
Abstract
Cap-dependent translation is a potential cancer-related target (oncotarget) due to its critical role in cancer initiation and progression. 4EGI-1, an inhibitor of eIF4E/eIF4G interaction, was discovered by screening chemical libraries of small molecules. 4EGI-1 inhibits cap-dependent translation initiation by impairing the assembly of the eIF4E/eIF4G complex, and therefore is a potential anti-cancer agent. Here, we report that 4EGI-1 also inhibits mTORC1 signaling independent of its inhibitory role on cap-dependent translation initiation. The inhibition of mTORC1 signaling by 4EGI-1 activates Akt due to both abrogation of the negative feedback loops from mTORC1 to PI3K and activation of mTORC2. We further validated that mTORC2 activity is required for 4EGI-1-mediated Akt activation. The activated Akt counteracted the anticancer effects of 4EGI-1. In support of this model, inhibition of Akt potentiates the antitumor activity of 4EGI-1 both in vitro and in a xenograft mouse model in vivo. Our results suggest that a combination of 4EGI-1and Akt inhibitor is a rational approach for the treatment of cancer.
Abbreviations
| eIF4E | = | eukaryotic translation initiation factor 4E |
| 4EBP1 | = | eIF4E-binding protein 1 |
| p70S6K | = | ribosomal p70 S6 kinase |
| mTORC1 | = | mammalian target of rapamycin complex 1 |
| mTORC2 | = | mammalian target of rapamycin complex 2 |
| PI3K | = | phosphatidylinositol 3-kinase |
| ER stress | = | endoplasmic reticulum stress |
Introduction
Cancer initiation and progression involves deregulation of cap-dependent translation.Citation1 Thus far, the best characterized translation factors in cancer development are in the eIF4F cap-binding initiation complex, which promotes cap-dependent translation by facilitating the interaction between the 5′ cap mRNA and the 40S ribosome subunit.Citation2 eIF4F promotes cancer pathogenesis by enhancing the translation of a subset of mRNAs such as cyclin D1, survivin, VEGF, and MMP9 that regulate cell proliferation, survival, angiogenesis, and metastasis, respectively.Citation3 The eIF4F complex consists of the 5′ cap mRNA-binding protein (eIF4E), an ATP-dependent RNA helicase (eIF4A), and a scaffolding protein (eIF4G).Citation4 Assembly and activity of the eIF4F complex depends mainly on the availability of eIF4E, which is controlled by increased eIF4E expression or elevated phosphorylation of the 4EBPs. Increased eIF4E expression has been related to progression of malignancy and low patient survival in multiple human cancers, including lymphomas, and cancers in the breast, colon, lung, prostate, and skin.Citation5-9 Recent studies have also associated the elevated phosphorylation of 4EBP1 with disease progression in ovarian, breast, and prostate cancers.Citation8,10-12 In addition, the oncogenic potential of eIF4E hyperactivity has been well addressed both in vitro and in vivo.Citation13-15 Together, these reports indicate the potential of both eIF4E and the eIF4F complex to be attractive therapeutic targets to treat human cancers.
4EGI-1, an inhibitor of eIF4E/eIF4G interaction, was initially discovered by screening chemical libraries of small molecules using a high throughput fluorescence polarization (FP) assay.Citation16 4EGI-1 interferes with the interaction of eIF4E with eIF4G, and thereby impairs the recruitment of 40S ribosomal subunit to the 5′ cap mRNA to inhibit cap-dependent translation initiation. Treatment of cancer cells with 4EGI-1 reduces the expression of eIF4F-regulated proteins such as cyclin D1, cyclin E, c-myc and Bcl-2, but has little effect on the expression of housekeeping proteins. In addition, 4EGI-1 has been shown to suppress growth and induce apoptosis of many myeloma and lung cancer cells in vitro and inhibit myeloma and breast cancer xenografts without apparent toxicity in vivo.Citation17-19 Other studies utilizing 4EGI-1 as an inhibitor of cap-dependent translation have significantly contributed to our understanding of the role of eIF4F complex in autism spectrum disorders, memory formation and consolidation and viral infection.Citation20-23
In this study, we investigated the effects of 4EGI-1 on PI3K/Akt/mTORC1 signaling due to its critical role in the regulation of cap-dependent translation initiation. We found that 4EGI-1 inhibits mTORC1 signaling and that this inhibitory role was independent from the inhibition of cap-dependent translation initiation. Surprisingly, we found that the inhibition of mTORC1 signaling by 4EGI-1 did not potentiate the inhibitory effect of 4EGI-1 on cell proliferation, but caused Akt activation in PI3K and mTORC2 dependent manner. We further validated that Akt activation by 4EGI-1 attenuates its antitumor activity both in vitro and in a xenograft mouse model in vivo.
Results
4EGI-1 inhibits mTORC1 signaling independent from its inhibition of cap-dependent protein translation
To evaluate the effects of 4EGI-1 on mTORC1 signaling, we examined the activation states of p70S6K and 4E-BP1, 2 downstream effectors of mTORC1, after treatment with 4EGI-1. As shown in , 4EGI-1 treatment significantly decreased the phosphorylation of p70S6K (p-p70S6K-389) and 4EBP1 (p-4EBP1–37/46, p-4EBP1–65, and p-4EBP1–70), indicating an inhibitory effect of 4EGI-1 on mTORC1 signaling. Phosphorylation of p70S6K declined at 8 h and remained low up to 24 h in both MCF7 and ZR-75–1 cells. Moreover, the phosphorylation of p70S6K substrate, S6 (pS6–240/244 and pS6–235/236), declined at 8 h followed by inhibition of S6K phosphorylation in both MCF7 and ZR-75–1 cells, suggesting that dephosphorylation of p70S6K in response to 4EGI-1 treatment results in the inhibition of p70S6K kinase activity. In MCF7 cells, the phosphorylation of 4EBP1 decreased at 12 h. However, in ZR-75–1 cells the phosphorylation of 4EBP1 declined much earlier at 4 h.
Figure 1. 4 EGI-1 inhibits mTORC1 signaling independent from its inhibition of cap-dependent protein translation. (A and B) Breast cancer cell lines MCF7 and ZR-75–1 were treated with DMSO or 50μM 4EGI-1 and collected at various time points. Phosphorylation status of downstream mTORC1 effectors was analyzed using protein gel blot with indicated antibodies. (C) Both MCF7 and ZR-75–1 cells were treated with DMSO, 50 μM 4EGI-1 or 50 μM Ribavirin for various times. Cell lysates were analyzed by immunoblotting with indicated antibodies. (D) Control and EIF4E-depleted MCF7 and ZR-75–1 cells were grown in 5% FBS and collected to perform western blot analysis with indicated antibodies.
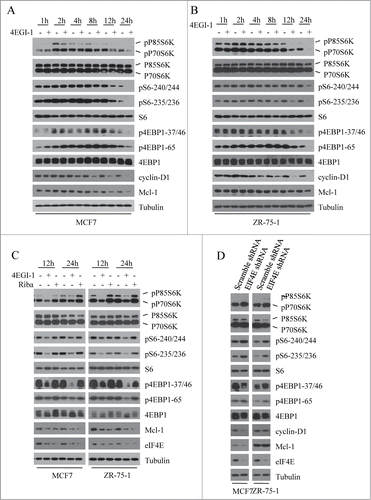
The best characterized role of 4EGI-1 is its function as an inhibitor of cap-dependent translation initiation. To investigate whether the inhibitory effect of 4EGI-1 on cap-dependent translation initiation leads to a decrease in mTORC1 signaling, we analyzed the effect of ribavirin, another inhibitor of cap-dependent translation initiation, on the mTORC1 signaling. Ribavirin suppressed cap-dependent translation initiation by directly interfering with the interaction of eIF4E and the 7-methylguanosine cap structure.Citation24 Unlike 4EGI-1, ribavirin had little effect on the phosphorylation of p70S6K, S6, and 4EBP1 in both MCF7 and ZR-75–1 cells, revealing that 4EGI-1 inhibits mTORC1 signaling independent from its ability to inhibit cap-dependent protein translation (). To further confirm this result, we evaluated the effect of eIF4E knockdown on mTORC1 signaling and found that eIF4E knockdown did not inhibit the phosphorylation of p70S6K, S6, and 4EBP1 but moderately elevated it (). These results clearly indicate that 4EGI-1 inhibits mTORC1 signaling independent from its inhibition of cap-dependent protein translation.
Inhibition of 4EBP1 phosphorylation does not further potentiate the inhibitory effect of 4EGI-1 on cell proliferation
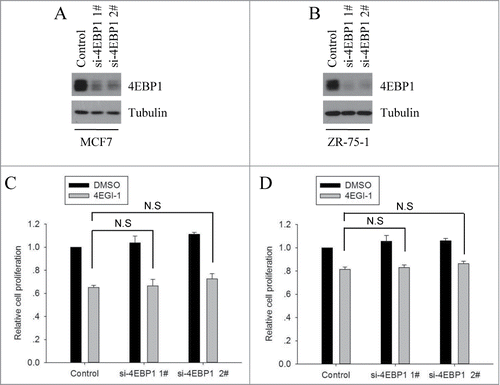
Hypo-phosphorylated 4E-BP1 disrupts eIF4F complex formation and inhibits cap-dependent translation by binding competitively to EIF4E. Besides, it is known that when compared to S6K1, 4E-BP1 plays a more determinant role in mTORC1-mediated cell proliferation and cancer progression.Citation25,26 The above results demonstrated that 4EGI-1 treatment inhibits the phosphorylation of 4EBP1. We presumed that the inhibition of 4EBP1 phosphorylation may further enhance the inhibitory effect of 4EGI-1 on cell proliferation. To test this hypothesis, we knocked down 4EBP1 expression using 2 different siRNAs in both MCF7 and ZR-75–1 cells () and compared the inhibitory effect of 4EGI-1 on cell proliferation to control cells. Clearly, 4E-BP1 knockdown did not restore the inhibitory effect of 4EGI-1 on cell proliferation (). These data show that the inhibition of 4EBP1 phosphorylation does not further potentiate its inhibitory effect on cell proliferation.
Figure 2. 4E-BP1 knockdown did not restore the inhibitory effect of 4EGI-1 on cell proliferation. MCF7 (A) and ZR-75–1 (B) cells were transfected with control siRNA or 2 different 4EBP1 siRNAs. After 72 h, the cells were collected and used for protein gel blot analysis. MCF7 (C) and ZR-75–1 (D) cells were transfected with control siRNA or 2 different 4EBP1 siRNAs. Twelve hours after transfection, cells were seeded into 96-well plates and cultured for an additional 24 h and then treated with DMSO or 4EGI for 48 h. After that, cell number was determined using the CCK8 assay and normalized to the corresponding control. The data presented are the mean from 3 independent experiments. N.S indicates that data between compared samples are not significant.
4EGI-1 increases Akt phosphorylation independent of its inhibitory role in cap-dependent translation initiation
The negative feedback loop from mTOR and S6K1 to PI3K and Akt has been well studied. A number of studies have shown that rapamycin treatment leads to the hyperactivation of Akt by inhibiting this negative feedback loop.Citation27,28 Because 4EGI-1 can inhibit mTORC1 signaling, we determined whether mTOR inhibition by 4EGI-1 could trigger Akt activation. For this, we performed a time course experiment and found that 4EGI-1 treatment significantly increased the phosphorylation of Akt (p-Akt-308 and p-Akt-473) at 12 h followed by inhibition of S6K phosphorylation in both MCF7 and ZR-75–1 cells ().
Figure 3. 4EGI-1 increases Akt phosphorylation independent from its inhibitory role on cap-dependent translation initiation. MCF7 (A) and ZR-75–1 (B) cells were treated with DMSO (-) or 50 μM 4EGI-1 (+) for the indicated times followed by preparation of whole cell protein lysates. The indicated proteins were detected using western blot analysis. Relative expression of pAKT(S473) and pAKT(T308) after the indicated times of treatment with 4EGI-1 was quantified from 3 independent experiments as shown in (a). Columns, mean percentage of 3 independent experiments; bars, SD. *P < 0.05 (C) MCF7 and ZR-75–1 cells were treated with DMSO (-), 50 μM 4EGI-1 (+) or 50 μM Ribavirin (+) for the given times and then harvested to prepare whole cell protein lysates followed by protein gel blot analysis using indicated antibodies. Control and EIF4E-depleted MCF7 and ZR-75–1cells (D) were grown in 5% FBS and harvested for the preparation of whole-cell protein lysates. Western blot analysis was performed with indicated antibodies.
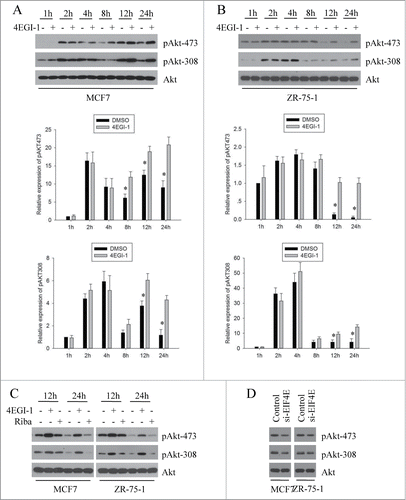
The experiments performed thus far, showed that 4EGI-1 inhibits mTORC1 signaling independent of its ability to inhibit cap-dependent protein translation. To further investigate whether the enhanced effect of 4EGI-1 on Akt phosphorylation depended on its inhibitory role on cap-dependent translation initiation, we compared the effect of 4EGI-1 and ribavirin on Akt. 4EGI-1 markedly increased the phosphorylation of Akt at 12 and 24 h after treatment (). However, ribavirin had little effect on Akt phosphorylation at both time points. To verify this result, we analyzed the effect of eIF4E knockdown on Akt phosphorylation and found that the phosphorylation of Akt was not enhanced by eIF4E knockdown (). Together, these data demonstrate that 4EGI-1 increases Akt phosphorylation independent of its inhibitory role on cap-dependent translation initiation.
4EGI-1-mediated Akt phosphorylation results from mTORC1 inhibition and is PI3K and mTORC2 dependent
The above data have shown that 4EGI-1 suppresses mTORC1 signaling and enhances Akt phosphorylation. To investigate the relationship between these 2 effects (suppression of mTORC1 signaling and induction of Akt phosphorylation), we analyzed 4EGI-1-mediated Akt phosphorylation after specific inhibition of mTORC1 with RAD001. If 4EGI-1-mediated Akt phosphorylation results from the inhibition of mTORC1, pretreatment with RAD001 would abrogate 4EGI-1-induced Akt phosphorylation. As shown in , in both MCF7 and ZR-75–1 cells, either 4EGI-1 or RAD001 enhanced the phosphorylation of Akt, but 4EGI-1 failed to further increase Akt phosphorylation in the presence of RAD001. These results demonstrate that the 2 effects are linked; inhibition of mTORC1 by 4EGI-1 leads to the induction of Akt phosphorylation.
Figure 4. 4EGI-1-mediated Akt feedback activation results from mTORC1 inhibition and is PI3K and mTORC2 dependent. Both MCF7 and ZR-75–1 cells were pretreated with 2 nM RAD001 (A), 5 μM LY294002 (B) or 0.25 μM PP242 (C) for 30–60 min, and then co-treated with 50 μM 4EGI-1. After 12 h, cell lysates were collected and immunoblotted with indicated antibodies. Both MCF7 (D) and ZR-75–1 (E) cells were transfected with control, Rictor or mTOR siRNA for 48 h and then treated with 50 μM 4EGI-1 for 12 h before harvesting cell lysates. The indicated proteins were detected by western blot analysis.

Rapamycin has been reported to induce Akt phosphorylation by inhibiting the negative feedback loop from mTOR and S6K1 to PI3K.Citation29 We therefore hypothesized that 4EGI-1 induced Akt phosphorylation in a similar fashion. To investigate this, we determined the effect of LY294002, an inhibitor of PI3K kinase, on 4EGI-1-mediated Akt phosphorylation. As shown in , LY294002 completely inhibited 4EGI-1-induced Akt phosphorylation in both MCF7 and ZR-75–1 cells, indicating that PI3K kinase activity is required for Akt feedback activation induced by 4EGI-1.
mTORC2 activity is required for the phosphorylation of Akt Ser-473. We next investigated the role of mTORC2 in 4EGI-1-induced Akt phosphorylation. As shown in , pretreatment of MCF7 and ZR-75–1 cells with PP242, an ATP-competitive mTOR kinase inhibitor, abrogated 4EGI-1-mediated Akt phosphorylation. To further confirm the role of mTORC2 in this process, we analyzed 4EGI-1-mediated Akt phosphorylation after impairing mTORC2 complex by silencing the expression of mTOR or Rictor. We found that the disruption of mTORC2 complex prevented Akt feedback activation induced by 4EGI-1 (). Unexpectedly, we found that knockdown of rictor minimally blocked Akt phosphorylation. We speculate that the residual mTORC2 activity after rictor depletion is sufficient for basal phosphorylation levels of Akt, but is not enough to mediate Akt feedback activation induced by 4EGI-1.
Taken together, these data suggest that 4EGI-1-mediated Akt feedback activation results from mTORC1 inhibition and is PI3K and mTORC2 dependent.
4EGI-1-induced Akt feedback activation counteracts its anticancer effects in vitro
Given that Akt is a key survival kinase, it is plausible that 4EGI-1-induced Akt phosphorylation may counteract its anticancer effects. Therefore, we examined the effects of 4EGI-1 together with MK-2206, a clinically available pan Akt inhibitor, on the growth of MCF7 cells. As shown in , the combination of 4EGI-1 and MK-2206 exhibited additive growth-inhibitory effects of the cells when compared to either agent alone, indicating that the induction of Akt phosphorylation in tumors treated with 4EGI-1 may attenuate its inhibitory effect on cell proliferation. We next compared the effect of 4EGI-1 together with MK-2206 on cyclin D1 expression (marker of G1 arrest) and levels of cleaved PARP (marker of apoptosis). As shown in , the combination treatment significantly suppressed the expression of Cyclin D1 and greatly increased the levels of cleaved PARP that in part could be responsible for the synergistic inhibitory effect of 4EGI-1 on cell proliferation.
Figure 5. 4EGI-1-induced Akt feedback activation counteracts its anticancer effects in vitro. Both MCF7 cells were seeded in 96-well plates. On the second day, they were treated with DMSO, 50 μM 4EGI-1, 0.1 μM MK2206 (A), 5 μM LY294002 (B) or 0.25 μM PP242 (C) individually or in combination for 24, 36 or 48 h. Cell proliferation was determined using the CCK8 assay. Relative proliferation was expressed as fold change vs. the corresponding control (DMSO). All experiments were repeated 3 times. *P < 0 .05, compared to the control. MCF7 cells were treated with DMSO, 50 μM 4EGI-1, 0.1 μM MK2206 (A), 5 μM LY294002 (B) or 0.25 μM PP242 (C) individually or in combination for 12 h and harvested for protein gel blot analysis using indicated antibodies. (D) Control, Rictor or mTOR siRNA transfected MCF7 cells were treated with DMSO or 50 μM 4EGI-1 for 24 and 48 h. Cell proliferation was measured by the CCK8 assay and the cell number was normalized to the corresponding control. The data presented are the mean from 3 independent experiments. *P < 0.05, when compared to the control. For western blot analysis, control, Rictor or mTOR siRNA transfected MCF7 cells were treated with DMSO or 50 μM 4EGI-1 for 12 h and then harvested to prepare whole cell protein lysates.
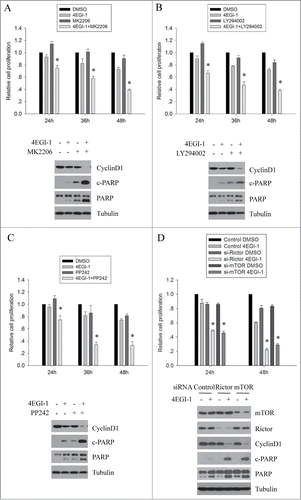
The above studies demonstrated that the induction of Akt phosphorylation by 4EGI-1 is dependent on PI3K and mTORC2. To further confirm that the induction of Akt phosphorylation counteracts the anticancer effects of 4EGI-1, we determined whether inhibition of PI3K or mTORC2 activity enhanced the anti-proliferative effect of 4EGI-1. As shown in , 4EGI-1 together with LY294002 or PP242 exhibited growth-inhibitory effects better than each agent separately. Moreover, similar to MK-2206, both LY294002 and PP242 markedly enhanced the inhibition of Cyclin D1 expression and induction of PARP cleavage in response to 4EGI-1 treatment. We subsequently examined the effect of silencing mTOR or Rictor expression on the anti-proliferative effect of 4EGI-1. The results showed that mTOR or Rictor depletion significantly enhanced the growth-inhibitory effects of 4EGI-1 on MCF7 cells (). Such silencing of mTOR or Rictor expression further reduced Cyclin D1 expression and enhanced PARP cleavage induced by 4EGI-1 treatment, demonstrating that mTORC2-mediated Akt activation in response to 4EGI-1 treatment counteracts the anticancer effects of 4EGI-1.
The combination index (CI) was calculated using the method of Chou and Talalay.Citation30,31 The CI values for combination treatment of 4EGI-1 and MK2206 against MCF7 cells for 24, 36 and 48h were in . After 24 h incubation, the CI values were less than 1 at any given concentration of 4EGI-1 combined with 0.1 μM Mk2206. In addition, after 36 or 48 h incubation, the CI values were also lower than 1 in 4EGI-1 combined with 0.1 μM MK2206 treatment, suggesting a synergistic effect in 4EGI-1 and MK2206 co-treatment.
Table 1. CI Analyses of Combination in MCF7 Cells
Together, these results suggest that 4EGI-1-induced Akt phosphorylation counteracts its anticancer effects.
Akt inhibition enhances the antitumor activity of 4EGI-1 in vivo
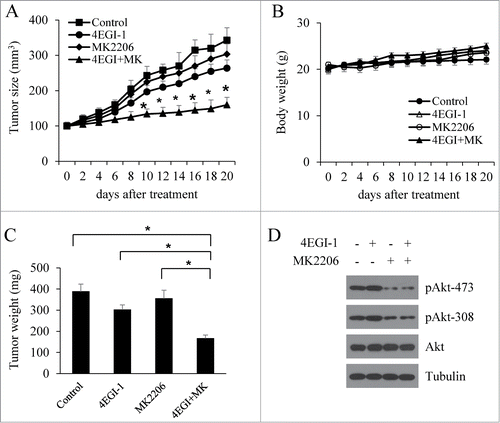
To explore whether the inhibition of reactivated Akt sensitized tumors to 4EGI-1 inhibition, we determined the growth of MCF7 xenografts after exposure to a combination of MK-2206 with 4EGI-1. When tumors reached approximately 100 mm3, the mice were randomly allocated to 4 groups and treated with vehicle control, 4EGI-1, MK-2206, or 4EGI-1 plus MK-2206 for 3 weeks. Consistent with the results from cell cultures, the inhibition of MCF7 xenograft growth by 4EGI-1 and MK-2206 was significantly greater than either agent alone (). During the 3-week treatment period, the tumor weights in mice that received the combination treatment were smaller compared to the groups that received either vehicle or single-agent treatment (). There was no obvious difference in mouse body weights among the groups during the 3-weeks (), indicating that the treatment was well tolerated. We next investigated whether the continuous 4EGI-1 treatment of MCF7 xenografts also induced Akt phosphorylation as observed in cell cultures. The results showed that 4EGI-1 treatment increased the phosphorylation of Akt in vivo andMK-2206 treatment inhibited the phosphorylation of Akt induced by 4EGI-1 in vivo (). These results clearly indicate that 4EGI-1-induced Akt phosphorylation abrogates its anticancer effects and that preventing Akt feedback activation enhances the antitumor activity of 4EGI-1 in vivo.
Figure 6. Akt inhibition enhances the antitumor activity of 4EGI-1 in vivo. Four groups of mice with MCF7 xenografts were treated with vehicle control, 4EGI-1 (50 mg/kg/day, intra-peritoneal injection) or MK2206 (100 mg/kg/two days, oral gavage) individually or in combination. Tumor growth (A) and body weights of mice (C) were monitored over a 20-day period. (B) Tumor weight after necropsy. Data are presented as means ± s.d. *P < 0.05, compared to the vehicle control. #P < 0.05, compared to the 4EGI-1 treatment. (D) Tumor samples were analyzed by protein gel blot with indicated antibodies.
Discussion
eIF4F plays a critical role in the transformation to malignant status and maintenance of transformed phenotypes. These processes are affected by promoting the translation of mRNAs implicated in all aspects of malignancy including growth factors, anti-apoptotic proteins, angiogenesis factors and degrading enzymes. It is reported that eIF4F function is enhanced by increased eIF4E expression or 4EBP1 phosphorylation. Hypo-phosphorylated 4EBP1 restricts the abundance of the eIF4F complex by competing with eIF4G to bind eIF4E. On the other hand, mTORC1 increases the formation of the eIF4F complex by directly phosphorylating 4E-BP1 and promoting its dissociation from eIF4E.Citation32 4EGI-1 has been reported to inhibit cap-dependent translation by specifically inhibiting the interaction of eIF4E and eIF4G. Given that both 4EGI-1 and mTORC1 are associated with the regulation of eIF4F function, we investigated the effects of 4EGI-1 on mTORC1 signaling. Our findings showed that 4EGI-1 effectively suppressed the phosphorylation of p70S6K, 4E-BP1 and S6 in both MCF7 and ZR-75–1 cells, indicating that 4EGI-1 inhibits mTORC1 signaling. However, neither ribavirin nor eIF4E knockdown decreased the phosphorylation of p70S6K, 4E-BP1 and S6, suggesting that 4EGI-1 modulates mTORC1 signaling independent of its ability to inhibit cap-dependent protein translation. Although our studies validate a link between 4EGI-1 and mTORC1 signaling, it is not clear how 4EGI-1 modulates mTORC1 signaling. Previous data has suggested that ER stress could modulate mTORC1 signaling.Citation33 Since 4EGI-1 has been reported to induce ER stress through “off-target” mechanisms, we investigated whether mTORC1 signaling inhibition could be due to an ER stress. However, in our experiments the inhibition of mTORC1 signaling was not reversed by 4-PBA treatment, which alleviates ER stress in both cell lines and animal models (data not shown), indicating that 4EGI-1 inhibits mTORC1 signaling independent of its function as an ER stress inducer. Further studies are required to address the mechanisms by which 4EGI-1 inhibits mTORC1 signaling.
Given that mTORC1 inhibition induces Akt activation by releasing the feedback inhibition of receptor tyrosine kinase signaling, we investigated whether 4EGI-1 mediated mTORC1 inhibition led to Akt activation and found that indeed 4EGI-1induces Akt phosphorylation. Further, our studies showed that 4EGI-1 induces Akt phosphorylation independent of its inhibitory role on cap-dependent translation initiation. To validate whether 4EGI-1- induced Akt phosphorylation is due to its inhibitory role on mTORC1 signaling, we examined 4EGI-1-induced Akt phosphorylation in the presence of an mTORC1 inhibitor and found that inhibition of mTORC1 by RAD001 suppressed 4EGI-1-induced Akt phosphorylation, demonstrating that 4EGI-1-mediated mTORC1 inhibition contributes to the induction of Akt activity. Not surprisingly, our next result showed that 4EGI-1-induced Akt phosphorylation depends on the activity of PI3K and mTORC2, which are both required for Akt Ser-473 phosphorylation. Indeed, the finding that 4EGI-1 induces Akt phosphorylation by releasing the mTOR-PI3K/Akt feedback loop is consistent with the enhancement of Akt activity by rapamycin.Citation34 Recently, multiple mechanisms have been reported to mediate rapamycin-induced feedback activation of PI3K/Akt; by enhancing the expression of the insulin receptor, IGF-1R, and their substrates, IRS-1 and IRS-2, which are downregulated by mTORC1 activation,Citation35-37 by inducing the expression of EGFR, HER2 and HER3Citation38,39 and by blocking PDGFβ-dependent feedback loop.Citation40 It is also reported that mTORC1 mediated phosphorylation stabilizes Grb10, leading to feedback inhibition of PI3K/Akt activation.Citation41,42 It is possible that 4EGI-1 induces Akt phosphorylation by one of above mentioned mechanisms similar to rapamycin. However, the precise mechanism is still unclear and further studies are required to elucidate this.
Akt activation is frequently associated with cancer cell proliferation and survival and PI3K/Akt pathway activation contributes to cancer cell drug resistance.Citation43-45 Given that 4EGI-1 treatment induced the feedback activation of Akt, we speculated that preventing the activation of Akt may enhance the antitumor effect of 4EGI-1. Indeed, as expected, our results showed that MK2206 in combination with 4EGI-1 had a synergistic effect on the inhibition of breast cancer cell proliferation. As mTOR pathway is involved in Cyclin D1 accumulation,Citation46 we further investigated the effect of 4EGI-1 and MK2206 combination on Cyclin D1 expression and found that the combination of 4EGI-1 and MK2206 enhanced the inhibition of Cyclin D1 expression and induction of cleaved PARP, indicating that the induced Akt activation by 4EGI-1 may counteract its anticancer efficacy by activating pathways that attenuate its effects on proliferation and apoptosis. The results also demonstrate that LY294002 or PP242 combined with 4EGI-1 exhibit additive inhibition of cell growth when compared to the effects of each single agent alone, thus confirming the hypothesis that the feedback activation of Akt antagonizes the antitumor effect of 4EGI-1. We next investigated the synergistic anticancer effect of 4EGI-1 and MK2206 combination in xenograft models of human breast cancers. Compared to the effect exerted by each compound separately, the effects of administering the combination showed significantly stronger inhibition of tumor growth in vivo. In addition, the combination treatment did not significantly affect the mice weight, indicating that the combination is well tolerated. Although our results demonstrate that 4EGI-1-induced Akt activation compromises its antitumor activity, the mechanism underlying the negative regulation of 4EGI-1 efficacies by Akt feedback activation is still unclear and need further investigation.
Material and Methods
Chemicals and reagents
4EGI-1 and LY294002 was provided by Calbiochem (Shanghai, China) and Cell Signaling Technology (Danvers, MA), respectively. RAD001, PP242 and MK-2206 were obtained from selleckchem.com (Shanghai, China). Antibodies against, P70S6K, pP70S6K(T389), S6, pS6 (S240/244), pS6 (S235/236), 4EBP-1, p4EBP-1 (T37/46), p4EBP-1 (S65), AKT, pAKT (S473), pAKT (T308), CyclinD1, PARP and cleaved PARP were purchased from Cell Signaling Technology. Antibodies against Tubulin were purchased from Santa Cruz biotechnology (Santa Cruz, CA).
Cell culture
MCF7 and ZR-75–1 breast cancer cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Hyclone, Logan, UT, USA). All treatments with 4EGI-1 were done in DMEM containing 5% FBS. Cells were cultured in an incubator at 37°C and 5% CO2.
To decrease the expression of EIF4E, MCF7 and ZR-75–1 cells were infected with appropriate amounts of lentiviral particles carrying EIF4E shRNA or control shRNA (lentivirus obtained from GeneChem Co., Shanghai, China) with 5–10 ug/ml polybrene. Twelve hours later, virus-containing medium was refreshed with fresh medium. After additional 48 h, stable EIF4E knockdown cells were selected in 1 ug/ml puromycin and pooled clones were used for further experiments.
Western blot analysis
Western blot analysis was performed as described previously.Citation47 Briefly, 20–100 ug of protein were resolved by 10% SDS-PAGE gel after measuring protein concentration using the BCA protein reagent (Pierce Chemical, Rockford, IL, USA) and then transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat milk for 2 h at room temperature and then incubated with primary antibodies overnight at 4°C, followed by incubation with HRP-conjugated anti-rabbit/mouse/goat IgG for 2 h at room temperature. Detection was performed using enhanced chemiluminescence (ECL) detection reagent.
siRNA and transient transfections
siRNA for 4EBP1 (HSS141934 and HSS141936), Rictor (HSS153834), and mTOR (HSS103827) were purchased from Invitrogen. MCF7 and ZR-75–1 breast cancer cells were transfected with 4EBP1, Rictor, or mTOR siRNA or negative control using LipofectamineTM 2000 (Invitrogen) according to the manufacturer's instructions. For western blot analysis, 48 h after transfection, the cells were treated with DMSO and 4EGI for 1–24 h and then lysed. For CCK-8 assay, 12 h after transfection, cells were seeded into 96-well plates and cultured for an additional 24 h and then treated with DMSO or 4EGI for 24–48h.
CCK-8 assay
Cells were seeded in 96-well plates at a density of 1500 cells per well and cultured in DMEM containing 5% FBS. Once attached, the cells were treated with the agents indicated and cultured for the indicated times. After indicated treatment(s), cell proliferation was determined with the Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Tokyo, Japan) assay according to the manufacturer's instructions. The absorbance of individual wells was determined at 450 nm. The OD value of the treatment group was normalized to the values from the untreated control group. All reactions were repeated 3 times. Data are presented as means ± s.d.
Chou-Talalay analyses
The synergistic effect of MK2206 with 4EGI-1 was examined usingmedian effect method as established by Chou and Talalay.Citation30,31 The combination index (CI) values reflect the ways of interaction between 2 drugs. The CI < 1 indicates a synergy, CI = 1indicates an additive effect, and CI > 1 indicates antagonism. The mean values of 3 independent experiments were used. The combination index analysis was carried on CompuSyn software.
Breast cancer xenografts and treatments
Five- to 6-week-old female BALB/c nude mice were obtained from Vital River Laboratory (Beijing, China) and implanted with estrogen pellets (E2, 0.36 mg/pellet, 60-day release) (Innovative Research of America, Sarasota, FL, USA). Two days after implantation, 1 × 107 MCF7 cells in serum-free medium were injected into the abdominal mammary fat pad of nude mice. When tumors reached an approximate size of 100 mm3, the mice were randomly allocated to 4 groups (n = 6/group) according to body weights and tumor volumes for the following treatments: vehicle control, 4EGI-1 (50 mg/kg/day, intra-peritoneal injection), MK2206 (100 mg/kg/2 days, oral gavage), and the combination of 4EGI-1 + MK2206. The tumor volumes and body weights of mice were monitored every 2 d Three weeks after initiation of the treatment, all mice were sacrificed and the tumors were weighed. Xenograft primary tumors were harvested and proteins were extracted for western Blot analysis. Data are presented as means ± s.d.
Statistical analysis
Statistical calculations were performed using SPSS 13.0. The data in this study were presented as means ± standard deviation (sd). Student's t test and x2 test were used when appropriated. P < 0.05 was judged to be statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Natural Science Foundation of China (No. 81470138 to SH.Li and No. 81372770 to JG.Zhou and Nos. 81172445, 81372140 to J.Wang) and National Science and Technology Major Project of the Ministry of Science and Technology of China (No. 2012ZX10003008002 to SH.Li).
References
- Konicek BW, Dumstorf CA, Graff JR. Targeting the eIF4F translation initiation complex for cancer therapy. Cell Cycle 2008; 7:2466-71; PMID:18719377; http://dx.doi.org/10.4161/cc.7.16.6464
- Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer 2010; 10:254-66; PMID:20332778; http://dx.doi.org/10.1038/nrc2824
- De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004; 23:3189-99; PMID:15094768; http://dx.doi.org/10.1038/sj.onc.1207545
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 2009; 136:731-45; PMID:19239892; http://dx.doi.org/10.1016/j.cell.2009.01.042
- Kerekatte V, Smiley K, Hu B, Smith A, Gelder F, De Benedetti A. The proto-oncogenetranslation factor eIF4E: a survey of its expression in breast carcinomas. Int J Cancer 1995; 64:27-31; PMID:7665244; http://dx.doi.org/10.1002/ijc.2910640107
- Rosenwald IB, Chen JJ, Wang S, Savas L, London IM, Pullman J. Upregulation of protein synthesis initiation factor eIF-4E is an early event during colon carcinogenesis. Oncogene 1999; 18:2507-17; PMID:10229202; http://dx.doi.org/10.1038/sj.onc.1202563
- Wang S, Rosenwald IB, Hutzler MJ, Pihan GA, Savas L, Chen JJ, Woda BA. Expression of the eukaryotic translation initiation factors 4E and 2alpha in non-Hodgkin's lymphomas. Am J Pathol 1999; 155:247-55; PMID:10393856; http://dx.doi.org/10.1016/S0002-9440(10)65118-8
- Graff JR, Konicek BW, Lynch RL, Dumstorf CA, Dowless MS, McNulty AM, Parsons SH, Brail LH, Colligan BM, Koop JW, et al. eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival. Cancer Res 2009; 69:3866-73; PMID:19383915; http://dx.doi.org/10.1158/0008-5472.CAN-08-3472
- Wang R, Geng J, Wang JH, Chu XY, Geng HC, Chen LB. Overexpression of eukaryotic initiation factor 4E (eIF4E) and its clinical significance in lung adenocarcinoma. Lung Cancer 2009; 66:237-44; PMID:19261348; http://dx.doi.org/10.1016/j.lungcan.2009.02.001
- Armengol G, Rojo F, Castellvi J, Iglesias C, Cuatrecasas M, Pons B, Baselga J, Ramon Y Cajal S. 4E-binding protein 1: a key molecular "funnel factor" in human cancer with clinical implications. Cancer Res 2007; 67:7551-5; PMID:17699757; http://dx.doi.org/10.1158/0008-5472.CAN-07-0881
- Rojo F, Najera L, Lirola J, Jimenez J, Guzman M, Sabadell MD, Baselga J, Ramon Y Cajal S. 4E-binding protein 1, a cell signaling hallmark in breast cancer that correlates with pathologic grade and prognosis. Clin Cancer Res 2007; 13:81-9; PMID:17200342; http://dx.doi.org/10.1158/1078-0432.CCR-06-1560
- Noske A, Lindenberg JL, Darb-Esfahani S, Weichert W, Buckendahl AC, Roske A, Sehouli J, Dietel M, Denkert C. Activation of mTOR in a subgroup of ovarian carcinomas: correlation with p-eIF-4E and prognosis. Oncol Rep 2008; 20:1409-17; PMID:19020722
- Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5' cap. Nature 1990; 345:544-7; PMID:2348862; http://dx.doi.org/10.1038/345544a0
- Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, Pandolfi PP. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med 2004; 10:484-6; PMID:15098029; http://dx.doi.org/10.1038/nm1042
- Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004; 428:332-7; PMID:15029198; http://dx.doi.org/10.1038/nature02369
- Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007; 128:257-67; PMID:17254965; http://dx.doi.org/10.1016/j.cell.2006.11.046
- Fan S, Li Y, Yue P, Khuri FR, Sun SY. The eIF4EeIF4G interaction inhibitor 4EGI-1 augments TRAIL-mediated apoptosis through c-FLIP Down-regulation and DR5 induction independent of inhibition of cap-dependent protein translation. Neoplasia 2010; 12:346-56; PMID:20360945
- Chen L, Aktas BH, Wang Y, He X, Sahoo R, Zhang N, Denoyelle S, Kabha E, Yang H, Freedman RY, et al. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget 2012; 3:869-81; PMID:22935625
- Descamps G, Gomez-Bougie P, Tamburini J, Green A, Bouscary D, Maiga S, Moreau P, Le Gouill S, Pellat-Deceunynck C, Amiot M. The cap-translation inhibitor 4EGI-1 induces apoptosis in multiple myeloma through Noxa induction. Br J Cancer 2012; 106:1660-7; PMID:22510748; http://dx.doi.org/10.1038/bjc.2012.139
- Hoeffer CA, Cowansage KK, Arnold EC, Banko JL, Moerke NJ, Rodriguez R, Schmidt EK, Klosi E, Chorev M, Lloyd RE, et al. Inhibition of the interactions between eukaryotic initiation factors 4E and 4G impairs long-term associative memory consolidation but not reconsolidation. Proc Natl Acad Sci U S A 2011; 108:3383-8; PMID:21289279; http://dx.doi.org/10.1073/pnas.1013063108
- McMahon R, Zaborowska I, Walsh D. Noncytotoxic inhibition of viral infection through eIF4F-independent suppression of translation by 4EGi-1. J Virol 2011; 85:853-64; PMID:21068241; http://dx.doi.org/10.1128/JVI.01873-10
- Hoeffer CA, Santini E, Ma T, Arnold EC, Whelan AM, Wong H, Pierre P, Pelletier J, Klann E. Multiple components of eIF4F are required for protein synthesis-dependent hippocampal long-term potentiation. J Neurophysiol 2013; 109:68-76; PMID:23054596; http://dx.doi.org/10.1152/jn.00342.2012
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 2013; 493:411-5; PMID:23263185; http://dx.doi.org/10.1038/nature11782
- Kentsis A, Topisirovic I, Culjkovic B, Shao L, Borden KL. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc Natl Acad Sci U S A 2004; 101:18105-10; PMID:15601771; http://dx.doi.org/10.1073/pnas.0406927102
- Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 2010; 328:1172-6; PMID:20508131; http://dx.doi.org/10.1126/science.1187532
- Yellen P, Saqcena M, Salloum D, Feng J, Preda A, Xu L, Rodrik-Outmezguine V, Foster DA. High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell Cycle 2011; 10:3948-56; PMID:22071574; http://dx.doi.org/10.4161/cc.10.22.18124
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 2004; 166:213-23; PMID:15249583; http://dx.doi.org/10.1083/jcb.200403069
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66:1500-8; PMID:16452206; http://dx.doi.org/10.1158/0008-5472.CAN-05-2925
- Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 2005; 65:7052-8; PMID:16103051; http://dx.doi.org/10.1158/0008-5472.CAN-05-0917
- Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58:621-81; PMID:16968952; http://dx.doi.org/10.1124/pr.58.3.10
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010; 70:440-6; PMID:20068163; http://dx.doi.org/10.1158/0008-5472.CAN-09-1947
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 2009; 10:307-18; PMID:19339977; http://dx.doi.org/10.1038/nrm2672
- Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol 2012; 22:274-82; PMID:22444729; http://dx.doi.org/10.1016/j.tcb.2012.02.006
- Leontieva OV, Demidenko ZN, Blagosklonny MV. Rapamycin reverses insulin resistance (IR) in high-glucose medium without causing IR in normoglycemic medium. Cell Death Dis 2014; 5:e1214; PMID:24810050; http://dx.doi.org/10.1038/cddis.2014.178
- Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptorinsulin receptor substrate-11487;phosphatidylinositol 3-kinase cascade. Mol Cancer Therapeut 2005; 4:1533-40; PMID:16227402; http://dx.doi.org/10.1158/1535-7163.MCT-05-0068
- Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007; 26:1932-40; PMID:17001314; http://dx.doi.org/10.1038/sj.onc.1209990
- Baumann P, Hagemeier H, Mandl-Weber S, Franke D, Schmidmaier R. Myeloma cell growth inhibition is augmented by synchronous inhibition of the insulin-like growth factor-1 receptor by NVP-AEW541 and inhibition of mammalian target of rapamycin by Rad001. Anti-Cancer Drugs 2009; 20:259-66; PMID:19240643; http://dx.doi.org/10.1097/CAD.0b013e328328d18b
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011; 19:58-71; PMID:21215704; http://dx.doi.org/10.1016/j.ccr.2010.10.031
- Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E, Baselga J, Guichard S, Rosen N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Disc 2011; 1:248-59; PMID:22140653; http://dx.doi.org/10.1158/2159-8290.CD-11-0085
- Li QL, Gu FM, Wang Z, Jiang JH, Yao LQ, Tan CJ, Huang XY, Ke AW, Dai Z, Fan J, et al. Activation of PI3KAKT and MAPK pathway through a PDGFRbeta-dependent feedback loop is involved in rapamycin resistance in hepatocellular carcinoma. PloS One 2012; 7:e33379; PMID:22428038; http://dx.doi.org/10.1371/journal.pone.0033379
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011; 332:1317-22; PMID:21659604; http://dx.doi.org/10.1126/science.1199498
- Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011; 332:1322-6; PMID:21659605; http://dx.doi.org/10.1126/science.1199484
- Rosich L, Montraveta A, Xargay-Torrent S, Lopez-Guerra M, Roldan J, Aymerich M, Salaverria I, Bea S, Campo E, Perez-Galan P, et al. Dual PI3KmTOR inhibition is required to effectively impair microenvironment survival signals in mantle cell lymphoma. Oncotarget 2014; 5:6788-800; PMID:25216518
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2:489-501; PMID:12094235; http://dx.doi.org/10.1038/nrc839
- Passacantilli I, Capurso G, Archibugi L, Calabretta S, Caldarola S, Loreni F, Delle Fave G, Sette C. Combined therapy with RAD001 e BEZ235 overcomes resistance of PET immortalized cell lines to mTOR inhibition. Oncotarget 2014; 5:5381-91; PMID:25026292
- Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle 2012; 11:4642-9; PMID:23187803; http://dx.doi.org/10.4161/cc.22937
- Wang H, Wu R, Yu L, Wu F, Li S, Zhao Y, Li H, Luo G, Wang J, Zhou J. SGEF is overexpressed in prostate cancer and contributes to prostate cancer progression. Oncol Rep 2012; 28:1468-74; PMID:22824926