
Abstract
Long non-coding RNAs (ncRNA) have recently been demonstrated to be expressed from a subset of enhancers and to be required for the distant regulation of gene expression. Several approaches to predict enhancers have been developed based on various chromatin marks and occupancy of enhancer-binding proteins. Despite the rapid advances in the field, no consensus how to define tissue specific enhancers yet exists. Here, we identify 2,695 long ncRNAs annotated by ENCODE (corresponding to 28% of all ENCODE annotated long ncRNAs) that overlap tissue-specific enhancers. We use a recently developed algorithm to predict tissue-specific enhancers, PreSTIGE, that is based on the H3K4me1 mark and tissue specific expression of mRNAs. The expression of the long ncRNAs overlapping enhancers is significantly higher when the enhancer is predicted as active in a specific cell line, suggesting a general interdependency of active enhancers and expression of long ncRNAs. This dependency is not identified using previous enhancer prediction algorithms that do not account for expression of their downstream targets. The predicted enhancers that overlap annotated long ncRNAs generally have a lower ratio of H3K4me1 to H3K4me3, suggesting that enhancers expressing long ncRNAs might be associated with specific epigenetic marks. In conclusion, we demonstrate the tissue-specific predictive power of PreSTIGE and provide evidence for thousands of long ncRNAs that are expressed from active tissue-specific enhancers, suggesting a particularly important functional relationship between long ncRNAs and enhancer activity in determining tissue-specific gene expression.
Introduction
Enhancers are genetic regulatory elements that can activate transcription of their target genes in a temporal and tissue specific manner.Citation1 Enhancers can regulate their target genes independent of distance and orientation. The underlying mechanism has been suggested to involve formation of long-range chromatin loops, bringing enhancers and promoters into proximity and allowing interaction of the necessary co-transcriptional factors.Citation1 Enhancers have long been thought to bind transcription factors and be primarily active at the DNA level. Recent work has identified long non-coding RNAs (ncRNA) transcribed from active enhancers as important players in enhancer activity and function,Citation2–4 emphasizing the role of transcription of functional molecules at regulatory regions in the genome. The involvement of long ncRNAs in mediating enhancer function is an expanding topic that has recently resulted in numerous publications on the molecular mechanisms of long ncRNA transcription in enhancer function.Citation5
Several approaches to predict enhancers have been developed based on various individual enhancer features. For instance binding of p300 has been used to predict enhancers genome-wide in several different cell lines.Citation6 Other studies have used the tendency of enhancers to reside within open chromatin surrounded by nucleosomes marked with mono-methylated lysine 4 at histone H3 (H3K4me1) as a means to predict enhancers.Citation7-9 Also, bidirectional ncRNA transcription has been used as a criterion to identify active enhancers genome-wide.Citation9 A recently developed approach for tissue-specific enhancer prediction, PreSTIGE (Predicting Specific Tissue Interactions of Genes and Enhancers) uses gene expression and H3K4me1 across a panel of cell lines to identify both tissue-specific enhancers and their targets ().Citation10
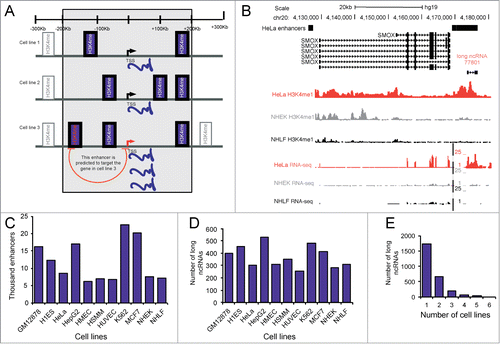
Figure 1. Overlap of long ncRNAs with predicted tissue-specific enhancers. (A) Overview of the modified PreSTIGE enhancer prediction used in this study. PreSTIGE predicts enhancers by first finding PCGs with tissue-specific increased expression. In the tissue in which the PCG has an increased expression and within the specified domain size surrounding the TSS of the PCG (200kb) PreSTIGE predicts enhancers based on the presence of cell type specific H3K4me1 domains. (B) Example of a long ncRNA that is specifically expressed in HeLa and overlaps a predicted HeLa specific enhancer. Black bars labeled HeLa enhancers show predicted enhancers at 2 sites in the locus. Also shown is the ENCODE annotated isoforms of long ncRNAs and PCGs. ENCODE data for selected representative cell lines are shown for H3K4me1 and RNA sequencing data deposited in the UCSC genome browser. (C) The number of PreSTIGE predicted cell type specific enhancers in 11 different cell lines. (D) The number of annotated long ncRNAs that overlap a tissue-specific enhancer predicted by PreSTIGE. (E) The number of cell lines in which a given long ncRNA overlaps a predicted enhancer.
Long ncRNAs show tissue-specific expression patterns, often restricted to a single cell line,Citation11,12 indicating specific regulatory roles in cellular functions. Expression of long ncRNAs has also been reported to be highly correlated with the expression of their neighboring protein-coding genes (PCG)Citation12 in agreement with the mounting evidence for long ncRNA involvement in mediating enhancer function.Citation3,4,9,13-18
The unique properties of the PreSTIGE enhancer prediction approach with respect to tissue specific enhancers prompted us to examine the overlap of these enhancers with annotated transcribed long ncRNAs and to evaluate the potential predictive power of PreSTIGE with respect to enhancer-associated long ncRNAs. Our results show that the tissue specific expression of long ncRNAs is associated with predicted enhancer activity for as many as one in 3 long ncRNA transcripts.
Results and Discussion
To identify tissue-specific enhancers across 11 cell lines (Supplementary Table 1), we used the PreSTIGE enhancer prediction approach.Citation10 PreSTIGE is a method that predicts enhancers by first finding PCGs with tissue-specific increased expression, and based on the assumption that these are targets of tissue-specific enhancers interrogates H3K4me1 domains in the vicinity. The predicted enhancers are therefore based on the presence of H3K4me1 domains in proximity to genes with tissue-specific expression. Predicted enhancers based on H3K4me1 can therefore be linked with the tissue-specific elevated expression of the putative target gene. The published application of PreSTIGE applies a domain size for enhancer activity of 100 kb and incorporates CTCF sites for expanding the borders of the domains around the TSS of the PCG.Citation10 We used a slightly modified version of PreSTIGE to address long ncRNA association with enhancer function, in which CTCF domains are not considered due to their potential involvement in enhancer functionCitation19-21 and the domain size is expanded to 200 kb surrounding the TSS (). These modifications are incorporated to account for the length that long ncRNAs occupy in the genome and based on recent evidence from ChIA-PET,Citation22 Hi-CCitation23 and 5CCitation19 that promoter-enhancer interactions typically occur over distances larger than 100 kb (average of 120 kb).Citation19 The comparison of different domain lengths has been done in the original report of the PreSTIGE algorithmCitation10 and shows that expanding the domain from 100 kb to 200 kb does increase the FDR (false discovery rate) slightly and increases the number of identified enhancers significantly. While expanding the domain size further leads to a further increase in the FDR, we settled for a domain size of 200 kb to accommodate inclusion of the enhancer promoter interactions within the typical distance of 120 kb.Citation19
One identified enhancer overlapping a long ncRNA specifically expressed in HeLa cells and predicted to regulate the SMOX gene is shown in . In the figure is shown the cell-type specific H3K4me1 peaks and cell-type specific expression of the long ncRNA and its predicted target by RNA sequencing data. According to PreSTIGE the SMOX gene is targeted by 5 different enhancers based on the specificity of expression of the SMOX gene and presence of H3K4me1 at the predicted enhancers. Only one of these overlaps an annotated long ncRNA, and therefore only this one enhancer of SMOX would be included in our subsequent analysis. The 4 remaining enhancers do not overlap long ncRNAs that have been annotated so far. This example illustrates that multiple enhancers can affect the same target leading to transcriptional regulation of the gene. That only one of the predicted enhancers overlaps a long ncRNA suggests that both long ncRNA dependent and independent mechanisms of enhancer function do exist.
Using the 200 kb domain PreSTIGE predicts a total of 131,917 cell-type specific enhancers across the 11 cell lines included in our analysis (). To address to which extent long ncRNA expression overlap active enhancers, we investigated the overlap between 9,505 ENCODE annotated long ncRNAsCitation12 and PreSTIGE predicted cell-type specific enhancers. We find that 2,695 long ncRNAs (28% of the analyzed ENCODE annotated long ncRNAs) overlap a predicted cell-type specific active enhancer in any of 11 cell lines used to establish the prediction algorithm (, Supplementary Table 1 and Supplementary Table 2). This number is prompting that cell-type specific enhancer function could specify to a certain degree the tissue-specific expression of long ncRNAs. One thousand, seven hundred and thirty-six long ncRNAs from 937 genomic regions were found to overlap with a predicted enhancer in one cell line only (), underlining the tissue-specific nature of both the algorithm for finding enhancers that are active and the function of long ncRNAs. Addition of more cell lines to the enhancer prediction algorithm does not increase the total number of predicted enhancers,Citation10 suggesting that the majority of tissue-specific enhancers are included in this analysis. Around 80% of the long ncRNAs overlapping predicted enhancers show evidence of transcription according to the data available from the ENCODE consortium.Citation12 Analyzing the complete annotation of long ncRNAs by ENCODE shows that 17% of the annotated long ncRNAs do not show evidence of expression in any of the 11 cell lines used in this study.
After establishing that a significant fraction of long ncRNAs overlap tissue-specific enhancers we addressed whether these long ncRNAs also show a tissue-specific expression pattern in agreement with the predicted enhancer activity. We intersected annotated long ncRNAs with predicted enhancers in each cell line, and using quantified expression values, fromCitation12 we established the relative expression of the long ncRNA at each enhancer for each cell line compared to the average across all 11 cell lines used in the study. All predicted enhancers with overlapping long ncRNA are included in Supplementary Tables 1 and 2. This analysis reveals a higher median expression of long ncRNAs associated with tissue-specific predicted enhancers for all cell lines (, and Supplementary ). We also observe significantly higher expression of long ncRNAs overlapping enhancers in 5 cell lines (GM12878, H1ES, HSMM, NHEK and HeLa) compared to the expression of all ncRNAs in the particular cell line (Wilcoxon rank-test). Shown is also the expression of each long ncRNA overlapping a tissue-specific enhancer for each cell line as relative values depicted in a heatmap to give an overview of the general expression preference ( and ). For the other cell lines in the analysis, long ncRNAs overlapping tissue-specific predicted enhancers show the highest expression, but results are also significant to a lesser extent in other cell lines (Supplementary ). A representation of the tissue-specific expression of the corresponding target PCGs is shown with heatmaps in Supplementary . Analysis of the gene ontologies of regulated PCGs shows highly significant enrichments of cell-type specific functional groups for GM12878 (Cell-To-Cell Signaling and Interaction, Hematological System Development and Function); H1ES (Embryonic Development, Developmental Disorder); HeLa (Reproductive System Disease); HepG2 (Carbohydrate Metabolism, Lipid Metabolism); K562 (Cardiovascular System Development and Function, Cell-mediated Immune Response); MCF7 (Breast or Ovarian Cancer) and NHEK (Dermatological Diseases and Conditions), further demonstrating the importance of tissue-specific enhancer predictions.
Figure 2. Long ncRNA expression correlates with predicted tissue-specific enhancers. In (A–D) are shown expression values for all long ncRNAs overlapping a predicted enhancer in (A) GM12878, (B) H1ES, (C) HSMM, and (D) NHEK. For each long ncRNA the relative expression compared to the average expression across all cell lines is shown as bar-plots (upper panels) or as heatmaps (lower panels). Heatmaps are normalized for each transcript such that blue shows the lowest expression and red shows the highest expression. Statistical analysis is done using Mann-Whitney-Wilcoxon test. a P-value < 2.2e-16, b P-value 7.9e-15.
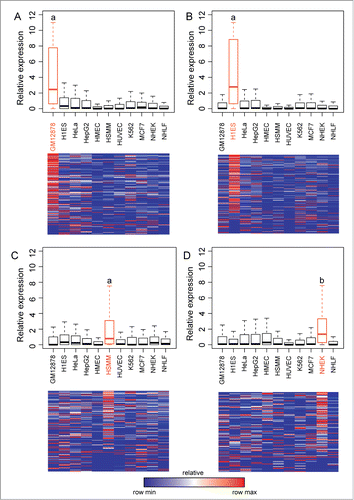
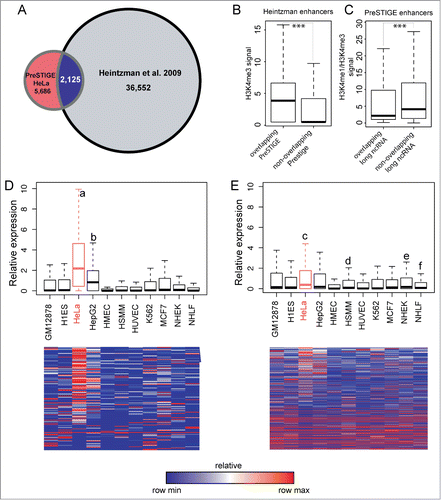
Figure 3. Comparison of PreSTIGE predicted enhancers to previous methods. (A) Venn diagram showing the overlap between enhancers in HeLa predicted byCitation7 using H3K4me1 and H3K4me3 profiles compared to HeLa tissue-specific enhancers predicted by PreSTIGE. (B) Quantification of H3K4me3 at enhancers predicted byCitation7 overlapping or not overlapping PreSTIGE predicted enhancers, respectively. *** P-value < 2.2e-16. (C) The ratio of H3K4me1 to H3K4me3 for PreSTIGE predicted enhancers overlapping or not overlapping a long ncRNA, respectively. *** P-value 0.00039. (D and E) As in . Shown are average relative expression values across 11 cell lines for all long ncRNAs overlapping predicted enhancers in HeLa cells as bar-plots (upper panels) or heatmaps (lower panels). (D) Expression at enhancers predicted by the modified PreSTIGE method. a P-value < 2.2e-16, b P-value 9.0e-10. (E) Expression at enhancers predicted by Heintzman et al., 2009. c P-value 9.5e-07, d P-value 3.9e-05, e P-value 6.4e-06 and f P-value 1.6e-06. Statistical analyses were done using Mann-Whitney-Wilcoxon test.
To address whether there is a correlation between the PCG and long ncRNA expression we calculated the Pearson correlation for each gene-pair across all cell lines. We find that 37.2% of gene pairs (PCG∼long ncRNA) are significantly correlated (P < 0.05) across all 11 cell lines, supporting our findings that the analyzed subset of tissue-specific enhancers expresses long ncRNAs dependent on their activity.
A pioneering study defined enhancers and promoters based on H3K4me1 and H3K4me3, respectively, and proposed that tissue-specific expression of genes is primarily due to differential enhancer activity while maintaining stable promoter activity.Citation7 While enhancers are predicted by the presence of H3K4me1 by PreSTIGE, our data show that transcription of long ncRNAs often occurs at sites marked with H3K4me1.
When comparing PreSTIGE predicted enhancers in HeLa, only 2,125 of 8,560 (24%) overlap with the 36,552 enhancers predicted byCitation7 demonstrating clearly different pools of enhancers being identified in the 2 studies (). One of the reasons could be, that the study byCitation7 considers H3K4me1 and H3K4me3 in defining enhancers and promoters as mutually exclusive, while PreSTIGE considers the tissue-specificity of H3K4me1 marks, irrespective of the presence of H3K4me3.Citation10 The observation that expressed long ncRNAs are being actively transcribed suggests a potential overlap of their promoters with H3K4me3, which would explain why these enhancer predictions have been omitted from the study by Heintzman et al.Citation7 When analyzing H3K4me3 at the enhancers predicted byCitation7 that overlap PreSTIGE predicted enhancers, we find a significantly higher signal than for those identified only byCitation7 (), supporting this idea. It has also been suggested that it is rather the ratio between the 2 H3K4 methylation marks that determines the properties of a regulatory element, leading occasionally to complex situations where enhancers can function as alternative promoters.Citation14 We calculated the ratio of H3K4me1 to H3K4me3 for PreSTIGE predicted enhancers and compared the values for those overlapping annotated long ncRNAs to those that do not. As shown in the enhancers overlapping an annotated long ncRNA show on average significantly lower H3K4me1/H3K4me3 ratio than those that do not overlap annotated long ncRNAs. This means, that enhancers overlapping annotated long ncRNAs have relatively higher H3K4me3 compared to H3K4me1. This observation is in line with the fact that long ncRNAs are expressed from these enhancers, and suggests that enhancers expressing long ncRNAs could have a characteristic epigenetic mark profile.
The average relative expression of the long ncRNAs overlapping PreSTIGE predicted tissue-specific enhancers in HeLa is significantly higher than in any other cell line examined (). While 1,217 of the enhancers predicted byCitation7 overlap long ncRNAs, their expression is less significant compared to PreSTIGE predicted enhancers (P-value 9.496e-07 vs P-value < 2.2e-16), while being transcribed not only in HeLa but also in HSMM, NHEK and NHLF (). These results suggest that the PreSTIGE method has a stronger predictive power than the previous methods, and that long ncRNA expression is associated with tissue-specific enhancers. While we find that the PreSTIGE method is very robust in predicting tissue-specific enhancers based on the association with long ncRNA transcription, the approach is limited to enhancers regulating tissue-specific gene expression. While the method reported byCitation7 would very likely find a general enhancer working in several cell lines, these would be missed by PreSTIGE. The enhancers predicted by PreSTIGE appear to include more true-positives likely because of the inclusion of expression of the potential target genes but at the same time dismisses many true enhancers due to activity across more cell lines.
While the results presented suggest that long ncRNA transcription is linked to tissue-specific enhancers, they do not tell us how they are mechanistically involved. The long ncRNAs expressed at enhancers have been suggested to be directly involved in mediating the enhancer activity on the regulated genes,Citation3,15,24 while other studies have predominantly found evidence for a correlation in expression.Citation2,4 How many of these enhancer transcribed long ncRNAs are mediating the enhancer activity and how many are expressed as a consequence of enhancer activation can not be definitely derived from our data, but should be addressed in future experiments. We find, however, that tissue-specific enhancer associated expression of long ncRNAs is characteristic for a subset of the predicted enhancers, implying a functional relationship.
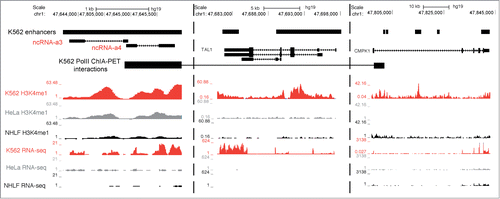
Several studies have reported the involvement of long ncRNAs in mediating enhancer function.Citation3,4,9,13-18 One of the PreSTIGE predicted enhancers in K562 cells has previously been described to transcribe activating long ncRNAs ncRNA-a3 and ncRNA-a4 regulating TAL1 and CMPK1 expression, respectivelyCitation3 (). While both activating long ncRNAs are identified as K562 specific, overlapping a K562 specific enhancer predicted by PreSTIGE, only juxtaposed TAL1 is assigned as the target of the enhancer due to its highly tissue-specific expression. While enhancer-like long ncRNAs can have several targets and mediate their effects over several megabases,Citation15 the experimental evidence derived from PolII ChIA-PET,Citation22 knock-down and reporter assaysCitation3 emphasizes in this case the regulation of CMPK1. This illustrates one of the complications of identifying long-range regulatory relationships between long ncRNAs and PCGs, and implies that an even larger fraction of long ncRNAs could be associated with tissue-specific enhancers. Long ncRNAs and enhancers can regulate gene expression over distances longer that 200 kb,Citation15 and these interactions would not be confidently identified using the reported approach due to the increased FDR with increased PCG enhancer domain size. While a subset of the predicted enhancers showing expression of long ncRNAs might be false-positives, the reported approach also excludes a potentially large number of true-positives due to the limitations in domain size in the PreSTIGE method.
Figure 4. PreSTIGE prediction of enhancers overlapping activating ncRNAs. A tissue-specific enhancer is predicted in K562 cells that overlap previously identified activating ncRNAs ncRNA-a3 and ncRNA-a4 shown to target TAL1 and CMPK1, respectively. Predicted enhancers are shown as black boxes (K562 enhancers). The ENCODE annotated transcripts are shown for discontinuous regions of the locus. See scale bar and coordinates. K562 PolII ChIA-PET data are from Li et al.Citation22 as deposited in the UCSC genome browser, and show interacting regions as experimentally determined. Additionally, H3K4me1 and RNA sequencing data from ENCODE are shown for representative cell lines.
Prediction of enhancers is a rapidly expanding area of research and numerous approaches have been proposed. Different approaches yield different predicted enhancers, and usually the target identification is left to guesswork or the simple assumption that the neighboring gene is being regulated.Citation5-8,25,26 The PreSTIGE method is one of the few approaches that integrate the prediction of targets in addition to the prediction of enhancers making it an important advance of enhancer prediction. Judged by the significant occurrence of active transcription of long ncRNAs at predicted tissue-specific enhancers it is reasonable to assume that the predictions are also more accurate than previous methodologies.
In conclusion, we demonstrate the tissue-specific predictive power of PreSTIGE and provide evidence for thousands of long ncRNAs that are expressed from tissue-specific enhancers, suggesting a particularly important functional relationship between long ncRNAs and enhancer activity in determining tissue-specific gene expression.
Methods
Enhancer prediction
PreSTIGE methodology was performed as described previously.Citation10 In brief, PreSTIGE is a method that predicts enhancers by first finding PCGs with tissue-specific increased expression, and based on the assumption that these are targets of tissue-specific enhancers searches for H3K4me1 domains within the specified domain size. Predicted enhancers based on H3K4me1 can therefore be linked with the tissue-specific elevated expression of the putative target gene. We used a slightly modified version of PreSTIGE to address long ncRNA association with enhancer function, in which CTCF domains are not considered and the domain size is expanded to 200 kb surrounding the TSS of the PCG. Additionally, all enhancers that were overlapping with their predicted targets were filtered out from the enhancer dataset used in the subsequent analysis.
ncRNA and enhancer overlap
Annotated long ncRNAs were obtained fromCitation12 and were further filtered for long ncRNAs that do not overlap PCGs. Long ncRNAs were intersected with cell type specific PreSTIGE enhancers, and vice versa, using BEDTools.Citation27 Minimal overlap for this analysis is one nucleotide.
Previously predicted enhancers in HeLa were obtained fromCitation7. Overlap of these enhancers with HeLa enhancers predicted by PreSTIGe, as well as intersection with long ncRNAs was performed using BEDToolsCitation27 with one nucleotide minimal overlap.
Gene ontology analysis
PreSTIGE predicted targets of enhancers that overlap long ncRNAs were subjected to gene ontology analysis. Data were analyzed through the use of QIAGEN's Ingenuity Pathway Analysis (IPA QIAGEN Redwood City www.qiagen.com/ingenuity).
Gene expression analysis
We obtained RNA sequencing data for long ncRNAs fromCitation12. Expression (RPKM) of each gene was normalized to the average expression across the 11 cell lines used in this study.
Calculating H3K4me1 and H3K4me3 levels in genomic regions
To determine the H3K4me1 and H3K4me3 levels for each predicted enhancer ENCODE HeLa broad.peaks were used. Enhancers with no histone mark peak were assigned pseudo signal value 0.5 in the case of H3K4me3 and 1 in the case of H3K4me1. In case one enhancer overlapped more than one peak then the mean signal value was calculated.
Statistical analysis
Statistical analysis was done using Student's t-test (for ) or using paired Mann-Whitney-Wilcoxon test prior to normalization of RNA sequencing data (in , and Supplementary ) comparing expression of the predicted long ncRNA overlapping enhancers set to all long ncRNAs.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
DV analyzed data, interpreted data and wrote the manuscript. EN analyzed data. OC and PCS developed the PreSTIGE method and modified enhancer prediction criteria. UAØ conceived the experiments, supervised research, interpreted data and wrote the manuscript. All authors read and approved the manuscript.
977641_Supplementary_Materials.zip
Download Zip (2.1 MB)Funding
EN is an Alexander von Humboldt postdoctoral fellow. Work in the author's laboratories is funded by the Federal Ministry of Germany through the Alexander von Humboldt Foundation Sofja Kovalevskaja Award to UAØ.
References
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell 2013; 49:825-37; PMID:23473601; http://dx.doi.org/10.1016/j.molcel.2013.01.038
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol 2010; 8:e1000384; PMID:20485488; http://dx.doi.org/10.1371/journal.pbio.1000384
- Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010; 143:46-58; PMID:20887892; http://dx.doi.org/10.1016/j.cell.2010.09.001
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010; 465:182-7; PMID:20393465; http://dx.doi.org/10.1038/nature09033
- Orom UA, Shiekhattar R. Long noncoding RNAs usher in a new era in the biology of enhancers. Cell 2013; 154:1190-3; PMID:24034243; http://dx.doi.org/10.1016/j.cell.2013.08.028
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009; 457:854-8; PMID:19212405; http://dx.doi.org/10.1038/nature07730
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108-12; PMID:19295514; http://dx.doi.org/10.1038/nature07829
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011; 470:279-83; PMID:21160473; http://dx.doi.org/10.1038/nature09692
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. An atlas of active enhancers across human cell types and tissues. Nature 2014; 507:455-61; PMID:24670763; http://dx.doi.org/10.1038/nature12787
- Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Cowper-Sal lari R, Lupien M, Markowitz S, Scacheri PC. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res 2014; 24:1-13; PMID:24196873; http://dx.doi.org/10.1101/gr.164079.113
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011; 25:1915-27; PMID:21890647; http://dx.doi.org/10.1101/gad.17446611
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 2012; 22:1775-89; PMID:22955988; http://dx.doi.org/10.1101/gr.132159.111
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011; 472:120-4; PMID:21423168; http://dx.doi.org/10.1038/nature09819
- Kowalczyk MS, Hughes JR, Garrick D, Lynch MD, Sharpe JA, Sloane-Stanley JA, McGowan SJ, De Gobbi M, Hosseini M, Vernimmen D, et al. Intragenic enhancers act as alternative promoters. Mol Cell 2012; 45:447-58; PMID:22264824; http://dx.doi.org/10.1016/j.molcel.2011.12.021
- Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, Shiekhattar R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 2013; 494:497-501; PMID:23417068; http://dx.doi.org/10.1038/nature11884
- Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 2013; 23:1210-23; PMID:23636943; http://dx.doi.org/10.1101/gr.152306.112
- Gomez JA, Wapinski OL, Yang YW, Bureau JF, Gopinath S, Monack DM, Chang HY, Brahic M, Kirkegaard K. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-gamma locus. Cell 2013; 152:743-54; PMID:23415224; http://dx.doi.org/10.1016/j.cell.2013.01.015
- Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 2013; 498:511-5; PMID:23728303; http://dx.doi.org/10.1038/nature12209
- Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature 2012; 489:109-13; PMID:22955621; http://dx.doi.org/10.1038/nature11279
- Merkenschlager M, Odom DT. CTCF and cohesin: linking gene regulatory elements with their targets. Cell 2013; 152:1285-97; PMID:23498937; http://dx.doi.org/10.1016/j.cell.2013.02.029
- Phillips-Cremins JE, Corces VG. Chromatin insulators: linking genome organization to cellular function. Mol Cell 2013; 50:461-74; PMID:23706817; http://dx.doi.org/10.1016/j.molcel.2013.04.018
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012; 148:84-98; PMID:22265404; http://dx.doi.org/10.1016/j.cell.2011.12.014
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009; 326:289-93; PMID:19815776; http://dx.doi.org/10.1126/science.1181369
- Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, Tsirigos A, Aifantis I. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell 2014; 158:593-606; PMID:25083870; http://dx.doi.org/10.1016/j.cell.2014.05.049
- Bonn S, Zinzen RP, Girardot C, Gustafson EH, Perez-Gonzalez A, Delhomme N, Ghavi-Helm Y, Wilczynski B, Riddell A, Furlong EE. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet 2012; 44:148-56; PMID:22231485; http://dx.doi.org/10.1038/ng.1064
- Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 2013; 339:1074-7; PMID:23328393; http://dx.doi.org/10.1126/science.1232542
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26:841-2; PMID:20110278; http://dx.doi.org/10.1093/bioinformatics/btq033