
Abstract
MicroRNAs (miRNAs) play an important role in tumorigenesis, but their role in tumor-induced immune suppression is largely unknown. STAT3 signaling, a key pathway mediating immune suppression in the tumor microenvironment, is responsible for the transcription of several important miRNAs. In this study, we observed that miR-146a, a known important regulator of immune responses, was downregulated by blocking activated STAT3 in hepatocellular carcinoma (HCC) cells. Furthermore, miR-146a inhibition in HCC cells not only altered the STAT3 activation–associated cytokine profile but also reversed HCC-induced NK cell dysfunction in vitro and improved the anti-tumor effect of lymphocytes in vivo. Importantly, ChIP and luciferase reporter assays confirmed that STAT3 directly bound to the miR-146a promoter and induced miR-146a expression. These findings indicated that miR-146a expression was regulated by aberrantly activated STAT3 in HCC cells and exerted negative effects on anti-tumor immune response, which resulted in the upregulation of cytokines such as TGF-β, IL-17, VEGF and downregulation of type I IFN to create an immunosuppressive microenvironment. This further insight into understanding the mechanism responsible for tumor-induced immune suppression highlights the potential application of miR-146a as a novel immunotherapeutic target for HCC.
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide.Citation1 Although improvements have been made in surgery and other treatments for HCC, the prognosis for HCC patients remains unsatisfactory due to the high rate of recurrence and metastasis. Tumor-induced immune suppression is accepted as an important mechanism that protects the tumor from the induction of an efficient anti-tumor immune response in the host.Citation2 As an oncogenic transcription factor, signal transducer and activator of transcription 3 (STAT3) has been noted to contribute to tumor-induced immune suppression in multiple tumor types,Citation3 including HCC.Citation4 Constitutive activation of STAT3 not only negatively regulates Th1 cytokines critical for potent anti-tumor immune responsesCitation5 but also activates many genes involved in immune suppression. Moreover, studies revealed that STAT3 could drive tumor-derived factors, such as IL-6, IL-10, and VEGF, to ensure persistent STAT3 activation in the tumor microenvironment through crosstalk between tumor cells and tumor-associated immune cells.Citation3,6,7 Activated STAT3 by this “positive feedback loop” further promotes the expression of growth factors and angiogenic factors, which then represses the effects of the host anti-tumor responses and accelerates tumor growth and metastasis. Conversely, inhibiting STAT3 not only enhances autophagy,Citation8 suppress epithelial-mesenchymal transition (EMT) and tumor metastasis of HCC,Citation9 but also induces robust anti-tumor innate and adaptive immune responses that impede tumor progression.Citation3-5,7 Therefore, identifying the malignant factors and understanding the molecular pathogenesis underlying STAT3-mediated immune suppression are critical for improving strategies for HCC therapy.
Studies on STAT3 targets were previously limited to protein-coding genes. Recently, some non-coding STAT3 targets were identified in liver and colon cancer.Citation10-12 In addition to the genomic mutation events that trigger the activation of oncogenes or the inactivation of tumor-suppressor genes, the dysregulation of microRNAs (miRNAs)—-a type of short, non-coding RNA, is also a common epigenetic event in HCC development.Citation13 miRNAs are involved in the post-transcriptional regulation of genes, either by degrading target mRNAs or by inhibiting the translation process,Citation14,15 and they play a critical role in many biological processes, including tumorigenesis.Citation16 During the initiation and progression of many malignancies, miRNAs modulate cell proliferation, survival, angiogenesis, invasion, and metastasis.Citation17-19 Among these identified miRNAs, miR-146a has been shown to be an important player regulating tumor progression in addition to its known modulatory effects on adaptive and innate immune responses. The initial evidence on the abnormalities of miR-146a in cancer originated from a study showing that miR-146a was upregulated in papillary thyroid carcinoma (PTC) as compared to normal thyroid tissue.Citation20 Bhaumik et al.Citation21 found that over-expression of miR-146a/b significantly downregulated TNF receptor-associated factor 6 (TRAF6) and IL-1 receptor associated kinase 1 (IRAK1) in the highly metastatic breast cancer cell line MDA-MB-231, which led to the inactivation of NF-κB and impaired the metastatic potential of these tumor cells. These findings not only suggest that miR-146a acts as a negative regulator of constitutive NF-κB activity in breast cancer cells but also indicate that miR-146a may be an effective molecule for suppressing breast cancer metastasis. Moreover, studies in mice deficient in miR-146a show that the loss of miR-146a favors the development of myeloid and lymphoid neoplasia, strongly supporting a role for miR-146a as a tumor suppressor for myelo–lymphoid cells.Citation22 However, in cervical cancer tissues, miR-146a is upregulated compared to normal cervix, and this enhanced expression increases the proliferation of cervical cancer cells, indicating that miR-146a is closely related with promoting cervical carcinogenesis and thus plays an oncogenic role in cervical cancer.Citation23 And, miR-146a plays a key role in enhancing angiogenic activity of endothelial cells in HCC,Citation24 in addition to increase the resistance to IFN-αCitation25 and antitumor drugsCitation26 during HCC therapy. Therefore, miR-146a may play different roles in diverse kinds of tumor cells.
In this study, we evaluated the relationship between the aberrant activation of STAT3 and miR-146a dysregulation in HCC and explored the underlying molecular mechanisms. We found the level of miR-146a expression in HCC cells affected not only the STAT3 activation–associated cytokine profile but also the HCC-induced suppression of anti-tumor immune responses. Importantly, further exploration of the relationship between STAT3 and miR-146a revealed that STAT3 directly activated miR-146a transcription. These findings suggest that STAT3 transcriptional modulating executes its immune suppressive effects partly by regulating miR-146a expression in HCC cells.
Results
Blocking of STAT3 decreased the expression of miR-146a
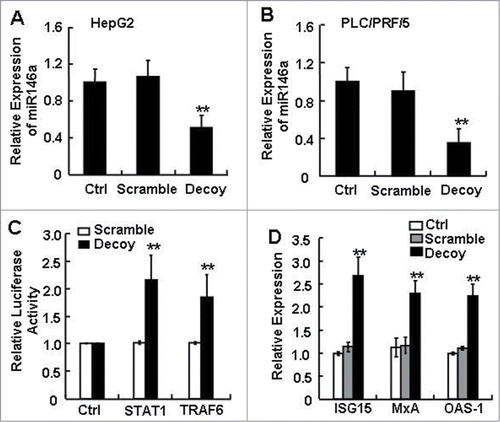
Increasing evidence supports that miR-146a contributes to the complex molecular mechanisms involved in controlling cell growth, differentiation, and survival as well as the processes related to cancer development and progression.Citation21-23 Interestingly, the function of STAT3, currently an attractive target for anticancer therapy, has also been linked to the survival, proliferation, angiogenesis, and immune suppression of HCC.Citation3,5-7 Since STAT3 was previously shown to regulate some miRNAs, we asked whether the constitutively activated STAT3 in HCC cells could be related to miR-146a expression, or if STAT3 had a potential modulatory effect on miR-146a expression. To address this question, we tested whether manipulating STAT3 using a Decoy ODN specifically targeting activated STAT3 would alter miR-146a expression in cultured HCC cells in vitro. As shown in , blocking STAT3 for 24 h suppressed miR-146a expression in both the HepG2 and PLC/PRF/5 HCC cell lines as compared to the Lipofectamine reagent control (Ctrl) or scramble ODN–treated HCC cells. Since miR-146a was known to target STAT1, and TRAF6 as well as to negatively regulate type I IFN signaling in SLE patients and miR-146a KO mice,Citation27,28 we next tested whether blocking STAT3 also influenced the expression of these miR-146a target genes in HCC cells. Blocking STAT3 significantly enhanced the activity of pmiR-Reporter vectors containing the 3′-UTR for STAT1 and TRAF6 mRNA, but not the control pmiR-Reporter empty vector (). Along with the STAT1 activation that occurred upon blocking STAT3, the mRNA levels of molecules downstream of the type I IFN pathway were also upregulated, including ISG15, MxA, and OAS-1 (). Collectively, these findings suggested that the expression of miR-146a and of molecules along miR-146a–targeted pathways were closely related to aberrant STAT3 activation in human HCC.
Figure 1. The expression level of miR-146a in human HCC cells was inhibited by blocking STAT3. After transfecting STAT3 decoy ODN (Decoy), scramble ODN (Scramble), or Lipofectamine reagent control (Ctrl) into HepG2 (A) and PLC/PRF/5 (B) cells for 24 h, miR-146a expression was measured by qPCR analysis. (C) Luciferase activity of pmiR-Reporter vector containing the mRNA 3′-UTR miR-146a target genes STAT1 and TRAF6 was detected using a dual-GloTM Luciferase assay system. (D) Levels of ISG15, MxA, and OAS-1 mRNA were analyzed by qPCR analysis. Data are representative of 3 independent experiments, and statistical significance was determined as **P < 0.01 and *P < 0.05 compared to Lipo-Ctrl.
miR-146a promoted the expression of STAT3 activation–associated cytokines in HCC cells
Dysregulation of many miRNAs, including miR-146a, favors oncogenesis and cancer progression.Citation20-23 To test whether miR-146a expression in HCC directly affected tumor growth by regulating cell proliferation, we modulated miR-146a expression in HepG2 cells using miR-146a mimics or inhibitors and evaluated cell growth and proliferation. As shown in , while blocking STAT3 inhibited the growth of HepG2 cells, treating HepG2 cells with miR-146a mimics or inhibitors did not significantly alter HepG2 proliferation, which was then confirmed by evaluating cell cycle (). These results suggested that the observed effect of miR-146a on tumor cells were not caused by a direct effect of miR-146a on tumor cell proliferation.
Figure 2. miR-146a promoted the expression of inflammatory cytokines associated with STAT3 activation in HCC cells. As described in the Materials and Methods section, HepG2 cells were transfected with negative control RNA (NC), miR-146a mimics (miR146a-Mim), miR-146a inhibitors (miR146a-Inh), or STAT3 decoy ODN (STAT3-Dec). (A) HepG2 proliferation was analyzed by MTT assay at the indicated time points. (B) Cell cycle was determined by flow cytometry. The levels of inflammatory cytokines associated with STAT3 activation were determined by qPCR (C) and ELISA (D) analysis. Data are representative of 3 independent experiments, and statistical significance was determined as **P < 0.01 and *P < 0.05 compared to NC.
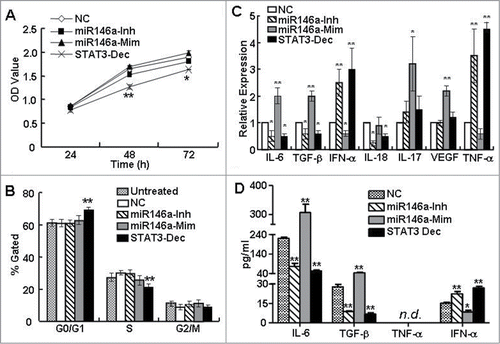
In the tumor microenvironment, aberrant STAT3 activation can suppress immune surveillance mechanisms by driving the production of tumor-derived proinflammatory and immunosuppressive cytokines.Citation3,29-31 Since various miRNAs are now considered to represent a new class of inflammatory mediators,Citation32,33 we investigated whether miR-146a indirectly regulated tumor growth by influencing the expression of cytokines important for immune surveillance of tumor growth. As shown in , inhibition of miR-146a using a specific inhibitor downregulated the mRNA expression of cytokines associated with STAT3 activation, such as the inflammatory cytokines IL-6 and IL-17 as well as the immunosuppressive factor TGF-β, but upregulated mRNA expression of the potent immune stimulator IFN-α. On the contrary, miR-146a overexpression using miR-146a mimics increased IL-6, IL-17, and TGF-β mRNA expression, but reduced IFN-α. We then confirmed that these changes also occurred at the protein level by ELISA analysis of the supernatant (). Since the changes in cytokine expression that occurred upon inhibiting or overexpressing miR-146a in HCC cells phenocopied the effects of blocking or activating STAT3, respectively, these results indicated that miR-146a expression might be downstream of STAT3 activation and be involved in creating a tumor microenvironment that further supported HCC progression.
STAT3 directly regulated miR-146a expression in HCC
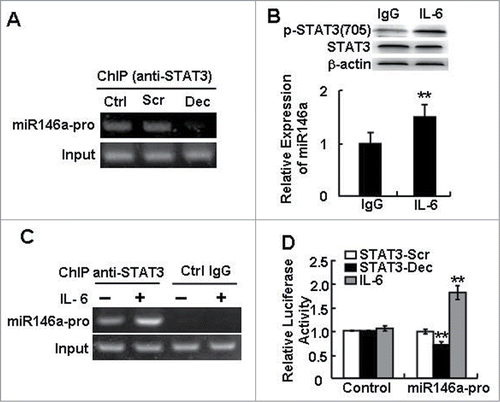
Based on the observations above, blocking STAT3 in HCC cells decreased miR-146a expression, and the effect of inhibiting miR-146a activity in HCC cells was similar to that of treating HCC cells with STAT3 decoy ODN. And the previous experiments showed the promoters of microRNAs contained STAT3 binding site, such as miR-17–92,12 miR-2110 and miR-23a.Citation11 We therefore tested whether STAT3 had a direct relationship to miR-146a expression. By ChIP assays using an antibody against p-STAT3705, we found that STAT3 directly bound to the miR-146a promoter and that blocking STAT3 could decrease the interaction between STAT3 and the miR-146a promoter (). By activating STAT3 with IL-6-a known inducer of STAT3 signaling for 24 h, the increase in phosphorylated STAT3 levels was accompanied by elevated miR-146a expression () and enhanced STAT3 binding to the miR-146a promoter (). Meanwhile, using a luciferase-based assay, we found that IL-6 stimulation increased the luciferase activity of the miR-146a promoter but that blocking STAT3 reduced this luciferase activity, confirming that STAT3 regulated the miR-146a promoter (). No chromatin enrichment by the STAT3 ChIP was observed in the negative control (IgG), verifying the specificity of the ChIP assay. Thus, these results demonstrated that STAT3 directly modulated miR-146a expression.
Figure 3. STAT3 directly regulated miR-146a expression in HCC. (A) A ChIP assay was performed 24 h after STAT3 decoy ODN (Dec) or scramble ODN (Scr) treatment to evaluate the recruitment of STAT3 on miR-146a promoter (miR146a-pro). (B) The level of phosphorylated STAT3 (p-STAT3) (Tyr705) in IL-6–stimulated (400 U/mL) HepG2 cells was examined by western blotting (upper), and a qPCR assay was used to detect the expression of miR-146a in HepG2 cells (lower). (C) After IL-6 stimulation, a ChIP-PCR assay was performed using an anti–p-STAT3705 antibody or rabbit IgG as a control. (D) The luciferase activity of the miR-146a promoter (miR146a-pro) in HepG2 cells was measured using a dual-GloTM Luciferase assay system. The ratio of firefly to Renilla luciferase activity with pGL3-TK-Luciferase transfection was set as 1. Data are representative of 3 independent experiments, and statistical significance was determined as **P < 0.01 and *P < 0.05 compared to control.
miR-146a contributed to human HCC-induced immune suppression in vitro
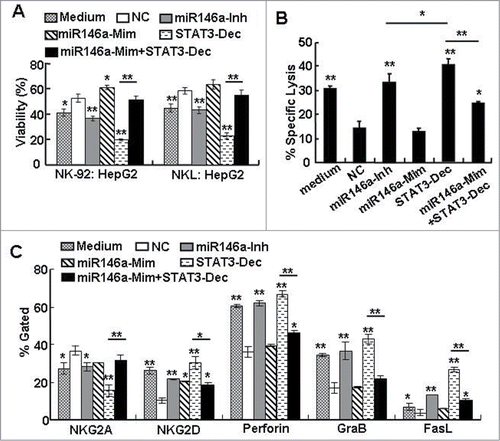
Tumor cells can escape from immune surveillance mechanisms by reprogramming immune cell functions through secreting cytokines such as IL-4, IL-10, and TGF-β.Citation34 One of the immune cells participating in immunosurveillance is the NK cell, which exerts its anti-tumor effects via cytotoxic and immune regulatory capacities and is critical in the induction of an effective adaptive anti-tumor immune response. And, previously we found targeting blockage of over activated-STAT3 in HCC can enhance NK cell cytotoxicity in vitro.Citation4 Therefore, in order to investigate whether the soluble mediators released as a result of miR-146a expression in HCC cells contributed to the escape of HCC from immune surveillance mechanisms, we performed an in vitro experiment, in which we incubated human NK cell lines (NK-92 or NKL) with supernatant from miR-146a mimic– or inhibitor–treated HepG2 cells for 12 h, and then examined the anti-tumor cytotoxic function of these NK cells on naïve HCC cells in a subsequent co-culture. The “Medium” group represents NK cells cultured in α-MEM alone without any supernatant from HCC cells. Compared to NK cells incubated with supernatant from HepG2 cells treated with the negative control (NC) RNA, treatment with supernatant from miR-146a inhibitor–treated HepG2 cells enhanced the NK cell–mediated cytotoxic anti-HCC effect by 14.82%–16.31%; conversely, treatment of NK cells with supernatant from miR-146a mimic–treated HepG2 cells increased the viability of HepG2 cells in co-culture by 9.57%–12.36% (). Moreover, we found that co-transfection of miR-146a mimics with STAT3 decoy ODN in HCC cells could restore the ability of the STAT3 decoy ODN–treated HCC cell supernatant to inhibit NK cell–mediated cytolysis (). Similar results were observed in NK-92 cell–mediated specific cell lysis against HCC cells by a CFSE/7-AAD flow cytometry assay () as well as in the levels of NK cytolysis–related molecules (NKG2A, NKG2D, FasL, perforin, granzyme B) in NK cells incubated with the various HCC cell supernatants (). As shown in , although the cytolytic ability of NK cells incubated with supernatant from miR-146a inhibitor–treated HepG2 cells was augmented compared to NK cells incubated with NC control supernatant, it was still lower than that of NK cells incubated with supernatant from STAT3 decoy ODN–treated HepG2 cells, indicating that other molecules downstream of STAT3 activation, but independent of miR-146a activity, were involved in HCC-induced immune suppression. Taken together, these findings suggested that miR-146a was involved in HCC-induced immune suppression, which was associated with STAT3 over-activation in HCC.
Figure 4. miR-146a contributed to human HCC–induced immune suppression in vitro. After HepG2 cells were transfected with negative control RNA (NC), miR-146a mimics (miR146a-Mim), miR-146a inhibitors (miR146a-Inh), or STAT3 decoy ODN (STAT3-Dec) as described in the Materials and Methods section, supernatant was collected and incubated with NK-92 or NKL cells in an in vitro culture for 12 h. (A) The inhibitory effect of these NK cells on the viability of naïve HepG2 cells was then analyzed by an MTT assay at an E:T ratio of 5:1. (B) The specific lysis of HepG2 cells by these NK-92 cells was detected using a 4-h CFSE/7-AAD flow cytometry assay at an E:T ratio of 5:1. (C) The molecules associated with NK cell cytolysis were examined by flow cytometry. The data in the histograms represent the statistical analysis of the percentage of positive cells. Medium, NK cells cultured in α-MEM alone without any supernatant from HCC cells. Data are representative of 3 independent experiments, and statistical significance was determined as **P < 0.01 and *P < 0.05 compared to NC.
Inhibiting miR-146a promoted the anti-tumor immune response in vivo
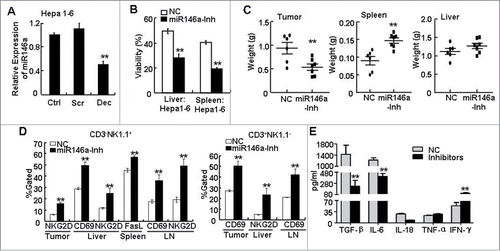
Based on our observations that miR-146a expression in HCC cells inhibited anti-tumor cytolytic NK cell function in vitro, we predicted that miR-146a expression regulated by STAT3 activation in HCC cells promoted tumor growth in vivo. To test this, a homograft mouse model was used to address whether miR-146a influenced the anti-tumor immune responses in vivo. First, we verified that blocking STAT3 for 36 h also downregulated miR-146a in Hepa 1–6 cells, a murine liver cancer cell line (). Next, 2 × 106 Hepa 1–6 cells transfected with miR-146a inhibitor or NC RNA were subcutaneously (s.c.) injected into the right posterior flank of C57BL/6 mice. Two weeks later, we found that liver and spleen lymphocytes from mice bearing miR-146a–inhibited tumor cells exhibited an enhanced anti-HCC effect that was at least 20% higher than that from NC-treated Hepa 1–6–bearing mice (). Meanwhile, mice bearing miR-146a inhibitor–treated Hepa 1–6 cells displayed a decreased tumor burden and increased spleen weight compared to mice bearing NC-treated Hepa 1–6 cells (). By flow cytometry analysis, we found that the proportion of NK and T cells in tumor samples derived from miR-146a-inhibiting group was 5.374 ± 0.459% and 22.134 ± 3.775%, respectively, which was significantly higher than NC-treated group (NK cells: 2.02 ± 0.622%; T cells: 5.78 ± 1.046%). Moreover, expression of the cytolysis molecules CD69, NKG2D, and FasL in CD3−NK1.1+ (, left) or CD3+NK1.1− (, right) lymphocytes from liver, spleen, tumor tissue, and axillary and inguinal LNs were upregulated in mice bearing miR-146a–inhibited tumor cells as assessed by flow cytometry, which was not only consistent with the enhanced lymphocyte cytolysis activity we observed in vitro but also further confirmed the improved anti-tumor function of host immune system. Additionally, ELISA analysis of cytokines in the serum showed that the immune system status was improved in mice bearing miR-146a–inhibited tumor cells, including decreased TGF-β, IL-6, and IL-18 as well as increased IFN-γ (). Collectively, these in vitro and in vivo data substantiated the importance of STAT3-induced miR-146a expression as a negative regulatory factor suppressing the anti-tumor immune response in both human HCC and murine models of HCC.
Figure 5. Inhibition of miR-146a promoted the anti-tumor immune response in vivo. (A) After transfecting STAT3 decoy ODN (Dec), scramble ODN (Scr), or Lipofectamine reagent control (Ctrl) into the murine liver cancer cell line Hepa 1–6 for 24 h, miR-146a expression was measured by qPCR analysis. (B–E) Hepa 1–6 cells transfected with miR-146a inhibitors (miR146a-Inh) or control RNA (NC) were injected s.c. into the right posterior flank of C57BL/6 mice (2 × 106 cells/mouse), and tumor-bearing mice were sacrificed after 2 weeks. (B) The inhibitory effect of freshly isolated lymphocytes from liver or spleen on the viability of naïve Hepa 1–6 cells was analyzed by MTT assay at an E:T ratio of 50:1. (C) The weight of tumor, spleen, and liver in the indicated tumor-bearing groups was measured. (D) Flow cytometry analysis was performed to examine CD69, CD25, NKG2D, and FasL levels in CD3−NK1.1+ and CD3+NK1.1− lymphocytes from tumor tissue, spleen, liver, and axillary and inguinal LNs. (E) TGF-β, IL-6, IL-18, TNF-α, and IFN-γ levels in serum were detected by ELISA assay. Data are representative of 3 independent experiments, and statistical significance was determined as **P < 0.01 and *P < 0.05 compared to control.
Discussion
As a key modulator of differentiation, miR-146a is dysregulated in various types of tumors.Citation20,21,23,35 Since previous studies indicated that STAT3 and miR-146a regulated similar processes, we attempted in this study to determine whether the constitutively activated STAT3 in HCC cells influenced miR-146a expression, and then we went on to explore whether miR-146a contributes to the process of HCC-induced immune suppression. We found that blocking STAT3 downregulated miR-146 expression in liver cancer (), which subsequently allowed for the upregulation of the known miR-146a targets, STAT1 and TRAF6, and their downstram signaling pathways ().Citation27,28,36 These results suggested that a relationship might exist between STAT3 activation and miR-146a expression, and that miR-146a might be involved in the biological properties of HCC.
In terms of whether miR-146a expression influenced the growth of HCC tumors, we initially ruled out the direct mechanism of miR-146a promoting HCC cell proliferation, as HCC cell proliferation was not affected by the introduction of a miR-146a inhibitor or mimic (). We then focused on testing ways in which miR-146a could indirectly influence HCC growth. A recent study demonstrated that miR-146a also acted as a modulator of inflammation via Toll-like and interleukin-1 receptor (TIR) signaling downstream of its effect on regulating IRAK1 and TRAF6.Citation36 Indeed, when we tested whether miR-146a could regulate inflammatory cytokine production in HCC cells, miR-146a inhibition downregulated inflammatory cytokines closely related with STAT3 activation in HCC cells, while the introduction of miR-146a mimics increased the levels of inflammatory cytokines, including IL-6 and IL-8, as well as the immunosuppressive factor TGF-β (); moreover, miR-146a expression negatively regulated IFN-α. Thus, these data revealed that miR-146a likely influenced the growth of HCC cells in an indirect way by contributing to HCC-induced immunosuppressive condition.
Since our data revealed that STAT3 was involved in regulating miR-146a expression, we further tested whether this regulation was via a direct or indirect mechanism. Similar to protein-coding genes, miRNA genes themselves are also subject to sophisticated regulation. The factors determining miRNA expression include genomic amplification, transcriptional regulation, processing, editing, and decay.Citation37 Among them, transcriptional regulation is a leading factor regulating miRNA expression; indeed, some reports have characterized miRNA transactivators or suppressors, such as c-MYC and p53.Citation38,39 Using a ChIP assay, we found that STAT3 directly bound to the miR-146a promoter in HCC cells and that blocking STAT3 obviously decreased this direct interaction (). Moreover, IL-6–induced activation of STAT3 increased miR-146a expression () and enhanced STAT3 binding to the miR-146a promoter () as well as the transcriptional activity of miR-146a promoter (), which was inhibited by blocking STAT3 with decoy ODN. Thus, our data reveal for the first time that STAT3 regulates miR-146a expression in HCC cells by a direct mechanism.
In the highly complex milieu of the tumor microenvironment, many proinflammatory and immunosuppressive cytokines are produced to suppress anti-tumor immune responses mediated by NK cells and/or cytotoxic T lymphocytes (CTLs). In testing whether inhibition of miR-146a could reverse HCC-induced immune suppression and lead to a robust NK cell–mediated anti-tumor response, which was observed as STAT3 in HCC was blocked by Decoy ODN 4, we found that incubating supernatant from miR-146a inhibitor–treated HepG2 cells enhanced NK cell cytolysis; increased the expression of the NK activation–associated molecules perforin, granzymes, IFNs, NKG2D, and CD69; and decreased inhibitory receptor (NKG2A) expression (). Thus, the similar anti-HCC effect observed after either STAT3 blockage or miR-146a inhibition provided further evidence to support a functional relationship between STAT3 and miR-146a. However, our data also suggested the possibility that other STAT3-regulated molecules independent of miR-146a were involved in HCC-induced immune suppression, as the cytolysis observed after miR-146a inhibition was still lower than that of NK cells incubated with supernatant from STAT3 decoy ODN–treated HepG2 cells. It would be important and interesting to identify these other STAT3-regulated mediators in future studies.
As miR-146a had previously been shown to play a significant role in tumor progression in both breast and cervical cancers,Citation21,23 we wondered whether miR-146a also played a role in hepatocarcinogenesis. We found that although altering miR-146a expression by transfecting miR-146a mimics or inhibitors into HepG2 cells had no effect on proliferation in the in vitro setting (), inhibiting miR-146 in the murine liver cancer cell line Hepa 1–6 decreased the growth of the homografted Hepa 1–6 tumors in vivo (). Importantly, lymphocytes harvested from mice bearing miR-146a inhibitor–treated Hepa 1–6 tumors showed an enhanced anti-tumor response, and cytolysis-related molecules were upregulated on NK and T cells (), recapitulating our in vitro observations on NK cells. Moreover, the immune system status was improved in mice bearing miR-146a inhibitor–treated Hepa 1–6 cells (). These findings thus confirmed that miR-146a exerted an important role in liver tumorigenicity and might be a potentially valuable molecular target for HCC immunotherapy.
In a recently published study, Rong et al. shows that miR-146a expression in HCC tissues is lower than that in adjacent non-cancerous hepatic tissues,Citation40 which is seemingly contradictory to what we would predict based on the data presented here. However, since HCC tissues also contain many infiltrating immune cells in which miR-146a is abundantly expressed, including T cells and regulatory T cells (Tregs),Citation41,42 we consider that the observed miR-146a levels in whole HCC tissues cannot accurately reflect the miR-146a expression in the HCC cells alone due to the potential contribution of these infiltrating immune cells.
The target of miR-146a responsible for mediating HCC-induced immunosuppression has still to be clarified in the future. However, the accumulated studies have revealed that miR-146a can induce the resistance to type I IFN by targeting STAT1,Citation25,27 and downregulate type I IFN expression by targeting IRAK1 and TRAF6.Citation43 Here, we also observed that STAT3 blockage upregulated STAT1 and TRAF6, as well as the downstream genes of type I IFN pathway. And type I IFN, which is critical for NK cell activation by blocking STAT3 in HCC cells,Citation4 could be increased by miR-146a inhibition accompanied with reversing HCC-mediated inhibitory effects on NK cells. These findings indicated that type I IFN signaling pathway might be a vital target for miR-146a-mediated HCC-induced immunosuppression. Furthermore, other molecules such as SMAD4 is reported to be one target of miR-146a, which can inhibit cell proliferation, promote apoptosis of gastric cancer,Citation44 and improve the sensitivity of HCC cells to the cytotoxic effects of IFN-α,25 suggesting miR-146a downregulation may contribute to the therapy of gastric cancer and HCC.
In summary, our results demonstrated that STAT3 in HCC cells could directly modulate miR-146a expression. In HCC cells, constitutively activated STAT3 not only resulted in the overproduction of inflammatory and immunosuppressive cytokines but also regulated the expression of miR-146a miRNA. Thus, STAT3-regulated miRNAs might act as mediators of STAT3-induced immunosuppression and promote the progression of liver cancer, and anti-tumor efficiency could likely be improved by interfering with the expression of these miRNAs, such as using nanoparticles delivery miR-146a inhibitors or miR-146a-sponge vector. These findings provide important insights into an immune suppression mechanism underlying hepatocarcinogenesis and shed new light on potential targeting strategies for HCC immunotherapy.
Materials and Methods
Cell lines
The human HCC cell lines HepG2 (The Cell Bank of Type Culture Collection of Chinese Academy of Sciences, TCHu 72) and PLC/PRF/5 (kindly supplied by Dr. Qu Xianjun, Department of Pharmacology, Shandong University) were grown in RPMI-1640 medium (GIBCO/BRL, Grand Island, NY, USA) with 10% FBS. The human natural killer (NK) cell line NK-92 was purchased from ATCC (CRL-2407) and maintained in α-MEM (GIBCO/BRL) supplemented with 12.5% horse serum (GIBCO), 12.5% FBS, 100 U/mL rhIL-2 (Changsheng, Changchun, China), 0.1 mM β-mercaptoethanol, and 0.02 mM folic acid. The human NK cell line NKL was generously provided by Dr. Jin Boquan (Fourth Military Medical University, China) and cultured in RPMI-1640 containing 10% FBS and 100 U/mL rhIL-2. The murine HCC cell line Hepa 1–6 was purchased from Chinese Academy of Sciences Typical Culture Collection cell bank (TCM39) and cultured in DMEM (GIBCO/BRL) with 10% FBS. The above cell lines were used within 6 months after receipt. Cells were never used above passage 10 and were cultured in a humidified incubator with 5% CO2 at 37°C.
STAT3 decoy/scramble oligonucleotide and RNA oligonucleotide
Using phosphorothioate chemistry, sense and antisense strands of STAT3 decoy or scramble oligonucleotide (ODN) were synthesized using the ExpediteTM Nucleic Acid Synthesis System (Takara Biotechnology). The STAT3 decoy ODN sequences were 5′-CATTTCCCGTAAATC-3′ and 5′-GATTTACGGGAAATG-3′, and the scramble ODN sequences were 5′-CATCTTGCCAATATC-3′ and 5′-GATATTGGCAAGATG-3′. The sense and antisense strands were annealed and purified by HPLC.Citation45 The specificity of STAT3 decoy ODN targeting STAT3 was confirmed previously.Citation4
The miR-146a mimics and matching negative control RNAs (dsRNA ODNs) as well as the miR-146a inhibitors and matching negative control RNAs (single-stranded chemically modified ODNs) were all purchased from Shanghai GenePharma Co., Ltd..
Cell transfection
Transient transfection was carried out with LipofectamineTM 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. One day before transfection, HCC cells were seeded to ensure >90 % confluency. For transfection of DNA oligonucleotides, 50 nM STAT3 decoy or scramble ODN were used with an ODN (μg) to LipofectamineTM 2000 (μL) ratio of 1:2.5. For transfection of RNA oligonucleotides, 50 nM miRNA mimics or 20 nM miRNA inhibitors was used with an RNA (nM) to LipofectamineTM 2000 (μL) ratio of 1:1. After ∼4–6 h of transfection, supernatant was removed and washed twice with 1 × PBS, and fresh medium containing serum was incubated with the cells for the indicated time periods.
NK cells treated with supernatant from tumor cells
After HCC cells were transfected with DNA oligonucleotides or RNA oligonucleotides for 6 h, the transfection medium was removed, and HCC cells were washed with 1× PBS to remove residual ODN. These HCC cells were then cultured in fresh medium (containing 10% FBS) for an additional 24 h. Supernatants were collected, and cells and debris were removed by centrifugation. NK cells were cultured in the supernatant for 12 h with a 1:1 ratio of supernatant to medium (v/v).
Cell proliferation and viability assay
For the cell proliferation assay, HepG2 cells were transfected with miR-146a mimics (50 nM), miR-146a inhibitors (20 nM), or control RNA. After 4 h, these treated HepG2 cells (6 × 103 cells/well) were seeded in 96-well plates. At the indicated time points, 20 μL (5 mg/mL) MTT (Sigma) was added to each well and incubated for another 4 h. After centrifugation, the MTT solution was removed, and 200 μL dimethyl sulfoxide (DMSO; Sigma) was added to dissolve the formazan crystals. Then, absorbance at 570–630 nm was determined using a Microplate Autoreader (Bio-Rad). For the cell viability assay, liver cancer cells were plated at density of 1 × 104 cells/well in 96-well plates. After 4 h, NK-92, NKL, or mouse lymphocytes were added into the plates at different E:T ratios. At the 12-h time point, cell viability was examined using the MTT method.
Cell cytotoxicity assay
Cell cytotoxicity against HepG2 cells was evaluated by flow cytometry. After labeling HCC cells with 5–6-carboxyfluorescein diacetate succinimidyl ester (CFSE, #C0051, Beyotime) for 15 min at 37°C, cells were washed and seeded in complete medium for adherent culture. Then, NK-92 cells were added with an E:T ratio of 5:1. HCC cells were incubated alone to measure basal levels of cell death. After 4 h, cells were collected, washed twice with 1 × PBS, and incubated with 7-amino actinomycin D (7-AAD, #KGA219, KeyGEN BioTECH) for 15 min at room temperature in the dark. Fluorescence data were acquired using a FACSCalibur flow cytometry system (BD Biosciences). The following formula was used to calculate Specific Lysis:% Specific Lysis = (Sample Ratio – Basal Ratio) × 100, where Ratio = % CFSE+7-AAD+ /% CFSE+.
Flow cytometry
Cell phenotypes were analyzed by flow cytometry. Cells were incubated with fluorescence-conjugated antibodies for 30 min at 4°C. For detection of intracellular cytokines, cells were stimulated with monensin (6 μM) and ionomycin (1 μg/mL) (Sigma) for 4 h in a 37°C, 5% CO2 incubator, and then washed, fixed, and permeabilized. Cells were then stained with a saturating amount of the fluorescence-conjugated antibodies for 1 h at 4°C. After washing with PBS, fluorescence from the stained cells was acquired using a FACSCalibur (BD Biosciences) and analyzed using WinMDI 2.0 software. The fluorescence-conjugated antibodies used in this study are described in Table S1.
For the cell cycle assay, at least 1 × 106 cells treated with the indicated treatments were harvested and washed in PBS. After fixation in 90% ethanol for 1 h at −20°C, cells were washed twice and resuspended in RNase solution (50 μg/mL) for 30 min at 37°C. Then, cells were stained with propidium iodide (PI) at room temperature for 15 min. Fluorescence was quantified on a flow cytometer, and the percentage of cells in each phase was calculated using ModFit software (BD Biosciences).
RNA isolation and quantitative real-time PCR (qPCR)
Quantitative real-time PCR (qPCR) was performed according to the manufacturer's instructions. Briefly, total RNA was isolated using TRIzol (Invitrogen). RNA quality and concentration were determined by spectrophotometric measurement of the A260/A280 ratio. cDNA was synthesized using the M-MLV reverse transcriptase (Invitrogen). qPCR was performed using the SYBR green real-time PCR Kit (TOYOBO) on an iQ5TM Real-Time PCR detection system (Bio-Rad). Relative mRNA expression levels of the gene of interest were calculated using the 2−ΔΔCt method. The primers are listed in Table S2. miRNA levels were quantified by qRT-PCR using specific Bulge-Loop™ miRNA qRT-PCR primers purchased from Guangzhou Ribobio with U6 small nuclear RNA as an internal normalized reference.
Western blotting
HepG2 cells were homogenized in a total protein extraction kit (BestBio) containing a dissolved protease inhibitor cocktail (Sigma) for 30 min in ice-cold lysis buffer, and the homogenate was centrifuged at 13,000 × g at 4°C for 15 min. Supernatants were mixed in Laemmli loading buffer, boiled for 5 min, and then subjected to SDS-PAGE. After transferring to PVDF membranes (Millipore), immunoblots were incubated with anti–p-STAT3, anti-STAT3 (Cell Signaling Technology), or anti–β-actin (Santa Cruz) mAbs, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies and visualization by Immobilon Western Chemiluminescent HRP Substrate (Millipore).
Chromatin Immunoprecipitation
Cells were seeded in 150-mm plates at a density of 2 × 106 cells/well. After a 24-h transfection, 1% formaldehyde was added into the plates for 15 min at room temperature. The Chromatin Immunoprecipitation (ChIP) assay was carried out with a ChIP Assay Kit (Millipore, #17–295) according to the manufacturer's instructions. Briefly, ChIP dilution buffer (containing a protease inhibitor) was added to the cell pellet to isolate chromatin, and the lysates were sonicated to shear DNA to an average length of 200–500 bp. The input was prepared by treating 5% of total chromatin with proteinase K, heating the samples at 65°C for 6 h to remove crosslinking, and precipitating out the pellet with ethanol. The final lysate was used for IP with anti-STAT3 (Cell Signaling Technology) or normal serum IgG. After overnight incubation with primary antibody at 4°C, protein A agarose beads were added to the mixture for isolating immune complexes. The complexes were washed and eluted from the beads with the elution buffer included in the ChIP Assay Kit. Crosslinks were reversed by incubating with 5M NaCl at 65°C, and ChIP DNA was purified by phenol–chloroform extraction and ethanol precipitation. Finally, the concentration of extracted DNA was determined on a NanoDrop spectrophotometer and analyzed by PCR using primers designed around the specific binding sites on the miR-146a promoter (miR146a-pro, 5′-GCTCACTGCAACCTCCAA-3′ and 5′-TGTCCCAGCCCT-GTAAAA-3′). The PCR products were electrophoresed on 2% agarose gels, and the relative light intensities of the bands were analyzed by AlphaEaseFC software. The resulting signals were normalized as (Amount of DNA in antibody-specific IP − DNA in IgG IP) / (Input DNA).
Luciferase reporter gene assay
For the reporter gene assay, HepG2 cells were plated at a density of 1 × 104 cells/well in 96-well plates (Costar) and transiently transfected with pGL3-STAT1-Promoter luciferase, pGL3-TRAF6-Promoter luciferase, or pGL3-miR146a-Promoter luciferase (200 ng/well, Cat# 15091, Addgene) as well as STAT3 decoy or scramble ODN in the presence of LipofectamineTM 2000. The Renilla expression vector pRL-TK (20 ng/well, Promega) was co-transfected into the cells to normalize the transfection efficiency. After 24 h, cells were washed and lysed, and a dual-GloTM Luciferase assay system (Promega, #E2920) was used to determine luciferase activity according to the manufacturer's instructions. The ratio of firefly to Renilla luciferase activity associated with pGL3-TK luciferase transfection was set as 1.
Homograft transplantation and in vivo studies
Hepa 1–6 cells (2 × 106 cells/mouse) treated with miR-146a inhibitors or control RNA were injected into the right posterior flank of 6-week-old male C57BL/6 mice, and these tumor-bearing mice were sacrificed 2 weeks later. Tumor, liver, and spleen weight were measured, and the lymphocytes from liver, spleen, tumor tissue, and axillary and inguinal lymph nodes (LNs) were isolated. All procedures were performed in accordance with Institutional Animal Care and Use Committee Protocols.
Isolation of mouse lymphocytes
Mouse liver, spleen, tumor tissue, and axillary and inguinal LNs were removed and pressed separately through a 200-gauge stainless steel mesh, and the cell suspension was collected. For liver lymphocyte isolation, cells were briefly centrifuged at 50 × g for 1 min, resuspended in 40% Percoll (GE Healthcare, Piscataway), gently overlaid on 70% Percoll, and centrifuged at 1260 × g for 30 min at 4°C. The lymphocyte cells at the interface between the Percoll solutions were aspirated and washed twice in PBS. For splenic and intratumoral lymphocyte isolation, the cell suspension was centrifuged at 890 × g for 10 min, and then the cells were subjected to red blood cell (RBC) lysis before being washed with PBS. To isolate lymphocytes from the axillary and inguinal LNs, cells were washed in PBS.
Enzyme-linked immunosorbent assay (ELISA)
IL-6, IL-18, TNF-α, TGF-β (MultiSciences Biotech Co., Ltd), IFN-α (PBL Interferon Source,), and IFN-γ (eBioscience) levels in cell culture supernatant and mouse serum were determined using ELISA kits according to each manufacturer's instructions.
Statistical analysis
All data are presented as the mean ± standard deviation (SD) of 3 or more independent experiments. Statistical analysis was performed using a paired Student's t-test. P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
977112_Supplementary_Materials.zip
Download Zip (4.8 KB)Funding
This study was supported by grants from National Basic Research Program of China (No. 2013CB531503), National Natural Science Foundation of China (No. 81172789,81373222) and National Mega Project on Major Infectious Diseases Prevention and Treatment (No. 2012ZX10002006).
References
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA: a cancer journal for clinicians 2010; 60:277-300; PMID:20610543; http://dx.doi.org/10.1017/S000983880999067X
- Lasaro MO, Ertl HC. Targeting inhibitory pathways in cancer immunotherapy. Curr Opin Immunol 2010; 22:385-90; PMID:20466529; http://dx.doi.org/10.1016/j.coi.2010.04.005
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9:798-809; PMID:19851315; http://dx.doi.org/10.1038/nrc2734
- Sun X, Sui Q, Zhang C, Tian Z, Zhang J. Targeting blockage of STAT3 in hepatocellular carcinoma cells augments NK cell functions via reverse hepatocellular carcinoma-induced immune suppression. Mol Cancer Ther 2013; 12:2885-96; PMID:24107450; http://dx.doi.org/10.1158/1535-7163.MCT-12-1087
- Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 2005; 11:1314-21; PMID:16288283; http://dx.doi.org/10.1038/nm1325
- Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med 2006; 203:1651-6; PMID:16801397; http://dx.doi.org/10.1084/jem.20051848
- Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 2004; 10:48-54; PMID:14702634; http://dx.doi.org/10.1038/nm976
- Su JC, Tseng PH, Hsu CY, Tai WT, Huang JW, Ko CH, Lin MW, Liu CY, Chen KF, Shiau CW. RFX1-dependent activation of SHP-1 induces autophagy by a novel obatoclax derivative in hepatocellular carcinoma cells. Oncotarget 2014; 5:4909-19; PMID:24952874
- Wang J, Yin D, Xie C, Zheng T, Liang Y, Hong X, Lu Z, Song X, Song R, Yang H, et al. The iron chelator Dp44mT inhibits hepatocellular carcinoma metastasis via N-Myc downstream-regulated gene 2 (NDRG2)/gp130/STAT3 pathway. Oncotarget 2014; 5(18): 8478–91; PMID: 25261367
- Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 2010; 39:493-506; PMID:20797623; http://dx.doi.org/10.1016/j.molcel.2010.07.023
- Wang B, Hsu SH, Frankel W, Ghoshal K, Jacob ST. Stat3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma, coactivator 1 α. Hepatology 2012; 56:186-97; PMID:22318941; http://dx.doi.org/10.1002/hep.25632
- Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circul Res 2009; 104:1184-91; PMID:19390056; http://dx.doi.org/10.1161/CIRCRESAHA.109.197491
- Braconi C, Henry JC, Kogure T, Schmittgen T, Patel T. The role of microRNAs in human liver cancers. Semin Oncol 2011; 38:752-63; PMID:22082761; http://dx.doi.org/10.1053/j.seminoncol.2011.08.001
- Ambros V. The functions of animal microRNAs. Nature 2004; 431:350-5; PMID:15372042; http://dx.doi.org/10.1038/nature02871
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281-97; PMID:14744438; http://dx.doi.org/10.1016/S0092-8674(04)00045-5
- Yue J, Tigyi G. MicroRNA trafficking and human cancer. Cancer Biol Ther 2006; 5:573-8; PMID:16760647; http://dx.doi.org/10.4161/cbt.5.6.2872
- Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer 2006; 6:259-69; PMID:16557279; http://dx.doi.org/10.1038/nrc1840
- Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. New Eng J Med 2005; 353:1793-801; PMID:16251535; http://dx.doi.org/10.1056/NEJMoa050995
- Gregory RI, Shiekhattar R. MicroRNA biogenesis and cancer. Cancer Res 2005; 65:3509-12; PMID:15867338; http://dx.doi.org/10.1158/0008-5472.CAN-05-0298
- He H, Jazdzewski K, Li W, Liyanarachchi S, Nagy R, Volinia S, Calin GA, Liu CG, Franssila K, Suster S, et al. The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci U S A 2005; 102:19075-80; PMID:16365291; http://dx.doi.org/10.1073/pnas.0509603102
- Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene 2008; 27:5643-7; PMID:18504431; http://dx.doi.org/10.1038/onc.2008.171
- Zhao JL, Rao DS, Boldin MP, Taganov KD, O’Connell RM, Baltimore D. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci U S A 2011; 108:9184-9; PMID:21576471; http://dx.doi.org/10.1073/pnas.1105398108
- Wang X, Tang S, Le SY, Lu R, Rader JS, Meyers C, Zheng ZM. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PloS One 2008; 3:e2557; PMID:18596939; http://dx.doi.org/10.1371/journal.pone.0002557
- Zhu K, Pan Q, Zhang X, Kong LQ, Fan J, Dai Z, Wang L, Yang XR, Hu J, Wan JL, et al. MiR-146a enhances angiogenic activity of endothelial cells in hepatocellular carcinoma by promoting PDGFRA expression. Carcinogenesis 2013; 34:2071-9; PMID:23671131; http://dx.doi.org/10.1093/carcin/bgt160
- Tomokuni A, Eguchi H, Tomimaru Y, Wada H, Kawamoto K, Kobayashi S, Marubashi S, Tanemura M, Nagano H, Mori M, et al. miR-146a suppresses the sensitivity to interferon-α in hepatocellular carcinoma cells. Biochem Biophys Res Commun 2011; 414:675-80; PMID:21982769; http://dx.doi.org/10.1016/j.bbrc.2011.09.124
- Zhuo L, Liu J, Wang B, Gao M, Huang A. Differential miRNA expression profiles in hepatocellular carcinoma cells and drug-resistant sublines. Oncol Rep 2013; 29:555-62; PMID:23229111
- Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, Huang X, Zhou H, de Vries N, Tak PP, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis and Rheumatism 2009; 60:1065-75; PMID:19333922; http://dx.doi.org/10.1002/art.24436
- Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, Garcia-Flores Y, Luong M, Devrekanli A, Xu J, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med 2011; 208:1189-201; PMID:21555486; http://dx.doi.org/10.1084/jem.20101823
- Leonardi GC, Candido S, Cervello M, Nicolosi D, Raiti F, Travali S, Spandidos DA, Libra M. The tumor microenvironment in hepatocellular carcinoma (review). Int J Oncol 2012; 40:1733-47; PMID:22447316
- Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, Biassoni R, Bottino C, Moretta L, Moretta A. Transforming growth factor β 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A 2003; 100:4120-5; PMID:12646700; http://dx.doi.org/10.1073/pnas.0730640100
- Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, Balsamo M, Conte R, Benelli R, Minghelli S, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res 2012; 72:1407-15; PMID:22258454; http://dx.doi.org/10.1158/0008-5472.CAN-11-2544
- Quinn SR, O’Neill LA. A trio of microRNAs that control Toll-like receptor signalling. Int Immunol 2011; 23:421-5; PMID:21652514; http://dx.doi.org/10.1093/intimm/dxr034
- Rusca N, Monticelli S. MiR-146a in Immunity and Disease. Mol Biol Int 2011; 2011:437301; PMID:22091404; http://dx.doi.org/10.4061/2011/437301
- Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 2013; 339:286-91; PMID:23329041; http://dx.doi.org/10.1126/science.1232227
- Lin SL, Chiang A, Chang D, Ying SY. Loss of mir-146a function in hormone-refractory prostate cancer. RNA 2008; 14:417-24; PMID:18174313; http://dx.doi.org/10.1261/rna.874808
- Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 2006; 103:12481-6; PMID:16885212; http://dx.doi.org/10.1073/pnas.0605298103
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 2010; 11:597-610; PMID:20661255
- He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. A microRNA component of the p53 tumour suppressor network. Nature 2007; 447:1130-4; PMID:17554337; http://dx.doi.org/10.1038/nature05939
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009; 458:762-5; PMID:19219026; http://dx.doi.org/10.1038/nature07823
- Rong M, He R, Dang Y, Chen G. Expression and clinicopathological significance of miR-146a in hepatocellular carcinoma tissues. Upsala J Med Sci 2014; 119:19-24; PMID:24172202; http://dx.doi.org/10.3109/03009734.2013.856970
- Curtale G, Citarella F, Carissimi C, Goldoni M, Carucci N, Fulci V, Franceschini D, Meloni F, Barnaba V, Macino G. An emerging player in the adaptive immune response: microRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood 2010; 115:265-73; PMID:19965651; http://dx.doi.org/10.1182/blood-2009-06-225987
- Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, Yoshimura A, Baltimore D, Rudensky AY. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 2010; 142:914-29; PMID:20850013; http://dx.doi.org/10.1016/j.cell.2010.08.012
- Li Y, Shi X. MicroRNAs in the regulation of TLR and RIG-I pathways. Cell Mol Immunol 2013; 10:65-71; PMID:23262976; http://dx.doi.org/10.1038/cmi.2012.55
- Xiao B, Zhu ED, Li N, Lu DS, Li W, Li BS, Zhao YL, Mao XH, Guo G, Yu PW, et al. Increased miR-146a in gastric cancer directly targets SMAD4 and is involved in modulating cell proliferation and apoptosis. Oncol Rep 2012; 27:559-66; PMID:22020746
- Leong PL, Andrews GA, Johnson DE, Dyer KF, Xi S, Mai JC, Robbins PD, Gadiparthi S, Burke NA, Watkins SF, et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc Natl Acad Sci U S A 2003; 100:4138-43; PMID:12640143; http://dx.doi.org/10.1073/pnas.0534764100