
Abstract
In response to genotoxic stress, cells protect their genome integrity by activation of a conserved DNA damage response (DDR) pathway that coordinates DNA repair and progression through the cell cycle. Extensive modification of the chromatin flanking the DNA lesion by ATM kinase and RNF8/RNF168 ubiquitin ligases enables recruitment of various repair factors. Among them BRCA1 and 53BP1 are required for homologous recombination and non-homologous end joining, respectively. Whereas mechanisms of DDR are relatively well understood in interphase cells, comparatively less is known about organization of DDR during mitosis. Although ATM can be activated in mitotic cells, 53BP1 is not recruited to the chromatin until cells exit mitosis. Here we report mitotic phosphorylation of 53BP1 by Plk1 and Cdk1 that impairs the ability of 53BP1 to bind the ubiquitinated H2A and to properly localize to the sites of DNA damage. Phosphorylation of 53BP1 at S1618 occurs at kinetochores and in cytosol and is restricted to mitotic cells. Interaction between 53BP1 and Plk1 depends on the activity of Cdk1. We propose that activity of Cdk1 and Plk1 allows spatiotemporally controlled suppression of 53BP1 function during mitosis.
Abbreviations
| 53BP1 | = | p53 binding protein 1 |
| ATM | = | ataxia telangiectasia mutated kinase |
| BRCA1 | = | breast cancer type 1 susceptibility protein |
| Cdk | = | cyclin dependent kinase |
| DDR | = | DNA damage response |
| Plk1 | = | Polo-like kinase 1 |
| H2AX | = | histone variant H2AX |
| IR – | = | ionizing radiation |
| MDC1 | = | mediator of DNA damage checkpoint protein 1 |
| NCS – | = | neocarzinostatin |
| NZ – | = | nocodazole |
| PTIP | = | PAX transactivation activation domain-interacting protein |
| RIF1 | = | Rap1-interacting factor 1 homolog |
| RNAi | = | RNA interference |
| RNF8 | = | RING finger protein 8 |
| RNF168 | = | RING finger protein 168 |
Introduction
Cells protect their genome integrity by a conserved DNA damage response pathway (DDR) that coordinates DNA repair with control of the cell cycle progression.Citation1 Depending on the type of genotoxic stress and the cell cycle phase when the DNA damage occurred, lesions are repaired either by an error prone non-homologous end-joining (NHEJ) or by homologous recombination (HR). Master regulator of DDR is the protein kinase ATM that rapidly after formation of DNA double strand breaks (DSB) phosphorylates histone H2AX at S139 (called γH2AX) in the chromatin flanking the DNA lesion.Citation2 Phosphorylation of H2AX is required for recruiting an adaptor protein MDC1 and also E3 ubiquitin ligases RNF8 and RNF168 to the site of DNA damage.Citation3–5 H2A and H2AX have recently been identified as major substrates of RNF168 and monoubiquitination at K13 and K15 is recognized by the ubiquitination-dependent recruitment (UDR) motif of 53BP1.Citation6,7 In addition, 53BP1 is recruited to the DNA damage foci through binding of its Tudor domain to the dimethylated histone H4K20-me2 and therefore 53BP1 is now recognized as a bivalent reader of posttranslationally modified mononucleosomes.Citation7,8 In contrast to 53BP1, the protein complex containing BRCA1, Abraxas and RAP80 is recruited to the DNA damage foci through the ubiquitin-interacting motif (UIM) of RAP80 that binds to K63 ubiquitinated histones.Citation9,10 Whereas BRCA1 is needed for DNA end resection and HR, 53BP1 facilitates repair of DNA lesions through NHEJ especially when localized in the heterochromatin or at telomeres.Citation11-13 Upon phosphorylation of the N-terminal domain by ATM, 53BP1 recruits RIF1 and PTIP that block resection of DNA ends and promote repair through NHEJ.Citation14-16 Thus there is now emerging evidence that recruitment of either 53BP1 or BRCA1 to the site of damage underlies the selection of particular DNA repair pathway.Citation17,18
DNA damage response is organized differently in interphase and in mitosis. Whereas the proximal phosphorylation-dependent events in DDR occur in interphase and mitotic cells, more distal responses that largely depend on histone ubiquitination are supressed during mitosis.Citation19-21 Irradiation of mitotic cells triggers activation of ATM, phosphorylation of H2AX and recruitment of MDC1 to the condensed chromatin.Citation19 In contrast, 53BP1 is not recruited to DNA lesions during mitosis until cells progress to the following G1 phase.Citation19,22,23 Here we show that 53BP1 is phosphorylated in its C-terminal domain by Plk1 and Cdk1 during mitosis and that this modification supresses the ability of 53BP1 to bind to ubiquitinated histones and to localize to DNA damage foci. We propose a model by which the suppression of 53BP1 function is strictly limited to mitosis.
Results
53BP1 is phosphorylated by Cdk1 and Plk1 in mitosis
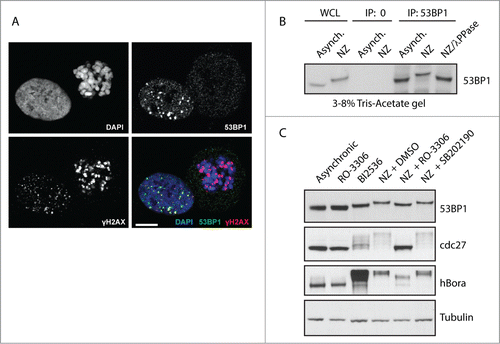
To compare the ability of 53BP1 to localize to the DNA damage foci in interphase and mitotic cells we irradiated exponentially growing U2OS cells with 3 Gy and analyzed them by immunofluorescence microscopy (). Irradiated mitotic cells were strongly positive for γH2AX signal at chromatin suggesting that ATM is normally activated following exposure to genotoxic stress in mitosis. In fact, γH2AX signal was much stronger in mitotic cells compared to the neighboring interphase cells. Increased sensitivity of mitotic cells to ionizing radiation probably reflects the degradation of PPM1D phosphatase (also known as Wip1) we previously reported.Citation24 In contrast, 53BP1 was excluded from the chromatin in mitotic cells and did not colocalize with γH2AX in mitotic cells after irradiation. We hypothesized that distinct behavior of 53BP1 in interphase and mitotic cells might be at least in part caused by its posttranslational modifications and thus we aimed to investigate the impact of mitotic posttranslational modifications on 53BP1 function. First, we immunopurified endogenous 53BP1 from exponentially growing or mitotic cells and noted a significant decrease in electrophoretic mobility of mitotic 53BP1 (). Since the change in electrophoretic mobility was completely reversed by treatment of 53BP1 by lambda phosphatase we concluded that 53BP1 is phosphorylated during mitosis (). This finding is in good agreement with recent phosphoproteomic screens that identified extensive phosphorylation of 53BP1 during mitosisCitation25,26. Next, we wondered what kinases are responsible for this modification of 53BP1. To this end, we arrested cells in mitosis by addition of nocodazole and compared the effect of treatment by several protein kinase inhibitors on the mobility shift (). Addition of RO-3306, a specific inhibitor of Cdk1, impaired the mobility shift of 53BP1 suggesting that 53BP1 is phosphorylated by Cdk1 during mitosis.Citation27,28 In contrast, treatment with SB202190, a selective inhibitor of mitogen-activated protein kinase p38 α (hereafter reported as p38α) did not significantly change the mobility of mitotic 53BP1. This data suggest that the majority of 53BP1 phosphorylation during mitosis depends on Cdk1 activity, although we cannot exclude some contribution of p38a on phosphorylation of 53BP1. In addition, inhibition of Plk1 by BI2536 inhibitor arrested cells in mitosis and increased the mobility of 53BP1 (). Change in the mobility of cdc27 and stabilization of hBora was used as control for effective inhibition of Cdk1 and Plk1, respectively.Citation29 We conclude that 53BP1 is phosphorylated by Cdk1 and Plk1 during mitosis.
Figure 1. 53BP1 is phosphorylated during mitosis by Cdk1 and Plk1. (A) U2OS cells were irradiated with 3 Gy, fixed after 1 h and probed for γH2AX, 53BP1 and DAPI and analyzed by confocal microscopy. Single focal plane is shown. Bar indicates 10 μm. (B) Endogenous 53BP1 was immunoprecipitated from exponentially grown cells (Asynch.) or from cells arrested in mitosis by NZ, separated on 3–8% Tris-Acetate gel and analyzed by immunoblotting. Where indicated immunopurified 53BP1 was incubated with λ-phosphatase for 15 min at 30°C. (C) U2OS cells were grown exponentially (Asynch.), treated for 12 h with RO-3306 to arrest them in G2, or with BI2536 or NZ to arrest them in mitosis. Mitotic cells treated with NZ were collected by shake-off and incubated for additional 60 min with DMSO or with RO-3306 or SB202190. Whole cell lysates were separated on 3–8% Tris-Acetate or 4–12% Bis-Tris gels and probed with indicated antibodies.
Plk1 phosphorylates 53BP1 during mitosis at Ser1618 in the UDR domain
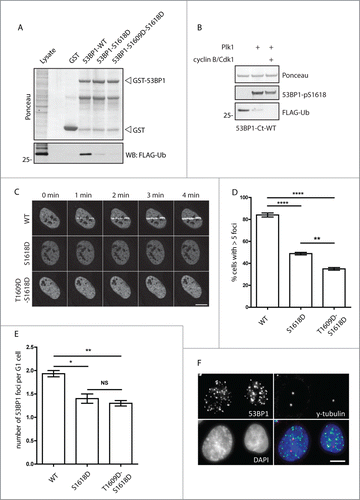
Plk1 plays essential role in control of mitotic spindle formation and cytokinesis.Citation30,31 In addition, Plk1 is required for recovery from the G2 checkpoint.Citation32,33 This is achieved by induction of the βTrCP-dependent degradation of Claspin, a cofactor of ATR required for activation of Chk1 and for establishing the checkpoint.Citation34,35 In addition, Plk1 has been reported to phosphorylate Chk2 in the FHA domain and to prevent its activation in mitosis.Citation23 Since Plk1 has a well-established role in negative regulation of the DDR response we aimed to investigate the effect of 53BP1 phosphorylation by Plk1. First, we identified putative phosphorylation sites based on known phosphorylation motif of Plk1 and conservation of the motif in the primary sequence of 53BP1.Citation36,37 Out of 6 identified conserved motifs only 2, namely S395 and S1618, were previously reported to be phosphorylated in phosphoproteomic screens.Citation25,26 We noted that S1618 is localized in the UDR motif immediately next to the L1619 residue that is essential for mediating the interaction of 53BP1 with ubiquitinated histone H2A and thus we focused on the ability of Plk1 to phosphorylate the C-terminal part of 53BP1.Citation7 We purified the truncated GST-53BP1 protein containing the Tudor domain, UDR motif and the tandem BRCT motifs and performed an in vitro kinase assay with active His-Plk1. Making use of a commercially available antibody against pS1618–53BP1 we found that Plk1 phosphorylated S1618 in vitro (). Importantly, the signal was completely lost in the 53BP1-S1618A mutant confirming the specificity of the antibody (). Whereas Plk1 did phosphorylate the wild-type 53BP1-C-term fragment, the autoradiography signal was reduced in the 53BP1-S1618A mutant (). This suggests that Plk1 can phosphorylate S1618 and possibly also other residues in the C-terminal part of 53BP1. Next, we tested whether Plk1 phosphorylates S1618 also in cells. We found that pS1618–53BP1 was highly enriched in cells synchronized in mitosis by nocodazole, whereas only basal levels were present in asynchronically growing cells (). The specificity of the pS1618–53BP1 antibody was validated by siRNA-mediated depletion of 53BP1 that caused a loss of the signal in mitotic cells (). In addition, signal of pS1618–53BP1 was strongly reduced in mitotic cells treated with Plk1 inhibitor and the same reduction was observed in cells depleted of Plk1 using RNAi (). From this we conclude that Plk1 phosphorylates S1618 of 53BP1 also in vivo.
Figure 2 (See previous page). Plk1 phosphorylates 53BP1 in the UDR domain. (A) Purified GST or GST-53BP1-C-term were incubated with His-Plk1 in the presence of 32P-γ-ATP and then separated on SDS-PAGE. Phosphorylation was detected by autoradiography or by immunoblotting with pS1618–53BP1 antibody. (B) Purified GST, GST-53BP1-C-term-WT or -S1618A were incubated with His-Plk1 and Phosphorylation was detected by autoradiography or by immunoblotting. (C) Unsynchronized cells (Asynch.) or cells arrested in mitosis by nocodazole or by Plk1 inhibitor (BI2536) were lyzed and probed with indicated antibodies. (D) U2OS cells were transfected with GAPDH or 53BP1 siRNA and grown asynchronically or arrested in mitosis by nocodazole. Arrowhead indicates the same position on the gel (E) U2OS cells were transfected by siRNA targeting GAPDH or Plk1. Nocodazole was added to cells transfected with GAPDH siRNA. Cells depleted of Plk1 spontaneously arrested in mitosis. Mitotic cells were collected by mitotic shake-off and analyzed by immunoblotting. (F) HeLa or U2OS cells were synchronized at G1/S transition by a double thymidine block, released to fresh media with nocodazole and collected in 2 h intervals. Media without nocodazole was used as control for cells that progressed to the following G1. (G) hRPE-TERT cells were grown exponentially or arrested in mitosis by nocodazole or BI2536 for 16 h and collected by mitotic shake-off. (H) Mitotic U2OS cells (NZ) were released to the fresh media and collected in 1 h intervals.
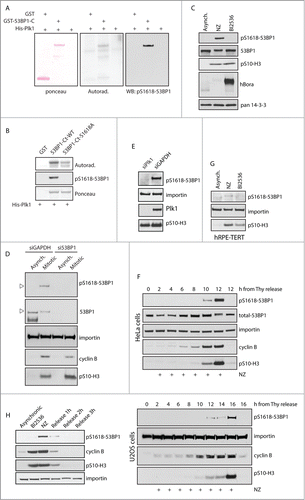
To study more closely the dynamics of pS1618–53BP1 phosphorylation, we synchronized cells at G1/S transition by thymidine, released them in fresh media supplemented with nocodazole and assayed the pS1618–53BP1 signal during progression to mitosis (). We have found that the occurrence of pS1618–53BP1 signal closely correlated with the positivity of pS10-histone H3 which is an established marker of mitosis. Similar pattern was observed in U2OS, HeLa and non-cancer hTERT-RPE1 cells suggesting that pS1618–53BP1 modification is not restricted to a particular cell type (). Further we assayed the dephosphorylation of 53BP1 during mitotic exit (). To this end, we synchronized cells in mitosis by nocodazole, collected them by shake off and released them to fresh media. The removal of pS1618–53BP1 modification correlated to disappearance of pS10-histone H3 as well as degradation of cyclin B and Plk1 during mitotic exit. We conclude that Plk1 phosphorylates S1618 specifically during mitosis.
Phosphorylated 53BP1 and Plk1 co-localize at kinetochores
Next we wondered in which subcellular compartment Plk1 phosphorylates 53BP1 during mitosis.Citation19,20,38 It is well established that active Plk1 is enriched at spindle poles and kinetochores during metaphase and translocates to the midbody during cytokinesis.Citation39 Kinetochore localization of 53BP1 has also been reported although its functional relevance still remains unclear.Citation40 First, we have used immunofluorescence microscopy and probed for endogenous 53BP1 in U2OS cells. Consistent with previous reports 53BP1 localized predominantly to the cell nucleus in interphase cells. In mitosis, 53BP1 was excluded from the condensed chromatin and the majority of 53BP1 was present diffusely in the cytosol (). In addition, substantial part of endogenous 53BP1 closely associated with the centromeric marker CREST in mitotic cells (). Specificity of the 53BP1 staining was confirmed by an RNAi-mediated depletion of 53BP1 that resulted in a complete loss of the kinetochore staining and a strong reduction of the diffuse staining (). Confocal microscopy showed that 53BP1 co-localized at kinetochores with Plk1 suggesting that active Plk1 may phosphorylate 53BP1 at kinetochores (). Indeed, we have found that 53BP1 phosphorylated at Ser1618 is present at kinetochores during mitosis (). Besides localization to kinetochores, pS1618–53BP1 was present diffusely throughout the mitotic cell. Importantly, the signal observed with the pS1618–53BP1 antibody disappeared in cells transfected with 53BP1 siRNA confirming the specificity of the antibody in immunofluorescence (). In addition, pS1618–53BP1 signal was strongly reduced after treatment with BI2536 or after siRNA-mediated depletion of Plk1 which is consistent with the key role of Plk1 in phosphorylating S1618 of 53BP1 (). Relative quantification of the immunofluorescence signal showed that 53BP1 is phosphorylated at Ser1618 exclusively in mitosis and the signal intensity reached the maximum during metaphase and anaphase and declined afterwards during telophase (). Thus dynamics of the pS1618–53BP1 phosphorylation closely correlates with the high activity of Plk1 during metaphase and decreased Plk1 activity due to the degradation of Plk1 by APC/Cdh1 complex during mitotic exit.Citation41 Due to the identified kinetochore localization of 53BP1 and its phosphorylated form pS1618–53BP1 we investigated whether 53BP1 might be involved in organization of a bipolar mitotic spindle and in control of the spindle assembly checkpoint. We have quantified the number of aberrant mitotic spindles in control and 53BP1-depleted cells and have found no significant differences (). Similarly, time lapse microscopy did not show any significant differences in timing of mitotic progression (data not shown). Consistent with a previous report we conclude that 53BP1 is not involved in control of the spindle assembly.Citation23 Instead, we propose that kinetochores may represent a compartment where Plk1 phosphorylates 53BP1 at S1618 and after release from the kinetochore phosphorylated 53BP1 may be present throughout the cell.
Figure 3. 53BP1 phosphorylated at S1618 colocalize with Plk1. (A) U2OS cells were transfected with GAPDH or 53BP1 siRNA, fixed after 48 h and probed for endogenous 53BP1 and CREST (marker of centromeres) using confocal microscopy. Images represent single focal planes. Insets show magnified regions of the same image. Bar indicates 10 μm or 1 μm in the insets. (B) Mitotic cells expressing EGFP-Plk1 were probed with 53BP1 or pS1618–53BP1 and CREST and analyzed by confocal microscopy as in A. (C) U2OS cells transfected with control, 53BP1 or Plk1 siRNA were analyzed after 48 h by confocal microscopy. Alternatively cells were treated with BI2536 (10 nM, 3 h). Images represent single focal plane and bar indicates 10 μm. (D) Exponentially growing U2OS cells were fixed and stained with pS1618–53BP1 antibody. Bar indicates 10 μm. Total cell fluorescence was quantified in interphase and mitotic cells. Each dot represents one cell. Error bars indicate mean and SD. (E) U2OS cells were transfected with GAPDH or 53BP1 siRNA, fixed and probed for tubulin and DAPI. Morphology of mitotic spindles was scored as bipolar or aberrant (monopolar and multipolar; n=3, error bars indicate SD). (F) U2OS stably expressing EGFP-53BP1 were grown exponentially (Asynch.), synchronized in G2 by RO-3306 or in mitosis by nocodazole and 53BP1 was immunoprecipitated by GFP-Trap. Bound proteins were analyzed by immunoblotting.
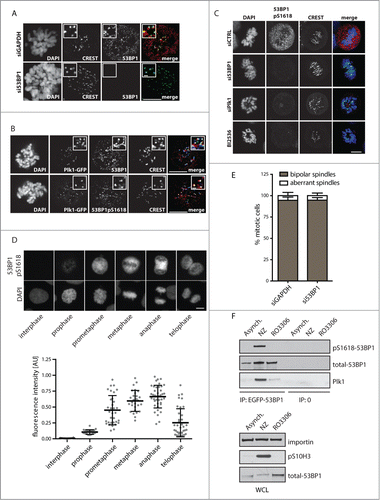
To study the interaction between 53BP1 and Plk1 we immunoprecipitated EGFP-53BP1 from cells synchronized in mitosis by nocodazole or from cells arrested in late G2 phase by treatment with Cdk1 inhibitor (). Whereas 53BP1 immunopurified from G2 cells interacted only weakly with Plk1, we found that this interaction was significantly increased in mitotic cells.
Cdk1 phosphorylates 53BP1 at S1678 and T1609
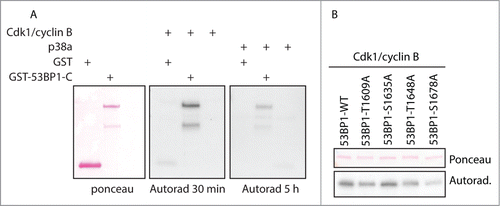
Cdk1/cyclin B is the major regulator of the mitotic progression and its activation triggers entry to mitosis whereas its activity declines after APC/C-dependent degradation of cyclin B after metaphase to anaphase transition. In addition, another prolin-directed protein kinase, p38α has been reported to be activated during mitosis although its physiological relevance remains unclear.Citation42,43 Here we compared the ability of Cdk1/cyclin B and p38a to phosphorylate the C-terminal part of 53BP1. Although both kinases were able to phosphorylate the 53BP1-C-term in vitro, it was much better substrate for Cdk1/cyclin B and comparably lower level of phosphorylation was observed with p38a (). Combined with the data with Cdk1 and p38a inhibitors () we conclude that in mitosis 53BP1 is physiologically phosphorylated by Cdk1/cyclin B rather than by p38a. Next, we mapped the Cdk1 phosphorylation sites in the C-terminal part of 53BP1 by in vitro kinase assay using a set of alanine mutants in the putative Cdk1 sites (). We found that phosphorylation of 53BP1-S1678A by Cdk1 was strongly reduced (approximately to 20%) compared to the wild-type C-terminal fragment of 53BP1. In addition, phosphorylation of the 53BP1-T1609A mutant was slightly but reproducibly lower compared to the wild-type 53BP1. S1635 and T1648 form consensus phosphorylation motifs SSP and STP, respectively, and after priming by Cdk1 phosphorylation they could potentially serve as docking sites for PBD of Plk1.Citation44 However, we found that 53BP1-S1635A and 53BP1-T1648A were phosphorylated by Cdk1 at comparable level to the wild-type 53BP1 and therefore these sites are unlikely to mediate binding of Plk1. These data together suggest that S1678 and T1609 are the major Cdk1 phosphorylation sites in the C-terminal part of 53BP1. Importantly, both S1678 and T1609 residues were identified in proteomic screens to be phosphorylated during mitosis in vivo.Citation26 Besides phosphorylation of the C-terminal part of 53BP1 reported here, Cdk1 was previously shown to phosphorylate S380 of 53BP1 forming a docking site for Plk1.Citation23
Figure 4. 53BP1 is phosphorylated by Cdk1/cyclin B. (A) Purified GST or GST-53BP1-C-term was phosphorylated in vitro by active Cdk1/cyclin B or p38a and phosphorylation was detected by autoradiography for 30 min or 5 h. (B) Various alanine mutants of GST-53BP1-C-term were phosphorylated in vitro by Cdk1/cyclin B.
53BP1 phosphorylation by Plk1 impairs its binding to ubiquitinated histones and localization to foci
Since S1618 resides in the UDR motif that has recently been reported to mediate the direct interaction of 53BP1 with monoubiquitinated histones, we wondered whether phosphorylation of this residue may affect the ability of 53BP1 to bind to modified histones.Citation7 To test this we established a pull down assay using the wild-type GST-53BP1-C-term fusion protein or its phosphorylation mimicking aspartate mutants and the extract from U2OS cells stably expressing FLAG-tagged ubiquitin that were treated with a topoisomerase II inhibitor etoposide. We found that the wild-type GST-53BP1-Cterm bound ubiquitinated histones in the extracts from cells exposed to genotoxic stress (). In contrast, the ability to bind ubiquitinated histones was strongly reduced or completely lost in GST-53BP1-Cterm-S1618D and GST-53BP1-Cterm-S1609D-S1618D mutants, respectively (). In addition, the binding of the wild-type GST-53BP1-Cterm to ubiquitinated histones was strongly reduced when the protein was phosphorylated by Plk1 and completely abolished when phosphorylated by Plk1 and Cdk1/cyclin B (). This suggests that Plk1 and Cdk1 collaborate on inhibition of 53BP1 interaction with the ubiquitinated chromatin.
Figure 5. Phosphorylation of 53BP1 by Plk1 and Cdk1 inhibits its binding to ubiquitinated histones. (A) Purified GST, GST-53BP1-C-WT, GST-53BP1-C-S1618D or GST-53BP1-C-S1618D-S1609D were incubated with extract from U2OS-FLAG-ubiqiuitin cells treated with etoposide. Pull down was done by glutathione sepharose and bound proteins were analyzed by immunoblotting. (B) Purified GST-53BP1-C-WT was phosphorylated in vitro by Plk1 or Plk1 and Cdk1/cyclin B or mock phosphorylated and incubated with extract from U2OS-FLAG-ubiqiuitin cells treated with etoposide as in A. (C) U2OS cells transfected with EGFP-53BP1-WT, -S1618D or –S1609D-S1618D were pre-treated with BrdU, laser micro-irradiated and recruitment of EGFP-tagged proteins to irradiated area was assayed by life imaging. (D) Cells from (C) were fixed 3 h after exposure to ionizing radiation (3 Gy) and DNA damage foci were analyzed by automated high-content microscopy and spot detection module. Percentage of cells with more than 5 53BP1 foci is shown. (n=3, error bars indicate SD) (E) U2OS cells transfected with EGFP-53BP1-WT, -S1618D or -S1609D-S1618D were grown exponentially and analyzed 48 h after transfection by automated high-content microscopy. Average number of 53BP1 foci was quantified in G1 cells gated by the intensity of the DAPI. (n=3, error bars indicate SD) (F) Exponentially growing U2OS cells were fixed 3 h after irradiation with 3 Gy and probed with antibodies against 53BP1 and γ-tubulin. Note no difference in 53BP1 foci formation in late G2 cell with separated centrosomes.
After exposure to genotoxic stress, E3 ubiquitin-protein ligase RNF168 mono-ubiquitinates histone H2A at K13 and K15.Citation6,45 In turn, H2A-K13/15 ubiquitination and H4K20 dimethylation are recognized by UDR and Tudor domains, respectively, and are essential for recruitment of 53BP1 to the DNA damage foci.Citation7,8 To determine how phosphorylation affects 53BP1 function, we compared the ability of wild-type and mutated 53BP1 to localize to the sites of DNA damage. First, we induced a local DNA damage by laser microirradiation and assayed the dynamics of EGFP-53BP1 recruitment to damaged chromatin in living cells. Whereas the wild-type 53BP1 quickly redistributed to the exposed area within 1 minute after irradiation, recruitment of 53BP1-S1618D and 53BP1-T1609D-S1618D mutants was strongly delayed showing only weak signal later than 5 minutes after irradiation ( and data not shown). Next we tested the ability of 53BP1 to localize to DNA damage foci induced by ionizing radiation (IRIF). We found that recruitment of 53BP1-S1618D to foci was reduced compared to the wild-type 53BP1 (). Consistent with the in vitro studies the ability to localize to foci was further decreased in the 53BP1-T1609D-S1618D mutant (). Formation of IRIFs was not completely blocked in the 53BP1-T1609D-S1618D mutant, which most probably reflects the ability of EGFP-53BP1 to oligomerize with the endogenous 53BP1 that was present in our experiment. Alternatively, yet other posttranslational modifications of 53BP1 may contribute to further decrease the binding capacity of 53BP1 to ubiquitin. One of such candidate modifications is phosphorylation of S1678 by Cdk1 as we observe that S1678D mutation partially inhibits binding to ubiquitinated histones in vitro (data not shown). Further, we tested the ability of mutant 53BP1 to localize to the OPT bodies, characteristic nuclear structures present in cells progressing through the G1 phase.Citation46,47 Similarly to decreased ability of 53BP1 carrying the phosphomimicking mutations in Plk1 and Cdk1 sites to IRIFs we find also impaired formation of the OPT bodies in unstressed conditions (). Finally, we wished to address when the Plk1-dependent inhibition of 53BP1 localization to DNA damage foci occurs in context of the cell cycle. Previously we have shown that Plk1 activity is triggered by phosphorylation at T210 and starts 5–6 h prior mitotic entry.Citation48 Therefore we wondered whether 53BP1 can localize to IRIFs in late G2 phase. We have found no differences in formation of the 53BP1 foci in cells with separated centrosomes (). This finding is in good agreement with the observed basal interaction between 53BP1 and Plk1, and with absent pS1618 phosphorylation of 53BP1 in G2 cells. We conclude that blocking the interaction of 53BP1 with damaged chromatin through phosphorylation of the UDR motif by Plk1 and Cdk1 is restricted to mitosis.
Plk1 activity supresses DNA repair in mitotic cells
To address the role of Plk1 in regulation of DNA repair during mitosis, we incubated mitotic cells with DMSO or Plk1 inhibitor and measured the level of DNA damage by comet assay at various time points after addition of a radiomimetic drug NCS (2 nM). Inhibition of Plk1 significantly reduced the level of DNA damage 1 h and 3 h after treatment with NCS which is consistent with increased DNA repair capacity in mitotic cells lacking Plk1 activity (). Next, we determined the level of DNA damage induced by NCS in mitotic cells by measuring γH2AX levels using flow cytometry (). This analysis revealed that DNA damage was efficiently induced in control mitotic cells and also in cells treated with Plk1 inhibitor. Interestingly, γH2AX levels decreased significantly faster in cells treated with Plk1 inhibitor further supporting the role of Plk1 in suppression of DNA repair in mitosis. Similar results were obtained when mitotic cells were treated with etoposide that induces DNA double strand breaks by inhibition of topoisomerase II suggesting that Plk1 inhibits repair of DNA lesions induced by distinct mechanisms (data not shown).
Figure 6. Inhibition of Plk1 increases DNA repair capacity in mitotic cells. (A) U2OS cells were synchronized in mitosis by NZ (16 h), incubated for additional 2 h with DMSO or BI2536 (50 nM) and treated with NCS (2 nM) for indicated times. DNA lesions were quantified by neutral comet assay. Plotted is average amount of DNA in tails, error bars indicate SD. Circles and triangles indicate individual cells. (B) Cells were treated as in (A), fixed at indicated times and γH2AX levels were measured by FACS (at least 104 cells per condition, n=4, error bars indicate SD). (C) U2OS cells were transfected with siRNA targeting coding region or 3-UTR region of 53BP1 and knock down was evaluated by immunoblotting. (D) U2OS-TR cells stably transfected with EGFP-53BP1-WT, -S1618D or –S1609D-S1618D were transfected with siRNA targeting 3-UTR region of 53BP1. After 48h expression of EGFP-53BP1 was induced by tetracycline for 12h, cells were irradiated with 3 Gy and fixed 8h afterwards. BRCA1 foci were analyzed by automated high-content microscopy. Average number of 53BP1 foci was quantified in G1 cells gated by the intensity of the DAPI and negative Cyclin A signal (n=3, error bars indicate SD). (E) RPE cells stably expressing EGFP-53BP1-WT or –S1609D-S1618D were treated and analyzed as in (D). (n=4, error bars indicate SD). (F) RPE cells stably expressing EGFP-53BP1-WT or –S1609D-S1618D were treated as in (D) and γH2AX-positive foci were analyzed by automated high-content microscopy. Average number of γH2AX- foci was quantified in G1 cells gated by the intensity of the DAPI and negative Cyclin A signal (n=3, error bars indicate SD).
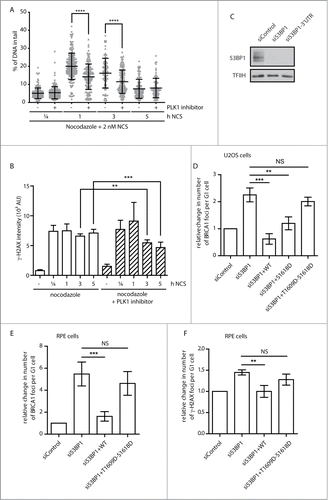
Next we wished to test how phosphorylation of 53BP1 can affect its function in DNA repair. To this end, we depleted endogenous 53BP1, reconstituted cells with siRNA non-targetable EGFP-53BP1 and assayed the response to DNA damage (). We noted that wild-type 53BP1 properly localized to the DNA damage-induced foci and prevented recruitment of BRCA1 to DNA damage foci in U2OS and RPE cells (). In contrast, cells reconstituted with 53BP1-S1618D or 53BP1-T1609D-S1618D formed BRCA1-positive foci suggesting that phosphorylation of 53BP1 impairs its ability to suppress the recruitment of BRCA1 to the foci (). Whereas most of DNA lesions are repaired during interphase in 53BP1-independent manner, a fraction of lesions (mostly in heterochromatin or at telomeres) requires 53BP1 for efficient repair by NHEJ pathway.Citation11-13 In agreement with this we found that cells reconstituted with 53BP1-T1609D-S1618D showed a delayed decrease of γH2AX levels after irradiation compared to cells reconstituted with wild-type 53BP1 (). We conclude that phosphorylation of 53BP1 by Plk1 and Cdk1 impairs its function in DNA repair.
Discussion
There is accumulating evidence that DNA damage response is differently organized during interphase and in mitotic cells. Following exposure to genotoxic stress, ATM is activated similarly in interphase and mitosis and phosphorylates histone H2AX in the chromatin flanking the DNA lesion. In fact, ATM has been reported to be activated also by a low level of DNA damage present in the condensed chromatin in unperturbed mitosis.Citation46,47 The ability of ATM to respond even to such minimal extent of DNA damage during mitosis may be facilitated by APCcdc20-dependent degradation of the opposing phosphatase Wip1 during mitosis.Citation24 Although ATM is activated normally during mitosis, the downstream events in the DDR that depend on chromatin ubiquitination are supressed in mitosis.Citation19,21 Here, we have shown that mitotic kinases Cdk1 and Plk1 phosphorylate 53BP1 within its UDR domain, block its interaction with ubiquitinated chromatin and interfere with its function in DNA repair. Previously we have established that Plk1 activity is regulated by phosphorylation of Thr210 within the T-loop of Plk1 by Aurora A and that initial activation of Plk1 occurs already in G2 and gradually increases toward mitosis.Citation48 Using immunofluorescence microscopy of exponentially grown cultures we found that phosphorylation of 53BP1 at S1618 is restricted to mitotic cells, suggesting that additional mechanisms regulating the modification of 53BP1 by Plk1 may exist. Indeed, interaction between 53BP1 and Plk1 was increased during mitosis. Mitotic phosphorylation of S380 in the N-terminal part of 53BP1 was previously reported to generate a docking site for the PBD domain of Plk1.Citation23 Whereas, Plk1 is activated at least 5–6 h prior mitosis, activation of Cdk1 triggers the onset of mitosis. Therefore, Plk1 interacts with 53BP1 only after its priming phosphorylation by Cdk1 during mitosis. In addition, Plk1 and 53BP1 co-localize at kinetochores from prophase to metaphase. How exactly 53BP1 is recruited to kinetochores still remains an open question. In this respect, it will be interesting to investigate whether localization of 53BP1 to kinetochores might be controlled by a recently described H4K20 monomethylation of the centromeric mononucleosomes.Citation49 The dynamics of pS1618–53BP1 correlates with the activity of Plk1 during mitosis and concurrently with the APCCdh1-dependent degradation of Plk1, pS1618–53BP1 levels rapidly decline during mitotic exit.Citation41 Protein phosphatase PP4 has recently been reported to dephosphorylate pS1618–53BP1 during mitotic exit.Citation50 Thus the interaction between 53BP1 and Plk1 and phosphorylation of the S1618 is tightly spatiotemporally regulated during mitosis.
Since the phosphomimicking 53BP1-S1618D mutant shows an intermediate phenotype, other posttranslational modifications in the C-terminal part of 53BP1 likely contribute to abolishing the recruitment of 53BP1 to DNA damage foci during mitosis. Indeed, we find that 53BP1-S1609D-S1618D mutant shows an increased defect in localization to DNA damage foci in cells and also in binding ubiquitinated histones in vitro. This suggests that Plk1 and Cdk1 both phosphorylate 53BP1 and collaborate on blocking its recruitment to damaged chromatin during mitosis. Although 53BP1-S1609A-S1618A mutant localizes to DNA damage foci in interphase cells, it fails to rescue the ability to localize to DNA damage foci during mitosis (data not shown). This suggests that recruitment of 53BP1 to damage foci is likely blocked by multiple mechanisms during mitosis. One of such mechanisms may involve control of histone ubiquitination in the chromatin flanking the DNA damage site. Ubiquitin ligase RNF8 has recently been identified as a substrate of Cdk1 and mitotic phosphorylation of RNF8 impairs its interaction with MDC1 and leads to the loss of histone ubiquitination during mitosis.Citation51 Combined expression of RNF8-T198A and 53BP1-T1609A-S1618A restored the ability of 53BP1 to localize to DNA damage foci in mitosis.Citation51 However, T198 of RNF8 is not evolutionary conserved and thus additional mechanisms may exist to regulate DDR in mitosis. For instance, level of the chromatin ubiquitination during mitosis may be controlled by deubiquitination. Multiple deubiquitinating enzymes were proposed to remove monomeric ubiquitin from K119 and K13/K15 of H2A and therefore it is likely that there is some redundancy in controlling histone deubiquitination.Citation52-54 Among these deubiquitinating enzymes USP3 and USP16 were reported to be regulated by mitotic events. USP3 associates with the chromatin and has the ability to deubiquitinate both K119 and K13/K15 of H2A.Citation52 USP16 is considered a major deubiquitinase of histone H2A and its depletion results in mitotic defects.Citation55 Moreover, function of USP16 is controlled by phosphorylation of S552 by Cdk1 during mitosis.Citation56 Consistent with the proposed role of USP16 in chromatin deubiquitination, its catalytically inactive mutant USP16-C205S was reported to associate with mitotic chromatin.Citation57 Similarly, we have observed that catalytically inactive mutant USP3-C168S is enriched at chromatin during mitosis (data not shown). Therefore it will be interesting to investigate the role of deubiquitinating enzymes in preventing histone ubiquitination during mitosis.
Localization of the total 53BP1 to kinetochores was reported previously, however its functional relevance has remained unclear.Citation40 Consistent with a previous report we did not observe any defects in formation of the bipolar mitotic spindle or in control of the spindle checkpoint in cells depleted of 53BP1.Citation23 Therefore, we propose that localization of 53BP1 to kinetochores enables its interaction with and phosphorylation by Plk1. In turn, phosphorylated 53BP1 may dissociate from kinetochores resulting in diffuse distribution throughout cytosol. Phosphorylation of the UDR motif by Plk1 and Cdk1 prevents the interaction of 53BP1 with the chromatin.
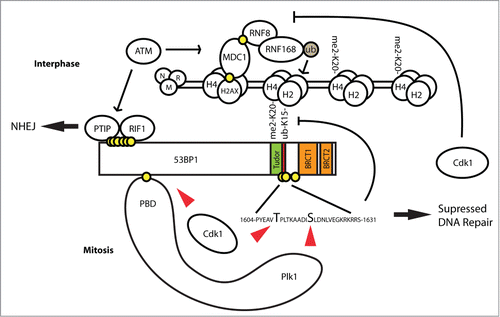
Condensed chromatin in mitosis represents a major challenge for DNA repair. Since mitotic DNA lesions cannot be repaired by an error free homologous recombination, the only repair pathway that may act during mitosis is non-homologous end joining.Citation21,58,59 However, erroneous joining of 2 DNA ends may result in chromosomal translocations with deleterious consequences. Therefore it might be beneficial for cells to supress DNA repair during mitosis and repair the lesions after progressing to the following G1 phase. In particular, fusions of telomeres may occur in prolonged mitotic arrest when integrity of the telomeres is partially affected and this may cause aneuploidy.Citation60 Indeed, inhibition of 53BP1 was reported to prevent fusion of telomeres in mitotic cells exposed to irradiation.Citation51 Our data are consistent with this recent report published while our manuscript was in preparation. Since 53BP1 plays important role in control of genome integrity during interphase, inhibition of its function has to be carefully regulated. We propose a model by which combined modification of 53BP1 by Cdk1 and Plk1 allows transient inhibition of 53BP1 binding to the chromatin specifically during mitosis (). Degradation of cyclin B and Plk1 in anaphase as well as direct dephosphorylation of pS1618 by protein phosphatases allows rapid reactivation of DNA repair during mitotic exit.
Figure 7. Model of 53BP1 inhibition by Plk1 and Cdk1 phosphorylation in mitosis. Following exposure of interphase cells to genotoxic stress, activation of ATM eventually leads to monoubiquitination of H2A by RNF168. Together with constitutive H4K20-me2 modification this allows recruitment of 53BP1 to DNA damage foci and its function in NHEJ repair. In mitosis, Cdk1 phosphorylates 53BP1 in the N-terminal part to generate a docking site for Plk1. In turn, Plk1 phosphorylates 53BP1 at S1618 within the UDR domain and disables binding of 53BP1 to H2A-Ub. In addition, Cdk1 phosphorylates S1609 and S1678 further inhibiting the ability of 53BP1 to bind to H2A-Ub. Mitotic 53BP1 is not phosphorylated by ATM in mitosis and its role in NHEJ is blocked.
Materials and Methods
Cell lines
Human U2OS, HeLa and hTERT-RPE1 were cultured in Dulbecco's modified Eagle's medium (D-MEM) supplemented with 10% foetal bovine serum (FBS), L-glutamine (2 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C under 5% CO2 atmosphere and 95% humidity. Cells were synchronized at G1/S transition by a double thymidine (2.5 mM) block (12 h-Thy/8 h-release/12 h-Thy), washed in PBS, released to fresh media supplemented with nocodazole to arrest cells in mitosis and collected in 2 h intervals. Alternatively, cells were synchronized in mitosis by 12 h cultivation in the presence of nocodazole, collected by shake-off, washed in PBS, released to fresh media and collected in 1 h intervals. Where indicated, cells were treated with BI-2536 (10 nM; Selleckchem), Ro 3306 (9 μM; Tocris Bioscience) or SB202190 (3 μM; Sigma) selective inhibitors of Plk1, Cdk1 and p38 kinases, respectively. Cells were transfected with Silencer Select siRNA (10 nM) targeting GAPDH (negative control), Plk1 (CAACCAAAGUCGAAUAUGA) or 53BP1 (GAAGGACGGAGUACUAAUA) using Lipofectamine RNAiMAX (Life Technologies) and 48 h after transfection nocodazole was added for additional 12 h to synchronize cells in mitosis. For reconstitution experiments, endogenous 53BP1 was depleted by transfection of siRNA targeting the 3-UTR region of 53BP1 transcript (AAAUGUGUCUUGUGUGUAA).Citation61 Plasmid DNA transfections were performed using GeneCellin Transfection Reagent (BioCellChallenge). U2OS cells stably expressing tetracycline repressor were cultured as described previously.Citation24
Antibodies
Following antibodies were used in this study: Cyclin B (sc-245), Cyclin A (sc-53230), BRCA1 (sc-6954), TFIIH (sc-293) and 53BP1 (sc-22760; Santa Cruz Biotechnology); pS10-histone H3 (06–570; Millipore-Upstate); anti-FLAG (F1804; Sigma); pS15-p53 (#9284S), γH2AX (#2577), pT210-Plk1 (#9062, clone D5H7) and pS1618–53BP1 (#6209, clone D4H11; Cell Signaling Technology); CREST (Cortex Biochem); anti-α-tubulin (clone TU-01) and anti-gamma-tubulin (clone TU-32, Exbio Praha). Alexa Fluor fluorescently labeled secondary antibodies were from Life Technologies. Secondary antibodies labeled with HRP were from Bio-Rad.
Plasmids
DNA fragment coding the EGFP was cloned into HindIII/KpnI sites of pcdna4/TO (Life Technologies) and human 53BP1 cDNA was ligated in frame with EGFP into KpnI/NotI sites. Fragment coding the C-terminal part of 53BP1 (amino acids 1483–1972; referred to as 53BP1-C-term) was cloned in BamHI/NotI sites of pGEX6P (GE Healthcare). Mutagenesis was performed using QuickChange II Site-directed mutagenesis kit (Agilent Technologies). Human USP3 cDNA (NM_006537, Open Biosystems) was cloned in frame in XhoI site of pcdna4/TO-EGFP and USP3-C168S mutant was generated by site directed mutagenesis. All mutants were verified by sequencing.
Protein purification and kinase assays
GST-tagged proteins were expressed in BL21-Gold (DE3) bacteria by induction with 0.5 mM IPTG for 5 h at 37°C and purified using Glutathione Sepharose 4 Fast Flow (GE Healthcare). Kinase assays were performed in 25 mM MOPS pH 7.2, 12.5 mM β-glycerol-phosphate, 25 mM MgCl2, 2 mM EDTA, 5 mM EGTA, 0.25 mM DTT supplemented with 100 μM ATP and 5 μCi 32P-γ-ATP. Purified GST, 53BP1-Ct-WT, 53BP1-Ct-S1618A (2 μg) were incubated with 100 ng His-Plk1 (17 nmol/min/mg; SignalChem) for 20 min at 37°C. Purified GST, 53BP1-Ct-WT, 53BP1-Ct-T1609A (2 μg) were incubated with 100 ng cyclin B/Cdk1 (16 nmol/min/mg; Biaffin) or 20 ng p38α (90 nmol/min/mg; Sigma) for 20 min at 37°C. Phosphorylation was visualized by autoradiography.
Pull-down assays
U2OS cells stably expressing FLAG-ubiquitin were treated with etoposide (10 μM) for 12 h and lyzed in IP-buffer [20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% TX-100, 10% glycerol] supplemented with 3 mM MgCl2, 5 mM N-Ethylmaleimide (Sigma) and protease inhibitor cocktail (Roche). Cell extract was sonicated 3× for 20 sec, spinned down 15.000 g for 10 min and DNA was removed by incubation with 5 U of Benzonase (EMD Millipore) for 1 h at 4°C. Cell extract was incubated with purified GST, 53BP1-Ct-WT, 53BP1-Ct-S1618D or 53BP1-Ct-S1609D-S1618D (2 μg) for 3 h at 4°C and bound proteins were pulled-down with glutathione sepharose beads (30 μl). Alternatively, 53BP1-Ct-WT was first phosphorylated in vitro by Plk1 or cyclin B/Cdk1 and then used for pull-down assay.
Immunoprecipitation
U2OS cells stably expressing EGFP-53BP1 were synchronized in G2 by Ro3306 or in mitosis by nocodazole, extracted in IP-buffer supplemented with PhosSTOP phosphatase inhibitor (Roche) and sonicated 3× for 20 sec. Cell extracts were clarified by spinning down 15.000 g for 10 min and incubated with GFP-Trap-A (Chromotek) for 1 h at 4°C. After washing with IP-buffer, bound proteins were eluted by SDS sample buffer, separated on 4–12% NUPAGE gradient gel and analyzed by immunoblotting.
Immunofluorescence and microscopic analysis
Cells were cultured on glass coverslips, fixed using 4% formaldehyde for 10 min, permeabilized by ice-cold methanol, blocked for 1 h with 3% BSA in PBS supplemented with 0.1% Tween-20 (PBST) and incubated with the primary antibodies for 1 h at room temperature. After washing with PBST, coverslips were incubated with Alexa Fluor conjugated secondary antibodies for 1 h at RT. DNA was stained with DAPI (Sigma) for 2 min and coverslips were mounted using Vectashield reagent (Vector Laboratories). For quantification of 53BP1-pS1619 signal intensity images were captured using a Leica AF 6000 System (DFC 350 FX R2 camera) equipped with 40×/0.75 DRY objective (HCX PL APO; Leica) and total cell fluorescence was measured using ImageJ. For each cell, the background signal of surrounding area without cells was subtracted. Confocal microscopy was carried out on Leica SP5 DMI 6000 equipped with 63×/1.40 objective (HC PL APO CS2 OIL; Leica) and each channel was scanned independently. Analysis of 53BP1, BRCA1 and γH2AX foci formation was performed using a high-content screening station (Scan^R; IX81 Olympus; ORCA-285 camera) equipped with a 40×/1.3 NA objective (RMS40X-PFO; Olympus) as described previously. Nuclei were identified based on DAPI signal. Average number of 53BP1, BRCA1 or H2AX foci was determined using a spot detection module. Cells transfected with wild-type or mutant EGFP-53BP1 were gated according to the EGFP signal. DNA damage by microirradiation and subsequent imaging was performed using Leica SP5 DMI 6000 equipped with 63×/1.40 objective (HC PL APO CS2 OIL; Leica) and temperature-controlled (37°C) chamber. Cells grown on μ-Slide 8-well (Ibidi) were pre-sensitized with BrdU (10 μM) for 24 h. Before imaging, culture media was replaced with colorless Leibovitz's L-15 medium (Invitrogen) supplemented with 10% FCS. Cells were microirradiated with a 405 nm laser (100% power) and imaged at 30 s intervals for 10 frames.
Flow cytometry
Cells were synchronized in mitosis by nocodazole in presence of BI-2536 (50 nM), collected by shake-off and treated with NCS (2 nM) for indicated time. Cells were harvested, fixed by ethanol and permeabilized using 0.2% Triton X-100. After blocking with 1% BSA in PBS, cells were incubated with primary antibodies in blocking solution for 2 h at room temperature, washed and incubated with Alexa Fluor conjugated secondary antibodies for 1 h at room temperature. Cells were washed, stained with DAPI and subjected to FACS analysis using LSRII (BD Biosciences) and FlowJo software (Tree Star) to determine intensity of γ-H2AX in mitotic cells.
Comet assay
Cells were synchronized in mitosis by nocodazole in presence or absence of BI-2536 (50 nM), collected by shake-off and treated with NCS (2 nM) for indicated time. Cells were harvested, washed by PBS, embedded to slides with low melting agarose and lysed in 2.5 M NaCl, 100 mM EDTA, 10 mM Tris-Cl pH 10 supplemented with 0.5% Triton X-100 overnight at 4°C. Next day, slides were immersed in cold neutral electrophoresis buffer (50 mM Tris, 150 mM sodium acetate pH 9) for 30 minutes and then subjected to electrophoresis in neutral electrophoresis buffer at 1 V/cm distance of electrodes for 45 minutes at 4°C. DNA was precipitated by ethanol and dried before staining with SYBR gold in PBS for 30 minutes at room temperature. Slides were washed and dried. Images were acquired by a Leica AF 6000 System (DFC 350 FX R2 camera) equipped with 10×/0.40 DRY objective (HCX PL APO). Comets were analyzed by ImageJ using OpenComet plugin and percentage of DNA in comet tails was quantified.
Statistical analysis
Statistical significance was evaluated by Prism software using a 2-tailed paired t-test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are thankful to Zuzana Horejsi (IMG, Prague) for critical reading of the manuscript, David Staňek (IMG, Prague) for making the Scan^R imaging station available and to Ondrej Horvat and Markéta Vančurová for technical support.
Funding
This work was supported by the Grant Agency of the Czech Republic (P305–12–2485). LM was supported also by Ministry of Education Youth and Sports (CZ-OPENSCREEN, LO1220).
References
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/10.1038/nature08467
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3:155-68; PMID:12612651; http://dx.doi.org/10.1038/nrc1011
- Stucki M, Clapperton J, Mohammad D, Yaffe M, Smerdon S, Jackson S. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005; 123:1213-26; PMID: 16377563; http://dx.doi.org/10.1016/j.cell.2005.09.038
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007; 131:887-900; PMID:18001824; http://dx.doi.org/10.1016/j.cell.2007.09.040
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009; 136:435-46; PMID:19203579; http://dx.doi.org/10.1016/j.cell.2008.12.041
- Mattiroli F, Vissers Joseph HA, van Dijk Willem J, Ikpa P, Citterio E, Vermeulen W, Marteijn Jurgen A, Sixma Titia K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012; 150:1182-95; PMID:22980979; http://dx.doi.org/10.1016/j.cell.2012.08.005
- Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CCY, Huang H, Landry M-C, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013; 499:50-4; PMID: 23760478; http://dx.doi.org/10.1038/nature12318
- Botuyan MV, Lee J, Ward IM, Kim J-E, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific Recognition of Histone H4-K20 by 53BP1 and Crb2 in DNA Repair. Cell 2006; 127:1361-73; PMID:17190600; http://dx.doi.org/10.1016/j.cell.2006.10.043
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007; 316:1194-8; PMID:17525340; http://dx.doi.org/10.1126/science.1139476
- Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 2007; 316:1202-5; PMID:17525342; http://dx.doi.org/10.1126/science.1139621
- Noon AT, Shibata A, Rief N, Lobrich M, Stewart GS, Jeggo PA, Goodarzi AA. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat Cell Biol 2010; 12:177-84; PMID:20081839; http://dx.doi.org/10.1038/ncb2017
- Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young Jordan TF, Tkáč J, Cook Michael A, Rosebrock Adam P, Munro M, Canny Marella D, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 2013; 49:872-83; http://dx.doi.org/10.1016/j.molcel.2013.01.001
- Dimitrova N, Chen Y-CM, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 2008; 456:524-8; PMID:18931659; http://dx.doi.org/10.1038/nature07433
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 2013; 339:711-5; PMID:23306439; http://dx.doi.org/10.1126/science.1230624
- Chapman JR, Barral P, Vannier J-B, Borel V, Steger M, Tomas-Loba A, Sartori Alessandro A, Adams Ian R, Batista Facundo D, Boulton Simon J. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 2013; 49:858-71; PMID:23333305; http://dx.doi.org/10.1016/j.molcel.2013.01.002
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 2013; 339:700-4; PMID: 23306437; http://dx.doi.org/10.1126/science.1231573
- Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014; 15:7-18; PMID:24326623; http://dx.doi.org/10.1038/nrm3719
- Zimmermann M, de Lange T. 53BP1: pro choice in DNA repair. Trends Cell Biol 2014; 24:108-17; PMID: 24094932; http://dx.doi.org/10.1016/j.tcb.2013.09.003
- Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to double-strand breaks during mitosis. J Cell Biol 2010; 190:197-207; PMID:20660628; http://dx.doi.org/10.1083/jcb.200911156
- Giunta S, Jackson SP. Give me a break, but not in mitosis: the mitotic DNA damage response marks DNA double-strand breaks with early signaling events. Cell Cycle 2011; 10:1215-21; PMID:21412056; http://dx.doi.org/10.4161/cc.10.8.15334
- Zhang W, Peng G, Lin S-Y, Zhang P. DNA damage response is suppressed by the high cyclin-dependent kinase 1 activity in mitotic mammalian cells. J Biol Chem 2011; 286:35899-905; PMID:21878640; http://dx.doi.org/10.1074/jbc.M111.267690
- Nelson G, Buhmann M, von Zglinicki T. DNA damage foci in mitosis are devoid of 53BP1. Cell Cycle 2009; 8:3379-83; PMID:19806024; http://dx.doi.org/10.4161/cc.8.20.9857
- van Vugt MATM, Gardino AK, Linding R, Ostheimer GJ, Reinhardt HC, Ong S-E, Tan CS, Miao H, Keezer SM, Li J, et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G2/M DNA damage checkpoint. PLoS Biol 2010; 8:e1000287; PMID:20126263; http://dx.doi.org/10.1371/journal.pbio.1000287
- Macurek L, Benada J, Müllers E, Halim VA, Krejčíková K, Burdová K, Pecháčková S, Hodný Z, Lindqvist A, Medema RH, et al. Downregulation of Wip1 phosphatase modulates the cellular threshold of DNA damage signaling in mitosis. Cell Cycle 2013; 12:251-62; PMID:23255129; http://dx.doi.org/10.4161/cc.23057
- Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci 2008; 105:10762-7; http://dx.doi.org/10.1073/pnas.0805139105
- Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 2010; 3:ra3-; PMID:20068231; http://dx.doi.org/10.1126/scisignal.2000475
- Vassilev LT. Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle 2006; 5:2555-6; PMID:17172841; http://dx.doi.org/10.4161/cc.5.22.3463
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci 2006; 103:10660-5; http://dx.doi.org/10.1073/pnas.0600447103
- Seki A, Coppinger JA, Du H, Jang C-Y, Yates JR, Fang G. Plk1- and β-TrCP–dependent degradation of Bora controls mitotic progression. J Cell Biol 2008; 181:65-78; PMID:18378770; http://dx.doi.org/10.1083/jcb.200712027
- Bruinsma W, Raaijmakers JA, Medema RH. Switching Polo-like kinase-1 on and off in time and space. Trends Biochem Sci 2012; 37:534-42; PMID:23141205; http://dx.doi.org/10.1016/j.tibs.2012.09.005
- Takaki T, Trenz K, Costanzo V, Petronczki M. Polo-like kinase 1 reaches beyond mitosis—cytokinesis, DNA damage response, and development. Curr Opin Cell Biol 2008; 20:650-60; PMID:19000759; http://dx.doi.org/10.1016/j.ceb.2008.10.005
- van Vugt MATM, Brás A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell 2004; 15:799-811; PMID:15350223; http://dx.doi.org/10.1016/j.molcel.2004.07.015
- Medema RH, Macurek L. Checkpoint control and cancer. Oncogene 2012; 31:2601-13; PMID:21963855; http://dx.doi.org/10.1038/onc.2011.451
- Mamely I, van Vugt MATM, Smits VAJ, Semple JI, Lemmens B, Perrakis A, Medema RH, Freire R. Polo-like kinase-1 controls proteasome-dependent degradation of claspin during checkpoint recovery. Curr Biol 2006; 16:1950-5; PMID:16934469; http://dx.doi.org/10.1016/j.cub.2006.08.026
- Mailand N, Bekker-Jensen S, Bartek J, Lukas J. Destruction of claspin by SCF[β]TrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell 2006; 23:307-18; PMID:16885021; http://dx.doi.org/10.1016/j.molcel.2006.06.016
- Nakajima H, Toyoshima-Morimoto F, Taniguchi E, Nishida E. Identification of a consensus motif for Plk (Polo-like Kinase) phosphorylation reveals Myt1 as a Plk1 substrate*. J Biol Chem 2003; 278:25277-80; PMID:12738781; http://dx.doi.org/10.1074/jbc.C300126200
- Santamaria A, Wang B, Elowe S, Malik R, Zhang F, Bauer M, Schmidt A, Silljé HHW, Körner R, Nigg EA. The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol Cell Proteomics 2011; 10:M110.004457; PMID:20860994; http://dx.doi.org/10.1074/mcp.M110.004457
- Heijink A, Krajewska M, van Vugt MA. The DNA damage response during mitosis. Mutat Res 2013; 750:45-55; PMID:23880065; http://dx.doi.org/10.1016/j.mrfmmm.2013.07.003
- Petronczki M, Lénárt P, Peters J-M. Polo on the rise—from mitotic entry to cytokinesis with Plk1. Dev Cell 2008; 14:646-59; PMID:18477449; http://dx.doi.org/10.1016/j.devcel.2008.04.014
- Jullien D, Vagnarelli P, Earnshaw WC, Adachi Y. Kinetochore localisation of the DNA damage response component 53BP1 during mitosis. J Cell Sci 2002; 115:71-9; PMID:11801725
- Lindon C, Pines J. Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. J Cell Biol 2004; 164:233-41; PMID:14734534; http://dx.doi.org/10.1083/jcb.200309035
- Cha H, Wang X, Li H, Fornace AJ. A functional role for p38 MAPK in modulating mitotic transit in the absence of stress. J Biol Chem 2007; 282:22984-92; PMID:17548358; http://dx.doi.org/10.1074/jbc.M700735200
- Lee K, Kenny AE, Rieder CL. P38 mitogen-activated protein kinase activity is required during mitosis for timely satisfaction of the mitotic checkpoint but not for the fidelity of chromosome segregation. Mol Biol Cell 2010; 21:2150-60; PMID:20462950; http://dx.doi.org/10.1091/mbc.E10-02-0125
- Elia AEH, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the polo-box domain. Cell 2003; 115:83-95; PMID:14532005; http://dx.doi.org/10.1016/S0092-8674(03)00725-6
- Gatti M, Pinato S, Maspero E, Soffientini P, Polo S, Penengo L. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle 2012; 11:2538-44; PMID:22713238; http://dx.doi.org/10.4161/cc.20919
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 2011; 193:97-108; PMID:21444690
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Solvhoj Pedersen R, Grofte M, Chan KL, Hickson ID, Bartek J, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 2011; 13:243-53; PMID:21317883; http://dx.doi.org/10.1038/ncb2201
- Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora a to promote checkpoint recovery. Nature 2008; 455:119-23; PMID:18615013; http://dx.doi.org/10.1038/nature07185
- Hori T, Shang W-H, Toyoda A, Misu S, Monma N, Ikeo K, Molina O, Vargiu G, Fujiyama A, Kimura H, et al. Histone H4 Lys 20 monomethylation of the CENP-A nucleosome is essential for kinetochore assembly. Dev Cell 2014; 29:740-9; PMID:24960696; http://dx.doi.org/10.1016/j.devcel.2014.05.001
- Lee D-H, Acharya Sanket S, Kwon M, Drane P, Guan Y, Adelmant G, Kalev P, Shah J, Pellman D, Marto Jarrod A, et al. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol Cell 2014; 54:512-25; PMID:24703952; http://dx.doi.org/10.1016/j.molcel.2014.03.020
- Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, Durocher D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 2014; 344:189-93; PMID:24652939; http://dx.doi.org/10.1126/science.1248024
- Sharma N, Zhu Q, Wani G, He J, Wang Q-e, Wani AA. USP3 counteracts RNF168 via deubiquitinating H2A and γH2AX at lysine 13 and 15. Cell Cycle 2014; 13:106-14; PMID:24196443; http://dx.doi.org/10.4161/cc.26814
- Mosbech A, Lukas C, Bekker-Jensen S, Mailand N. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J Biol Chem 2013; 288:16579-87; PMID:23615962; http://dx.doi.org/10.1074/jbc.M113.459917
- Delgado-Díaz MR, Martín Y, Berg A, Freire R, Smits VAJ. Dub3 controls DNA damage signalling by direct deubiquitination of H2AX. Mol Oncol 2014; 8(5):884-93; PMID:24704006
- Joo H-Y, Zhai L, Yang C, Nie S, Erdjument-Bromage H, Tempst P, Chang C, Wang H. Regulation of cell cycle progression and gene expression by H2A deubiquitination. Nature 2007; 449:1068-72; PMID:17914355; http://dx.doi.org/10.1038/nature06256
- Xu Y, Yang H, Joo H-Y, Yu J-H, Smith Iv AD, Schneider D, Chow LT, Renfrow M, Wang H. Ubp-M serine 552 phosphorylation by cyclin-dependent kinase 1 regulates cell cycle progression. Cell Cycle 2013; 12:3408-16
- Cai S-Y, Babbitt RW, Marchesi VT. A mutant deubiquitinating enzyme (Ubp-M) associates with mitotic chromosomes and blocks cell division. Proc Natl Acad Sci 1999; 96:2828-33; http://dx.doi.org/10.1073/pnas.96.6.2828
- Simoneau A, Robellet X, Ladouceur A-M, D’Amours D. Cdk1-dependent regulation of the Mre11 complex couples DNA repair pathways to cell cycle progression. Cell Cycle 2014; 13:1078-90; PMID:24553123; http://dx.doi.org/10.4161/cc.27946
- Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005; 434:598-604; PMID:15800615; http://dx.doi.org/10.1038/nature03404
- Hayashi MT, Cesare AJ, Fitzpatrick JAJ, Lazzerini-Denchi E, Karlseder J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol 2012; 19:387-94; PMID:22407014; http://dx.doi.org/10.1038/nsmb.2245
- Knobel PA, Belotserkovskaya R, Galanty Y, Schmidt CK, Jackson SP, Stracker TH. USP28 is recruited to sites of DNA damage by the tandem BRCT domains of 53BP1 but plays a minor role in double-strand break metabolism. Mol Cell Biol 2014; 34:2062-74; PMID:24687851; http://dx.doi.org/10.1128/MCB.00197-14