. In metabolic shift, CSCs showed a greater reliance on glycolysis for energy supply compared with the parental cells. In mitochondrial resetting, the quantity and function of mitochondria of CSCs were modulated by the biogenesis of the organelles, and the round-shaped mitochondria were distributed in a peri-nuclear manner similar to those seen in the stem cells. In addition, we blocked the glycolytic pathway, increased the ROS levels, and depolarized mitochondrial membranes of CSCs, respectively, and examined the effects of these metabolic factors on CSC properties. Intriguingly, the properties of CSCs were curbed when we redirected the quintessential metabolic reprogramming, which indicates that the plasticity of energy metabolism regulated the balance between acquisition and loss of the stemness status. Taken together, we suggest that metabolic reprogramming is critical for CSCs to sustain self-renewal, deter from differentiation and enhance the antioxidant defense mechanism. Characterization of metabolic reprogramming governing CSC properties is paramount to the design of novel therapeutic strategies through metabolic intervention of CSCs.
Abbreviations
| GLUT1 | = | glucose transporter 1 |
| HK | = | hexokinase |
| GPI | = | glucose-6-phosphate isomerase |
| PDK | = | pyruvate dehydrogenase kinase |
| PDH | = | pyruvate dehydrogenase |
| POLG | = | mitochondrial DNA polymerase gamma |
| TFAM | = | mitochondrial transcription factor A |
| PGC-1α | = | peroxisome proliferator-activated receptor gamma coactivator 1α |
| ND1 | = | NADH dehydrogenase subunit 1 |
| COX | = | cytochrome c oxidase |
| ATP6 | = | ATP synthase 6 |
| Cu/ZnSOD | = | copper/zinc superoxide dismutase |
| MnSOD | = | manganese superoxide dismutase |
| GR | = | glutathione reductase |
Introduction
Energy metabolism is an important physiology function for cell survival or progression. Different types of cells may use different metabolic pathways. For example, fully differentiated adult cells rely more on oxidative phosphorylation (OXPHOS) for supply of ATP, while cancer cells rely more on anaerobic glycolysis than on OXPHOS for energy supply even in normoxic condition. This special cancer metabolism was first recognized by Otto Warburg as early as the 1920s.Citation1 Several recent studies have shown that stem cells, including embryonic stem cells (ESCs) and adult stem cells, depend mostly on anaerobic metabolism for ATP supply, a phenomenon similar to that observed in cancer cells.Citation2 This metabolic shift of stem cells was also found in induced-pluripotent stem cells (iPSCs).Citation3 Interestingly, during this process that the somatic cells are converted into iPSCs, the upregulation of the expression of glycolytic genes was found to be antecedent to the expression of pluripotent markers.Citation4 This implies that metabolic switch is essential for the reprogramming processes of iPSCs. In addition to a quick production of ATP, glycolysis can provide metabolites to feed into pentose phosphate pathway and offers a source of building blocks and reducing equivalents for biosynthesis to meet the anabolic demands of stem cells for growth.Citation5-8
Cancer stem cells (CSCs) or tumor-initiating cells refer to a subset of tumor cells that are at the apex of the hierarchy and construct phenotypic diversity within tumor masses.Citation9 These CSCs exhibit self-renewal and differentiation capacity to persist in tumors as a distinct population and may cause relapse and metastasis of cancers. It remains to be clarified whether the energy metabolism of CSCs is similar to that in normal stem cells. Palorini et al. found that energy metabolism of a CSC-like osteosarcoma cell line 3AB-OS is similar to that of normal stem cells characterized by an increase of anaerobic glycolysis.Citation10 Higher production of lactate, a glycolytic byproduct, was also found in CSC-like ALDHbright breast cancer cells.Citation11 However, Vlashi et al. showed that CSCs of glioma relied mainly on OXPHOS for energy supply.Citation12 This suggests that the CSCs may exhibit unique metabolic adaptation to oxidative stress, hypoxia, normoxia and microenvironment.
Furthermore, reduction-oxidation (redox) homeostasis is another critical issue for the balance between self-renewal and differentiation of stem cells and CSCs, respectively.Citation2,3,13-19 As compared with differentiated cells, normal mammary epithelial stem cells and hemopoietic stem cells have lower levels of reactive oxygen species (ROS), which are important for their self-renewal and deterrence from differentiation.Citation13-15 Stem cells were reported to have higher expression levels of antioxidant genes to dispose of the ROS and protect cells from oxidative damage.Citation16,17 Increase in the ROS levels may coerce lineage-specific differentiation of stem cells.Citation18 Similar to the findings of normal stem cells, breast CSCs were found to have lower levels of ROS compared with non-tumorigenic cells.Citation19 Cellular ROS may come from different sources, and the majority are from the respiratory chain of mitochondria. Although stem cell research has attracted clinicians and scientists in the past 2 decades, little is known pertaining to the role of mitochondrial metabolism in the regulation of CSC properties.
Accumulating evidence shows that nasopharyngeal carcinoma (NPC) cells with stem-like characteristics are able to self-renew, propagate the tumor, and resist to standard anticancer therapies.Citation20-22 In light of these observations, we used NPC as a model to validate whether CSCs can withstand therapy and spawn metastasis by virtue of their distinct metabolic signatures. The ultimate goal of this study was to thoroughly dissect the key events in energy metabolism of CSCs and provide clues to tackle CSCs by metabolic interventions. Perturbing the survival skills of CSCs through blockade of metabolic pathways might increase the efficacy of the treatment of cancers.
Results
NPC CSCs undergo metabolic shift
Using a behavior selection approach, radioresistant cancer cells were first harvested and their radioresistant phenotype was verified (). The clones with strong radioresistant capacity formed compact spheres which expressed the stem cell self-renewal markers, stage-specific embryonic antigen-3 (SSEA3), and antigen-4 (SSEA4) (). Expression of the ABC-transporter ABCG2 was also increased (). Radioresistant cells capable of forming tumor spheres contained more CSC-like cells and possessed a higher percentage of side population cells (17.5%) when compared to their differentiated counterparts ().
Figure 1. Isolation of NPC CSC clones by behavior selection. (A) Survival fractions of TW01 radioresistant clones and parental cells. TW01 radioresistant clone had significantly higher survival fraction than did parental cells. Immunofluorescent images reveal (B) specific embryonic antigen-3 (SSEA3), SSEA4, and (C) ABCG2 expressed in TW01 tumor spheres. Scale bars in (B) and (C) indicate 10 μm. (D) TW01 parental cells contained 0.1% side population cells. (E) TW01 tumor sphere cells possessed 17.5% side population cells. In (D and E), left plot shows cells incubated with Hoechst 33342; right plot shows cells pre-incubated with fumitremorgin C to block the efflux of Hoechst 33342. (F) Expressions of SSEA3 and SSEA4 quantified by flow cytometry were higher in TW01 CSCs than in TW01 parental cells. (G) Cell morphology of TW01 parental and CSC cells. Phase-contrast images on the left side reveal epithelial type of TW01 parental cells and mesenchymal type of CSCs. Scale bars indicate 100 μm. Immunofluorescence images on the right side indicat the transition of epithelial (E-cad) and mesenchymal (Vim) markers in TW01 parental cells and CSCs. Scale bars indicate 20 μm. (H) Western blot confirmed the EMT of TW01 CSCs at the protein level, with α-tubulin (α-tub) as the loading control. Epithelial marker (E-cad: E-cadherin) was downregulated, while mesenchymal markers (N-cad: N-cadherin and Vim: vimentin) were upregulated in TW01 CSCs compared with the parental cells. (I) Transwell cell invasion assay depicted greater invasion capacity of TW01 CSCs compared with that of the parental cells. (J) Soft agar assay indicated that TW01 CSCs had higher clonogenic formation capacity than that in the parental cells. (PT: parental cells, RR: radioresistant clones, CSC: CSCs isolated by behavior selection; *, P < 0.05; **, P < 0.01).
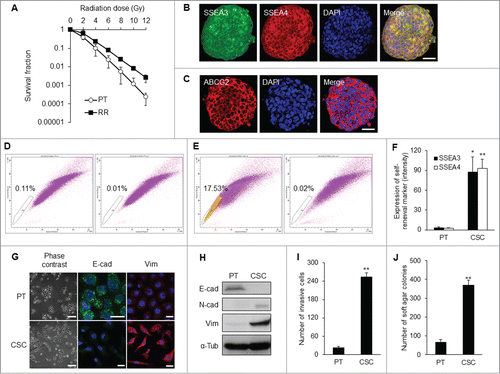
CSCs obtained from behavior selection displayed elevated levels of self-renewal markers SSEA3 and SSEA4 relative to parental cells (). CSCs also exhibited mesenchymal type of morphology, which was distinctively different from the epithelial type of parental NPC cells (). This EMT process was accompanied by down-regulation of epithelial marker E-cadherin and concomitant upregulation of mesenchymal markers, including N-cadherin and vimentin (; Fig. S1A). CSCs manifested higher invasion capacity than did parental NPC cells (). The in vitro tumorigenicity assay also revealed that CSCs formed more colonies in soft agar when compared with their differentiated counterparts ().
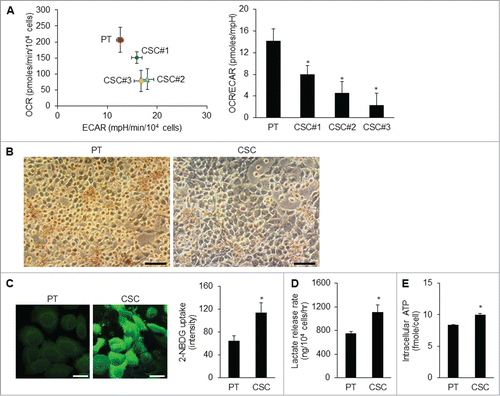
To unravel the metabolic hallmarks of CSCs, we investigated possible pathways of energy metabolism in CSCs. The rates of OCR (O2 consumption rate)/ECAR (extracellular acidification rate) of different CSC clones were lower than those of parental cells (; Fig. S1B), implying that these CSCs did not rely much on mitochondrial respiration for energy supply. In agreement with the OCR, the decrease of cytochrome c oxidase (CCO) staining substantiated the reduction of Complex IV activity of the respiratory chain in CSCs compared with the parental cells (; Fig. S1C). We then utilized 2-NBDG, a fluorescent deoxyglucose analog, to monitor glucose uptake and found that the 2-NBDG uptake of CSCs was higher than that of the parental cells (; Fig. S1D). Likewise, CSCs excreted larger amounts of lactate as a byproduct of anaerobic glycolysis than did the parental cells (). Nevertheless, the ATP levels of the CSCs were not lower than those of the parental cells (). The high glucose uptake and lactate production rate of CSCs suggests that the glycolytic flux was high enough to maintain the energy status of CSCs.
Figure 2. Metabolic shift in CSCs. (A) The O2 consumption rate (OCR) was lower and the extracellular acidification rate (ECAR) was higher in TW01 CSCs compared with those of the parental cells. The values of OCR and ECAR were determined after normalization to cell number. Right plot represents the quantitative results of OCR/ECAR ratios which indicated lower mitochondrial respiration in 3 TW01 CSC clones than that in TW01 parental cells. TW01 CSC clone #3 with the lowest OCR/ECAR ratio was chosen for further metabolic characterization in this study. (B) Cytochrome c oxidase (CCO) activity assay in TW01 parental cells and CSCs was performed through histochemical staining. Faint brown color in cells indicates low CCO activity of TW01 CSCs. Scale bars indicate 100 μm. (C) Immunofluorescence imaging observed by a confocal microscope (left; scale bars indicate 20 μm) and quantified by flow cytometry (right) of 2-NBDG uptake denoted the higher glucose uptake in TW01 CSCs compared with that in TW01 parental cells. (D) TW01 CSCs had higher levels of lactate production than did TW01 parental cells. (E) Intracellular ATP content in TW01 CSCs was higher than that in the parental cells. (*, P < 0.05).
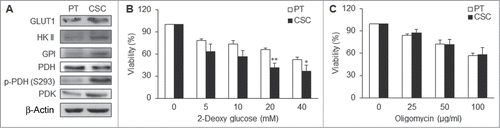
In agreement with the high glycolytic flux of the CSCs, the glycolysis-related proteins or enzymes were upregulated in CSCs. Glycolysis-related enzymes, including glucose transporter 1 (GLUT1), hexokinase II (HK II) and glucose-6-phosphate (GPI), were also elevated in CSCs compared with the parental cells (; Fig. S2A). Pyruvate dehydrogenase kinase (PDK), which elicited the switch from aerobic metabolism to glycolysis, was also upregulated in CSCs (; Fig. S2A). On the contrary, pyruvate dehydrogenase (PDH) was down-regulated and inactivated (phosphorylation of PDH on serine residue 293) in CSCs compared with the parental cells (; Fig. S2A). To further examine the dependence of cells on glycolysis or mitochondrial respiration for ATP supply, cell viability was evaluated by addition of 2-deoxyglucose (2-DG), a glycolytic inhibitor, or oligomycin, an inhibitor of Complex V, to impair mitochondrial respiration. The results showed that cell viability was significantly decreased after treating CSCs with 2-DG () but no such change by treatment with oligomycin (). All these findings implied that NPC CSCs predominately rely on glycolysis to meet their energy demand and survival.
Figure 3. CSCs were dependent on glycolysis for major supply of energy. (A) GLUT1, HK II, GPI, p-PDH (S293), and PDK were upregulated while PDH was down-regulated in TW01 CSCs. (B) 2-deoxyglucose (2-DG) was used to measure the extent of dependence on glycolysis for ATP of TW01 CSCs and the parental cells, respectively. Lower cell viability was detected in CSCs after treatment with the glycolytic inhibitor 2-DG compared with the parental cells. (C) Oligomycin was used to measure the extent of dependence on mitochondrial respiration and OXPHOS of TW01 CSCs and the parental cells. No significant differences of cell viability were observed between the parental cells and CSCs after oligomycin treatment. (*, P < 0.05; **, P < 0.01).
Mitochondrial resetting in CSCs
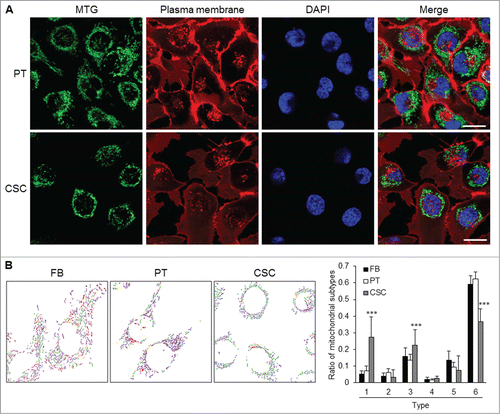
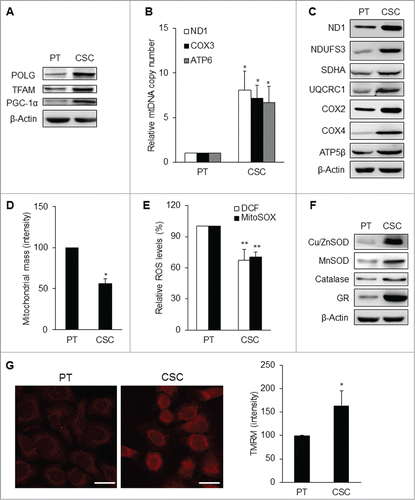
Morphological features of the mitochondrial network within the parental cells and CSCs were considerably different in distribution and shape. Regarding the intracellular distribution of mitochondria, the organelles were distributed throughout the cytoplasm in parental cells, while mitochondria were located at the peri-nuclear region in CSCs (). As to the ultra-structural morphology of mitochondria, CSCs displayed a significantly higher percentage of small globules and linear tubules, with a concomitant decrease in branched tubules compared with somatic cells and parental cancer cells (). The composition of mitochondrial subtypes did not show significant differences between parental cancer cells and somatic cells ().
Figure 4. Mitochondrial architecture within CSCs. (A) Distribution of mitochondria was revealed by MitoTracker Green (MTG) staining. Cytoplasmic distribution of mitochondria was observed in TW01 parental cells. Mitochondria of TW01 CSCs were scattered away from the plasma membrane and clustered in the peri-nuclear region. DAPI was used to stain the nucleus and CellMask™ Plasma Membrane Stain was utilized to label the plasma membrane. Scale bars indicate 20 μm. (B) Left plots indicate the composition of mitochondrial subtypes in normal skin fibroblasts (FB), TW01 parental cells and CSCs. Individual mitochondria were labeled as follows: small globules (type 1, blue), swollen globules (type 2, yellow), linear tubules (type 3, green), twisted tubules (type 4, orange), loops (type 5, red), branched tubules (type 6, purple). Right histogram quantified the average ratio of type 1 to type 6 mitochondrial subtypes in indicated cells, respectively. Statistically significant differences were determined when compared with TW01 parental cells. (***, P < 0.001).
Interestingly, although mitochondrial respiratory function was attenuated in CSCs, mitochondrial biogenesis was still active. The expression levels of mitochondrial biogenesis-related genes like mitochondrial DNA polymerase gamma (POLG), mitochondrial transcription factor A (TFAM), and peroxisome proliferator-activated receptor gamma coactivator (PGC-1α), were all increased in CSCs (; Fig. S2B). The contents of mitochondrial DNA (mtDNA) and mitochondrial respiratory enzyme subunits were significantly elevated in CSCs as compared with those of the parental cells (; Fig. S2C). However, the mitochondrial mass of CSCs was lower than that of the parental cells (; Fig. S2D). Moreover, CSCs had lower levels of intracellular H2O2 and mitochondrial superoxide anions (; Figs. S2E and 3A), which seemed to be a results of high levels of antioxidant enzymes including copper/zinc superoxide dismutase (Cu/ZnSOD), manganese superoxide dismutase (MnSOD), catalase, glutathione reductase (GR), and reduced glutathione (; Figs. S2F and S3A). The mitochondrial membrane potential (Δψm) of CSCs was also higher than that of the parental cells (; Fig. S3B). These findings indicate that CSCs underwent mitochondrial resetting to possess unique metabolic signatures similar to the characteristics of stem cells.
Figure 5. Mitochondrial resetting in CSCs. (A) Mitochondrial biogenesis transcription factors were upregulated in TW01 CSCs. β-actin was used as the loading control. (B) The copy number of mtDNA was upregulated in TW01 CSCs compared with the parental cells. (C) Western blot analysis of the expression of mitochondrial respiratory enzyme subunits, including those of Complex I (ND1 and NDUFS3), Complex II (SDHA), Complex III (UQCRC1), Complex IV (COX2 and COX4), and Complex V (ATP5β) in TW01 parental cells and CSCs. ND1 and COX2 indicate mtDNA-encoded respiratory enzymes and the others represent nuclear DNA-encoded polypeptides of respiratory enzymes. β-actin was used as the loading control. (D) By nonyl acridine orange (NAO) fluorescent dye staining, lower mitochondrial mass was found in TW01 CSCs compared with that in TW01 parental cells. (E) Intracellular H2O2 and mitochondrial superoxide anions were comparatively lower in TW01 CSCs. H2DCFDA was used to measure the intracellular level of H2O2 and MitoSOX was used to measure the levels of superoxide anions in mitochondria. (F) Antioxidant enzymes were upregulated in TW01 CSCs. β-actin was used as the loading control. (G) Immunofluorescence imaging observed by a confocal microscope (left; scale bars indicate 20 μm) and quantified by flow cytometry (right) of TMRM depicted the higher mitochondrial membrane potential in TW01 CSCs compared with TW01 parental cells. (*, P < 0.05; **, P < 0.01).
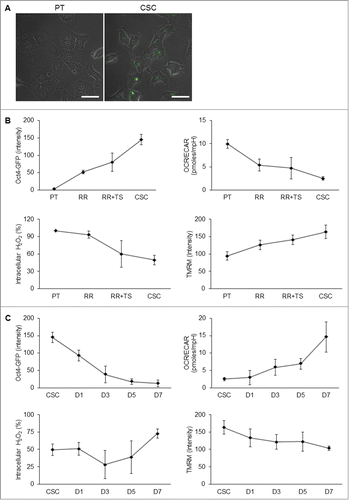
CSCs reprogram and de-reprogram into precise bioenergetic states upon derivation and differentiation processes
To track the metabolic changes during selection and differentiation process, we infected lentivirus vector harboring Oct4 promoter-driven GFP reporter to observe the stemness status of NPC cells (). As expected, Oct4-GFP expression was gradually increased during behavior selection process of CSCs (, upper left panel). We then measured the metabolic profiles of these cells. The results showed that the OCR/ECAR ratio was decreased (, upper right panel), intracellular ROS levels were decreased (, lower left panel), and mitochondrial membrane potential was increased (, lower right panel) during the selection process. On the contrary, the expression of Oct4-GFP was attenuated during the spontaneous differentiation process (, upper left panel). The OCR/ECAR ratio (, upper right panel) and intracellular ROS levels (, lower left panel) were increased but mitochondrial membrane potential was decreased after differentiation (, lower right panel). These metabolic profile changes signified the importance of modulation of a precise bioenergetic state of CSCs in order to acquire and maintain the self-renewal state and proceed to cell differentiation.
Figure 6. Transition of metabolic states upon derivation and differentiation of CSCs. (A) To validate the stem cell competence, TW01 cells were first infected with Oct4 promoter-driven GFP reporter to indicate the stemness. Images indicate that CSCs expressed GFP fluorescence, whereas the parental cells did not. Scale bars indicate 20 μm. (B) The upper left plot shows that increasing Oct4-GFP intensity was detected by flow cytometry from PT, RR, RR+TS, to CSC cells, indicating the elevated percentage of CSCs in the selection process. Other plots indicate the changes of the OCR/ECAR ratio (upper right), intracellular ROS (lower left), and mitochondrial membrane potential (lower right) of indicated cells during the selection process. During the differentiation processess, (C) upper left plot shows gradually decreasing Oct4-GFP intensity of CSCs from day 1 to day 7. Other plots indicate the changes of the OCR/ECAR ratio (upper right), intracellular ROS (lower left), and mitochondrial membrane potential (lower right) of CSCs during the differentiation process. RR: TW01 parental cells after irradiation selection; RR+TS: TW01 parental cells after irradiation and tumor sphere selection; CSC: TW01 parental cells after full behavior selection. D1, D3, D5, D7, indicate TW01 CSC underwent differentiation on day 1, day 3, day 5, and day 7, respectively.
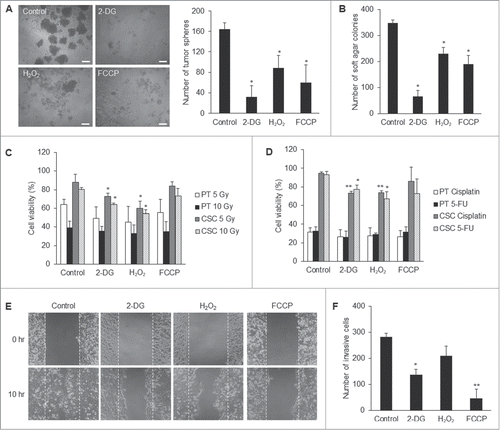
Dysfunction of metabolic reprogramming leads to impairment of CSC properties
To find out whether metabolic reprogramming contributes to CSC properties, we modulated the metabolic states of CSCs comparable to that of the parental cells. By titration of 2-DG, H2O2, and FCCP, respectively, we were able to optimize the dose of each compound to manipulate the levels of lactate production rate, intracellular ROS, and mitochondrial membrane potential of CSCs to match those of the parental cells (Figs. S4A–C). These treatments did not induce apoptosis of non-CSCs such as normal skin fibroblasts and parental NPC cells (Fig. S4D). 2-DG, H2O2, and FCCP respectively retarded anchorage-independent growth of CSCs in soft agar assay and anoikis resistance in tumor sphere assay (). CSCs exhibited greater radioresistant and chemoresistant capacity relative to parental cells (). However, treatment of CSCs with 2-DG or H2O2 increased their radiosensitivity and chemosensitivity, suggesting that 2-DG and H2O2 may serve as sensitizers of CSCs to therapeutic agents and irradiation, respectively (). Moreover, 2-DG and FCCP significantly reduced the migration and invasion capacity of CSCs (). These results have strengthened the importance of the metabolic signatures of CSCs and suggest that metabolic intervention may be able to directly affect the survival advantages of CSCs.
Figure 7. Suppression of CSC properties via manipulating the core metabolism of CSCs. (A) 2-DG, H2O2, and FCCP treatments, respectively, significantly reduced the tumor sphere formation of TW01 CSCs. The left panel shows phase-contrast images of tumor sphere formation. Scale bars indicate 100 μm. The right histogram shows quantitative data of tumor sphere formation. (B) Treatments with 2-DG, H2O2, and FCCP, respectively, hindered significantly the clonogenic formation of TW01 CSCs as assessed by soft agar assay. 2-DG and H2O2 increased (C) the radiosensitivity and (D) the chemosensitivity of TW01 CSCs. Relative percentages of viable cells of TW01 parental cells and CSCs were compared to the basal levels of parental cells or CSCs without any treatment, including 2-DG, H2O2, FCCP, radiotherapy or chemotherapy. Statistically significant differences were determined when compared to the control groups of indicated cells treated with radiotherapy or chemotherapy but without 2-DG, H2O2, and FCCP treatment. Treatment of TW01 CSCs with 2-DG and FCCP, respectively, significantly repressed (E) the migration capacity of TW01 CSCs measured by a wound healing migration assay and (F) invasion capacity of TW01 CSCs in the transwell cell invasion assay. (*, P < 0.05; **, P < 0.01).
Discussion
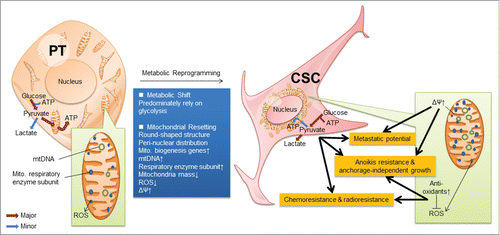
The origin of CSCs remains controversial. It may originate from the pre-existing stem cells bearing the intrinsic stemness or from differentiated neoplastic cells after acquiring extrinsic stemness properties.Citation23-27 In this study, we utilized the behavior selection method to isolate the pre-existing CSCs. These CSCs displayed the metabolic shift and mitochondrial resetting into precise bioenergetic states and lost their unique metabolic phenotypes after differentiation ( and ). Although cancer cells produce energy mainly by glycolysis, CSCs exhibit more pronounced Warburg effect compared with differentiated neoplastic cells (; Fig. S1). Therefore, blocking the energy production by 2-DG significantly reduced the viability of NPC CSCs (). This observation is consistent with the phenotypes of many malignant tumor cells and stem cells.Citation3,10,11,28 This pronounced Warburg effect may channel plenty of glycolytic intermediates into the pentose phosphate pathway to furnish nucleotide precursors required for anabolic metabolism and growth of CSCs.
Figure 8. Schematic representation of the reprogramming of CSCs. CSCs displayed metabolic reprogramming, which includes metabolic shift from mitochondrial respiration to glycolysis as well as mitochondrial resetting. This resetting shows that mitochondria in CSCs are round-shaped structure and in a peri-nuclear distribution, containing abundant mtDNA and mitochondrial respiratory enzyme subunits due to the upregulation of mitochondrial biogenesis. Manipulation of the core metabolism of CSCs into a differentated state compromised their stemness properties.
In addition, NPC CSCs underwent a subtle mitochondrial resetting including redistribution and reconstruction of mitochondria after reprogramming. In terms of mitochondrial redistribution, the organelles were changed from wide distribution over the cell periphery to a peri-nuclear pattern (), possibly because most of the mitochondrial genes reside in the nucleus. The peri-nuclear distribution is beneficial for the translocation of mitochondria-targeting polypeptides to form functional mitochondria.Citation29 It can also enhance the efficiency of the transport of ATP from mitochondria to the nucleus.Citation29 By further scrutiny of mitochondrial architecture within CSCs, we unveiled that representative mitochondria of CSCs inclined to small globular and simple tubular shapes, while branched network of elongated mitochondria were reduced in CSCs compared with non-tumorigenic cells (). These small fragmented globular mitochondria in CSCs may undergo mitophagy or fusion to form simple tubular mitochondria. Nevertheless, CSCs still contain less branched tubules of mitochondria which came from inter-mitochondrial end-to-end or side-to-side fusion, implying that CSCs are lack of mature and functionally active mitochondria. The mitochondrial composition and cellular distribution of CSCs are closer to those of iPSCs and ESCs.Citation3 Upon reprogramming process, the mitochondria of iPSCs were changed from an elongated tubular morphology to a round-shaped structure and peri-nuclear polarization.Citation3 Following spontaneous differentiation, the mitochondrial morphology of differentiated iPSCs manifests elongated tubular shape and fully-developed cristae similar to those of somatic cells.Citation3 These observations suggest that mitochondria of CSCs exhibited ultra-structural morphology and intracellular distribution similar to those of stem cells. Besides, the mitochondrial fission was also reported to be capable of inducing glycolytic reprogramming in the transformation of normal fibroblasts to cancer-associated fibroblasts by virtue of NF-κB activation.Citation30 The relationship between mitochondrial fission and glycolytic shift in CSCs merits further investigation.
For reconstruction of mitochondria, the quantity and quality of mitochondria in CSCs are dictated by the balance between mitochondrial biogenesis and mitochondrial turnover. Mitochondrial biogenesis is crucial for replenishing damaged and degraded mitochondria. Transcription of mtDNA requires nuclear DNA-encoded TFAM and POLG with its accessory subunit POLG2.Citation31 PGC-1 family members, PGC-1α and PGC-1β, and NRF1 drive mitochondrial gene expression. Moreover, PGC-1α binds to NRF1 to activate TFAM and enhance the expression of NRF1 to construct positive feedback loops.Citation32,33 These biogenesis-related factors were upregulated in NPC CSCs (; Fig. S2B) and resulted in an increase of mtDNA copy number () and more polypeptides constituting mitochondrial respiratory enzymes (; Fig. S2C). These results are in line with the report that the increase of mtDNA content is positively correlated with the tumor stages in head and neck cancers.Citation34 Increased mitochondrial biogenesis may be an adaptation of tumor cells to cope with mtDNA mutations and decline of respiratory function in late-stage cancers.Citation34 Nevertheless, the post-translational modifications on respiratory enzyme complexes are crucial for the control of their activities. Various post-translational modifications including acetylation, phosphorylation, ADP-ribosylation, nitrosylation, and oxidation of constituent polypeptides of respiratory enzyme complexes, have been reported to be associated with alterations of Complex I, Complex IV, and Complex V activities.Citation35-43 The NAD+-dependent deacetylase activity of sirtuin-3 (Sirt3), has been demonstrated to be able to regulate mitochondrial oxidative metabolism through deacetylation of mitochondrial enzymes and proteins.Citation44,45 Skeletal muscle of Sirt3 knockout mice manifested attenuated oxygen consumption.Citation45 We speculate that downregulation of Sirt3 may explain why CSCs contain abundant mtDNA and COX4 protein in mitochondria but their respiration rates appear to be restrained in regular culture condition (data not shown). To cope with the stringent growth condition, quiescent CSCs may employ post-translational modifications on specific mitochondrial enzyme complexes to transform themselves into more active states. These reversible post-translational modifications may allow CSCs to sustain more metabolic plasticity in response to perpetual or acute energy requirement.
Another prominent metabolic features of NPC CSCs are the low ROS level and high Δψm (; Figs. S2E and S3), which are consistent with previous reports of CSCs.Citation46,47 Enhanced antioxidant defense mechanisms (; Fig. S2F and S3A) leading to low ROS levels may confer survival advantage for the CSCs such as radioresistance and chemoresistance ().Citation19 As mitochondrial respiration is the major intracellular sources of ROS, high dependence on glycolysis in CSCs may also protect themselves from accumulating ROS (; Fig. S1). Glycolysis may also enhance the antioxidant defenses through providing NADPH by glucose-6-phosphate dehydrogenase (G6PD) for the re-generation of the key small-molecular-weight antioxidant, reduced glutathione, by glutathione reductase (; Figs. S2F and S3A). ROS can lead to mitochondrial depolarization, thus low ROS in CSCs may maintain the membrane integrity and high Δψm of mitochondria (; Fig. S3B).Citation48 Considering that mitochondrial depolarization is required for fusion events, CSCs may also manifest a hyperpolarized mitochondria owing to their non-fused and fragmented mitochondrial hallmark (). Furthermore, down-regulation of mitochondrial uncoupling proteins (UCPs) may also contribute to the high Δψm and low ROS levels, which leads to the maintenance of the malignancy of CSCs.Citation47 Besides, it was reported that Δψm is a key factor for ESCs to the differentiation or formation of teratoma. High Δψm ESCs were found to be prone to developing teratoma, while reducing the Δψm could inhibit tumor growth.Citation49
We could turn off the metabolic switch by treatment of CSCs with 2-DG (Fig. S4A), increasing ROS levels by H2O2 (Fig. S4B), and depolarizing mitochondrial membranes by FCCP (Fig. S4C), respectively. Importantly, these alterations in core metabolism suppressed the CSC properties, such as resistance to anticancer therapy, self-renewal capacity, tumor initiation capacity, and metastatic potential in NPC CSCs (). These findings indicate that CSCs may utilize their unique metabolic signatures to sustain their stemness state and strengthen their defense mechanism (). Loss of these metabolic signatures not only affects the plasticity of CSCs to adjust themselves to cope with the extrinsic and intrinsic stress but also compels CSCs to differentiate by directly tackling the nexus of CSC properties.
In this study, we have provided the proof-of-concept from the in-depth studies of possible targeting of CSCs by metabolic interventions. We uncovered that certain metabolic signatures distinguished CSCs from their differentiated progenies. The regulation of metabolic plasticity enables self-renewal and differentiation of CSCs. This reprogramming of energy metabolism delicately regulates the stemness properties and the survival advantages of CSCs. Further studies are warranted to elucidate how the metabolic reprogramming is regulated in CSCs, which may enlighten future development of novel strategies for treatment of cancers.
Materials and Methods
Cell culture
Human NPC cell lines TW01 and HONE-1 were used in this study.Citation50,51 These cancer cells were cultured in complete Dulbecco's Modified Eagle Medium (DMEM), supplemented with 1% sodium pyruvate, 1% non-essential amino acids, 1% penicillin (10,000 U/mL) and streptomycin sulfate (10,000 μg/mL), and 10% fetal bovine serum (FBS) (all purchased from Invitrogen, Carlsbad, CA) at a 37°C incubator with humidified 5% CO2.
Derivation and differentiation of CSCs
To acquire a high ratio of CSCs from NPC parental cells, we developed a behavior selection method as described previously.Citation21 Briefly, the parental NPC cells were selected sequentially by irradiation treatment, sphere formation, and side population selection. Firstly, irradiation selection was performed as described previously.Citation52 Radioresistant clones were selected after 4 rounds of 11 Gy irradiation at 37.9 mGy/s by using a Rad Source RS 2000 X-ray biological irradiator (Rad Source Technologies, Inc., Suwanee, GA), and the selected radioresistant cells were then subjected to tumor sphere selection as described previously.Citation53 Cells were seeded into a petri-dish coated with 0.4% soft agar containing serum-free DMEM. The soft surface prevented the cells from attaching to the dish and formed tumor spheres after 10 d. Finally, CSCs were isolated with side population selection by using the cells obtained from tumor sphere selection. The side population selection was performed as described previously.Citation20 The cell density was adjusted to 106 cells/mL with DMEM supplemented with 2% FBS and 5 μM Hoechst 33342 (Sigma-Aldrich, St. Louis, MO) and the cells were allowed to grow for 90 min at 37°C. Control cells were incubated with 10 μM fumitremorgin C (FTC, Sigma-Aldrich) for 30 min at 37°C prior to and during Hoechst 33342 staining. After washing twice with PBS, propidium iodide (PI) (2 μg/mL, Sigma-Aldrich) staining was used to exclude dead cells. Cells were kept at 4°C in the dark and then subjected to isolation by a BD FACSAria flow cytometer (BD Biosciences, San Jose, CA). A subpopulation of cells would extrude the Hoechst dye through ATP-binding cassette (ABC) transporters and demonstrated low fluorescence expression. However, FTC can hamper the ABC transporters leading to efflux inhibition. The subpopulation cells would then manifest high fluorescence when treated with FTC to hinder the efflux of Hoechst dye. We thus compared the fluorescence pattern of dot plots with and without the addition of FTC to designate and isolate this subpopulation cells (so-called side population). To initiate spontaneous differentiation, we used CSCs to form tumor spheres under non-adherent conditions and the spheres were then transferred onto a dish coated with a complete medium according to a protocol for stem cell in vitro differentiation.Citation54 Cells within the spheres would start migrating out and undergoing differentiation. Production and infection of Oct4-GFP viruses were performed as described previously.Citation21
Measurement of oxygen consumption and extracellular acidification rate
The oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR) were measured with a Seahorse XF-24 extracellular flux analyzer as described by Qian and van Houten.Citation55 Before assay, the medium had been replenished with un-buffered DMEM for 30 min at 37°C to stabilize the pH and temperature. OCR denotes mitochondrial respiration rate and ECAR represents the rate of lactate production by glycolysis of the cells.
Cytochrome c oxidase (CCO) staining
To measure the CCO activity, we performed the CCO activity staining based on the utilization of 3,3’-diaminobenzidine (DAB) as an acceptor of electrons from cytochrome c. Cells were washed twice with the SP buffer (7.5% sucrose in 50 mM phosphate buffer), and were incubated with 1 mg/mL cytochrome c solution (Sigma-Aldrich) at 37°C. After 20 min, cells were washed twice with the SP buffer and were then incubated with 2.7 mg/mL DAB solution at 37°C for 2 hr. The images of stained cells were captured by a Dino-Lite microscope eye-piece camera (Dino-Lite, Naarden, Netherlands).
Glucose uptake assay
For glucose uptake assay, a fluorescent glucose analog was used to monitor glucose uptake in live cells. Cells were first grown overnight and replenished with a glucose-free medium. To the medium, a final concentration of 100 μM of 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG, Invitrogen) was added and the cells were incubated for 2 hr at 37°C. The fluorescence intensity of the 2NBDG transported to the cells was analyzed with a flow cytometer (Cytomics FC500, Beckman Coulter, Fullerton, CA) and captured by the Olympus FV10i confocal microscope (Olympus America Inc., Center Valley, PA).
Measurement of lactate production rate
Cells were washed with PBS and replenished with a fresh medium for 5 hr of incubation at 37°C. We then transferred 10 μL medium to each of the 96-well dishes and the medium was mixed with the lactate reagent (Trinity Biotech Plc., Bray, Ireland). The absorbance at 540 nm was recorded on an Infinite® 200 multimode microplate reader (Tecan Group Ltd., Männedorf, Switzerland). The lactate production rate was calculated based on the absorbance and was normalized by the cell number and incubation time.
Measurement of the intracellular ATP content
The intracellular ATP content was measured by using the adenosine 5′-triphosphate (ATP) Bioluminescent Somatic Cell Assay Kit (Sigma-Aldrich) as described previously.Citation56 To release the intracellular ATP, the cells were trypsinized and 50 μl of the cell suspension was then mixed with 150 μl Somatic Cell Releasing Reagent. Half of the mixture was subsequently transferred into a black OptiPlate-96F 96-well plate (Perkin-Elmer, Waltham, MA), which contained 100 μl of the ATP Assay Mix. The luminescence intensity was analyzed by a VictorCitation2 1420 Multi-label counter (Perkin-Elmer). The intracellular ATP content was calculated from the intensity of luminescence and was normalized by the cell number.
Morphological subtyping of mitochondria
Cells were replenished with a fresh medium containing 100 nM MitoTracker Green (Invitrogen) for 20 min incubation at 37°C and then in the PBS containing 5 μg/ml CellMaskTM Deep Red plasma membrane stain (Invitrogen) for 10 min incubation at 37°C. Cell images were taken with the Olympus FV10i confocal microscope (Olympus America Inc.) and analyzed by Micro-P software according to the area, axial, and length/width of individual mitochondrion.Citation57 The Micro-P algorithm classified mitochondrial morphology into 6 representative subtypes: type 1: small globules – small round shaped and fragmented globules; type 2: swollen globules – large round and irregular-shaped globules; type 3: straight tubules – linear tubules with varied lengths; type 4: twisted tubules – curved tubules with varied lengths; type 5: loops – donuts and horseshoes-shaped tubules; and type 6: branched tubules – branched tubules with varied lengths.Citation57
Determination of mitochondrial mass
Cells were pre-incubated with a 2.5 μM nonyl acridine orange (NAO, Invitrogen) for 10 min at 25°C in the dark and harvested in a solution containing 5 mM KCl, 140 mM NaCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 5 mM HEPES buffer (pH 7.4). The fluorescence intensity was analyzed on a flow cytometer (Cytomics FC500, Beckman Coulter).
Mitochondrial membrane potential (Δψm)
Cells were pre-incubated with 100 nM of the tetramethylrhodamine methyl ester (TMRM) fluorescent dye (Invitrogen) for 30 min at 37°C. The fluorescence intensity was recorded on the Cytomics FC500 flow cytometer (Beckman Coulter) and captured by the Olympus FV10i confocal microscope (Olympus America Inc.).
Measurement of reactive oxygen species (ROS) and antioxidants
Cells were incubated with DMEM containing 40 μM 2′,7′-dichlorofluorescin diacetate (H2DCFDA, Invitrogen) or MitoSOX Red for 15 min at 37°C. H2DCFDA was used for the determination of intracellular H2O2, while MitoSOX Red was used to measure the mitochondrial levels of superoxide anions. The fluorescence intensities of FL1 channel for H2DCFDA staining and FL2 channel for MitoSOX Red staining, respectively, were recorded on the Cytomics FC500 flow cytometer (Beckman Coulter) and captured by using the Olympus FV10i confocal microscope (Olympus America Inc.). For the evaluation of the cellular level of reduced glutathione (GSH), cells were stained with 8 μg/mL VitaBright-48 (VB48; ChemoMetec A/S, Allerød, Denmark) for 5 min at 37°C.Citation58 Fluorescent intensity of VB48 was captured by using laser excitation at 405 nm and emission at 475/25 nm of Olympus FV10i confocal microscope (Olympus America Inc.).
Statistical analysis
All data are presented as mean ± SD. Statistical analysis was performed by using Student's t test and a difference between groups with P < 0.05 is considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authorship
YA Shen, YJ Chen, and YH Wei contributed to the conception and design of experiments, and wrote the first draft of this manuscript. YA Shen, YH Wei, CY Wang, and YT Hsieh contributed to the acquisition, analysis, and interpretation of the reported data. All authors approved the final version of this manuscript.
Supplementary_Materials.zip
Download Zip (1.2 MB)Acknowledgments
The authors acknowledge Mackay Medical College and the technical support provided by Microarray & Gene Expression Analysis Core Facility in National Yang-Ming University (NYMU)-VGH Genome Research Center. The Gene Expression Analysis Core Facility was supported by the National Research Program for Genomic Medicine, National Science Council.
Funding
This study was financially supported by grants from the National Science Council, Taiwan (NSC-96–2320-B-010-035-MY2, NSC-99-2314-B-010-006-MY3,NSC100-2320-B-715-003-MY3, NSC-101-2321-B-010-015-MY3, MOST103-2321-B-715-001), Department of Health, Taipei City (Grant No. 10001-62-030, 099XDAA00121), and National Yang-Ming University, the Aim for the Top University Plan of the Ministry of Education.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Warburg O. On the origin of cancer cells. Science 1956; 123:309-14; PMID:13298683; http://dx.doi.org/10.1126/science.123.3191.309
- Chen CT, Shih YR, Kuo TK, Lee OK, Wei YH. Coordinated changes of mitochondrial biogenesis and anti-oxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008; 26:960-8; PMID:18218821; http://dx.doi.org/10.1634/stemcells.2007-0509
- Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J. The senescence-related mitochondrialoxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 2010; 28:721-33; PMID:20201066; http://dx.doi.org/10.1002/stem.404
- Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 2011; 14:264-71; PMID:21803296; http://dx.doi.org/10.1016/j.cmet.2011.06.011
- Vander Heiden MG, Cantley LC, Thompson CB. Thompson, understanding the warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029-33; PMID:19460998; http://dx.doi.org/10.1126/science.1160809
- Folmes CD, Nelson TJ, Terzic A. Energy metabolism in nuclear reprogramming. Biomark Med 2011; 5:715-29; PMID:22103608; http://dx.doi.org/10.2217/bmm.11.87
- Chung S, Arrell DK, Faustino RS, Terzic A, Dzeja PP. Glycolytic network restructuring integral to the energetics of embryonic stem cell cardiac differentiation. J Mol Cell Cardiol 2010; 48:725-34; PMID:20045004; http://dx.doi.org/10.1016/j.yjmcc.2009.12.014
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008; 7:11-20; PMID:18177721; http://dx.doi.org/10.1016/j.cmet.2007.10.002
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3:730-7; PMID:9212098; http://dx.doi.org/10.1038/nm0797-730
- Palorini R, Votta G, Balestrieri C, Monestiroli A, Olivieri S, Vento R, Chiaradonna F. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J Cell Biochem 2014; 115:368-79; PMID:24030970; http://dx.doi.org/10.1002/jcb.24671
- Cioce M, Valerio M, Casadei L, Pulito C, Sacconi A, Mori F, Biagioni F, Manetti C, Muti P, Strano S, et al. Metformin-induced metabolic reprogramming of chemoresistant ALDHbright breast cancer cells. Oncotarget 2014; 5:4129-43; PMID:24980829
- Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian C, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A 2011; 108:16062-7; PMID:21900605; http://dx.doi.org/10.1073/pnas.1106704108
- Zhang H, Trachootham D, Lu W, Carew J, Giles FJ, Keating MJ, Arlinghaus RB, Huang P. Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism. Leukemia 2008; 22:1191-9; PMID:18385754; http://dx.doi.org/10.1038/leu.2008.74
- Naka K, Muraguchi T, Hoshii T, Hirao A. Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells. Antioxid Redox Signal 2008; 10:1883-94; PMID:18627347; http://dx.doi.org/10.1089/ars.2008.2114
- Toyokuni S. Novel aspects of oxidative stress associated carcinogenesis. Antioxid Redox Signal 2006; 8:1373-7; PMID:16910784; http://dx.doi.org/10.1089/ars.2006.8.1373
- Saretzki G, Walter T, Atkinson S, Passos JF, Bareth B, Keith WN, Stewart R, Hoare S, Stojkovic M, Armstrong L, et al. Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells 2008; 26:455-64; PMID:18055443; http://dx.doi.org/10.1634/stemcells.2007-0628
- Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, Wahjudi PN, Setoguchi K, Wang G, Do A, et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J 2011; 30:4860-73; PMID:22085932; http://dx.doi.org/10.1038/emboj.2011.401
- Crespo FL, Sobrado VR, Gomez L, Cervera AM, McCreath KJ. Mitochondrial reactive oxygen species mediate cardiomyocyte formation from embryonic stem cells in high glucose. Stem Cells 2010 28:1132-42; PMID:20506541
- Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009; 458:780-3; PMID:19194462; http://dx.doi.org/10.1038/nature07733
- Wang J, Guo LP, Chen LZ, Zeng YX, Lu SH. Identification of cancer stem cell-like side population cells in human nasopharyngeal carcinoma cell line. Cancer Res 2007; 67:3716-24; PMID:17440084; http://dx.doi.org/10.1158/0008-5472.CAN-06-4343
- Shen YA, Lin CH, Chi WH, Wang CY, Hsieh YT, Wei YH, Chen YJ. Resveratrol impedes the stemness, epithelial-mesenchymal transition, and metabolic reprogramming of cancer stem cells in nasopharyngeal carcinoma through p53 activation. Evid Based Complement Alternat Med 2013; 2013:590393; PMID:23737838
- Wang WJ, Wu SP, Liu JB, Shi YS, Huang X, Zhang QB, Yao KT. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res 2013; 73:1219-31; PMID:23269272; http://dx.doi.org/10.1158/0008-5472.CAN-12-1408
- Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature 2012; 488:527-30; PMID:22854777; http://dx.doi.org/10.1038/nature11344
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012; 488:522-6; PMID:22854781; http://dx.doi.org/10.1038/nature11287
- Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, Clevers H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 2012; 337:730-5; PMID:22855427; http://dx.doi.org/10.1126/science.1224676
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704-15; PMID:18485877; http://dx.doi.org/10.1016/j.cell.2008.03.027
- Scaffidi P, Misteli T. In vitro generation of human cells with cancer stem cell properties. Nat Cell Biol 2011; 13:1051-61; PMID:21857669; http://dx.doi.org/10.1038/ncb2308
- Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol 2008; 18:165-73; PMID:18296052; http://dx.doi.org/10.1016/j.tcb.2008.01.006
- Lonergan T, Bavister B, Brenner C. Mitochondria in stem cells. Mitochondrion 2007; 7:289-96; PMID:17588828; http://dx.doi.org/10.1016/j.mito.2007.05.002
- Guido C, Whitaker-Menezes D, Lin Z, Pestell RG, Howell A, Zimmers TA, Casimiro MC, Aquila S, Ando’ S, Martinez-Outschoorn UE, et al. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 2012; 3:798-810; PMID:22878233
- Clayton DA. Nuclear-mitochondrial intergenomic communication. BioFactors 1998; 7:203-5; PMID:9568247; http://dx.doi.org/10.1002/biof.5520070307
- Goffart S, Wiesner RJ. Regulation and co-ordination of nuclear gene expression during mitochondrial biogenesis. Exp Physiol 2003; 88:33-40; PMID:12525853; http://dx.doi.org/10.1113/eph8802500
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999; 98:115-24; PMID:10412986; http://dx.doi.org/10.1016/S0092-8674(00)80611-X
- Kim MM, Clinger JD, Masayesva BG, Ha PK, Zahurak ML, Westra WH, Califano JA. Mitochondrial DNA quantity increases with histopathologic grade in premalignant and malignant head and neck lesions. Clin Cancer Res 2004; 10:8512-5; PMID:15623632; http://dx.doi.org/10.1158/1078-0432.CCR-04-0734
- Bao J, Sack MN. Protein deacetylation by sirtuins: delineating a post-translational regulatory program responsive to nutrient and redox stressors. Cell Mol Life Sci 2010; 67:3073-87; PMID:20680393; http://dx.doi.org/10.1007/s00018-010-0402-y
- Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, Hüttemann M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem 2005; 280:6094-100; PMID:15557277; http://dx.doi.org/10.1074/jbc.M411335200
- Lai Y, Chen Y, Watkins SC, Nathaniel PD, Guo F, Kochanek PM, Jenkins LW, Szabó C, Clark RS. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J Neurochem 2008; 104:1700-11; PMID:17996029; http://dx.doi.org/10.1111/j.1471-4159.2007.05114.x
- Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res 2007; 101:1155-63; PMID:17916778; http://dx.doi.org/10.1161/CIRCRESAHA.107.155879
- Lin TK, Hughes G, Muratovska A, Blaikie FH, Brookes PS, Darley-Usmar V, Smith RA, Murphy MP. Specific modification of mitochondrial protein thiols in response to oxidative stress: a proteomics approach. J Biol Chem 2002; 277:17048-56; PMID:11861642; http://dx.doi.org/10.1074/jbc.M110797200
- Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, Wang X, MacArthur PH, Shoja A, Raghavachari N, et al. Nitrite augments tolerance to ischemiareperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med 2007; 204:2089-102; PMID:17682069; http://dx.doi.org/10.1084/jem.20070198
- Acin-Perez R, Gatti DL, Bai Y, Manfredi G. Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: coupled mechanisms of energy metabolism regulation. Cell Metab 2011; 13:712-9; PMID:21641552; http://dx.doi.org/10.1016/j.cmet.2011.03.024
- Scholz TD, Balaban RS. Mitochondrial F1-ATPase activity of canine myocardium: effects of hypoxia and stimulation. Am J Physiol 1994; 266:H2396-403; PMID:8024001
- Territo PR, Mootha VK, French SA, Balaban RS. Ca2+ activation of heart mitochondrial oxidative phosphorylation: role of the F0F1-ATPase. Am J Physiol Cell Physiol 2000; 278:C423-35; PMID:10666039
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 2007; 27:8807-14; PMID:17923681; http://dx.doi.org/10.1128/MCB.01636-07
- Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, Kahn CR. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A 2011; 108:14608-13; PMID:21873205; http://dx.doi.org/10.1073/pnas.1111308108
- Dayem AA, Choi HY, Kim JH, Cho SG. Role of oxidative stress in stem, cancer, and cancer stem cells. Cancers 2010; 2:859-84; PMID:24281098; http://dx.doi.org/10.3390/cancers2020859
- Ye XQ, Wang GH, Huang GJ, Bian XW, Qian GS, Yu SC. Heterogeneity of mitochondrial membrane potential: a novel tool to isolate and identify cancer stem cells from a tumor mass? Stem Cell Rev 2011; 7:153-60; PMID:20306158; http://dx.doi.org/10.1007/s12015-010-9122-9
- Levraut J, Iwase H, Shao ZH, Vanden Hoek TL, Schumacker PT. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol 2003; 284:H549-58; PMID:12388276
- Schieke SM, Ma M, Cao L, McCoy JP Jr, Liu C, Hensel NF, Barrett AJ, Boehm M, Finkel T. Mitochondrial metabolism modulates differentiation and teratoma formation capacity in mouse embryonic stem cells. J Biol Chem 2008; 283:28506-12; PMID:18713735; http://dx.doi.org/10.1074/jbc.M802763200
- Lin CT, Chan WY, Chen W, Huang HM, Wu HC, Hsu MM, Chuang SM, Wang CC. Characterization of seven newly established nasopharyngeal carcinoma cell lines. Lab Invest 1993; 68:716-27; PMID:7685844
- Glaser R, Zhang HY, Yao KT, Zhu HC, Wang FX, Li GY, Wen DS, Li YP. Two epithelial tumor cell lines (HNE-1 and HONE-1) latently infected with Epstein-Barr virus that were derived from nasopharyngeal carcinomas. Proc Natl Acad Sci U S A 1989; 86:9524-8; PMID:2556716; http://dx.doi.org/10.1073/pnas.86.23.9524
- Chang JT, Chan SH, Lin CY, Lin TY, Wang HM, Liao CT, Wang TH, Lee LY, Cheng AJ. Differentially expressed genes in radioresistant nasopharyngeal cancer cells: gp96 and GDF15. Mol Cancer Ther 2007; 6:2271-9; PMID:17671084; http://dx.doi.org/10.1158/1535-7163.MCT-06-0801
- Chen SF, Chang YC, Nieh S, Liu CL, Yang CY, Lin YS. Nonadhesive culture system as a model of rapid sphere formation with cancer stem cell properties. PLoS One 2012; 7:e31864; PMID:22359637; http://dx.doi.org/10.1371/journal.pone.0031864
- Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, Amit M, Soreq H, Benvenisty N. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol Med 2000; 6:88-95; PMID:10859025
- Qian W, Van Houten B. Alterations in bioenergetics due to changes in mitochondrial DNA copy number. Methods 2010; 51:452-7; PMID:20347038; http://dx.doi.org/10.1016/j.ymeth.2010.03.006
- Wu YT, Lee HC, Liao CC, Wei YH. Regulation of mitochondrial FoF1ATPase activity by Sirt3-catalyzed deacetylation and its deficiency in human cells harboring 4977bp deletion of mitochondrial DNA. Biochim Biophys Acta 2013; 1832:216-27; PMID:23046812; http://dx.doi.org/10.1016/j.bbadis.2012.10.002
- Peng JY, Lin CC, Chen YJ, Kao LS, Liu YC, Chou CC, Huang YH, Chang FR, Wu YC, Tsai YS, et al. Automatic Morphological Subtyping Reveals New Roles of Caspases in Mitochondrial Dynamics. PLoS Comput Biol 2011; 7:e1002212; PMID:21998575; http://dx.doi.org/10.1371/journal.pcbi.1002212
- Skindersoe ME, Rohde M, Kjaerulff S. A novel and rapid apoptosis assay based on thiol redox status. Cytometry A 2012; 81:430-6; PMID:22407950; http://dx.doi.org/10.1002/cyto.a.22032