
Abstract
Deregulated Wnt/β-catenin signaling promotes colorectal cancer (CRC) by activating expression of the c-MYC proto-oncogene (MYC). In the nucleus, the β-catenin transcriptional co-activator binds T-cell factor (TCF) transcription factors, and together TCF/β-catenin complexes activate MYC expression through Wnt responsive DNA regulatory elements (WREs). The MYC 3’ WRE maps 1.4-kb downstream from the MYC transcription stop site and binds TCF4/β-catenin transcription complexes to activate MYC. However, the underlying mechanisms for how this element operates are not fully understood. Here, we report that the TCF family member, TCF3, plays an important role in regulating MYC expression in CRCs. We demonstrate that TCF3 binds the MYC 3′ WRE to repress MYC. When TCF3 is depleted using shRNAs, the MYC 3′ WRE is more available to bind TCF4/β-catenin complexes. Stimulating downstream Wnt/β-catenin signaling by inhibiting GSK3β causes an exchange of TCF3 with TCF4/β-catenin complexes to activate MYC. Finally, this transcription factor switch at the MYC 3′ WRE controls MYC expression as quiescent cells re-enter the cell cycle and progress to S phase. These results indicate that a dynamic interplay of TCF transcription factors governs MYC gene expression in CRCs.
Abbreviations
| TCF | = | T-cell factor |
| Lef | = | Lymphoid enhancer-binding factor |
| MYC | = | myelocytomatosis |
| WRE | = | Wnt responsive DNA element |
| CRC | = | colorectal cancer |
| APC | = | adenomatous polyposis coli |
| TLE | = | Transducin-like enhancer of split |
| HDAC | = | histone deacetylase |
| GSK3β | = | glycogen synthase kinase 3 β |
| LiCl | = | lithium chloride |
| ChIP | = | chromatin immunoprecipitation |
| RT | = | reverse transcription |
| qPCR | = | quantitative PCR |
Introduction
Colorectal cancer (CRC) is the third leading cause of cancer-related deaths in the US, and it is predicted that approximately 50,000 individuals will die from this disease in 2014.Citation1 Mutations in components of the Wnt/β-catenin signaling pathway are prevalent in spontaneous CRCs with up to 80% of the cases containing a lesion in the Adenomatous polyposis coli (APC) gene and another 5% containing a lesion in the CTNNB1 gene that encodes β-catenin.Citation2 The consequence of these mutations is inappropriate accumulation of the β-catenin transcriptional co-activator in the nucleus and misexpression of β-catenin target genes.Citation3,4
As a transcriptional co-activator, β-catenin lacks a DNA binding domain, and it must therefore interact with sequence-specific transcription factors to activate gene expression. The T-cell factors/Lymphoid enhancer-binding factors (TCF/Lefs; hereafter referred to as TCFs) are a major class of transcription factors that control the nuclear response to Wnt/β-catenin signaling. In the presence of extracellular Wnt ligand, TCF/β-catenin complexes bind Wnt responsive DNA elements (WREs) and recruit histone aceytltransferases to modify the chromatin architecture of target gene promoters into a transcriptionally permissive state.Citation5,6 In the absence of Wnt, TCFs instead bind transcriptional corepressor complexes, such as Groucho/Transducin-like enhancer of split (Gro/TLE; hereafter TLE), that utilize associated histone deacetylases (HDACs) to repress target gene expression.Citation5,7 Thus, according to a transcriptional switch model, TCFs function as a platform, which exchange co-repressors with co-activators to regulate expression of Wnt/β-catenin target genes.
The 4 TCF family members in vertebrates are TCF1 (also known as TCF7), LEF1, TCF3 (also known as TCF7L1), and TCF4 (also known as TCF7L2).Citation5,7 TCF4 is highly expressed in intestinal epithelial cells, and deletion of Tcf4 in mice ablates the proliferative compartment of the intestinal crypts.Citation8-10 In human colorectal cancer cells, expression of a dominant negative form of TCF4, which retains its HMG box DNA binding domain but lacks its amino-terminal β-catenin interacting domain, causes cell cycle arrest.Citation11 These studies indicate that TCF4 functions to promote cellular proliferation, although it is not clear whether it functions as a tumor suppressor or an oncogene.Citation9,11-13 TCF3 has been most studied in embryonic stem cells and in the adult skin where it has been shown to primarily repress expression of Wnt target genes.Citation14,15 Deletion of Tcf3 within the intestinal epithelium of juvenile mice lacked an apparent phenotype, indicating that this TCF family member is not required for intestinal development or homeostasis.Citation16 Outside of one report that found that TCF3 contributed to the butyrate-resistant phenotype of a CRC cell line,Citation17 the role for TCF3 in human CRCs has not been extensively studied.
The c-MYC proto-oncogene (MYC) is a critical downstream effector of oncogenic Wnt/β-catenin signaling in the intestines.Citation18-25 To identify WREs that control MYC expression in human CRC cells, we previously conducted 2 genome-wide screens to map β-catenin binding sites.Citation26,27 These screens found a robust β-catenin binding site 1.4-kb downstream from the MYC transcription stop site, which we showed demarcated a 600-bp WRE that overlapped a previously identified DNAse I hypersensitivity site in CRC cells.Citation26-29 Using the human HCT116 cell line as a model, we showed that TCF4/β-catenin complexes assembled at this MYC 3′ enhancer and coordinated a chromatin loop with the MYC proximal promoter to activate MYC expression.Citation30 When these cells were synchronized and then released into the cell cycle, TCF4/β-catenin complexes bound the MYC 3′ WRE, and induced histone acetylation to activate MYC expression.Citation28 As cells transitioned into S phase, both TCF4 and β-catenin vacated the MYC 3′ WRE and MYC expression was repressed.Citation28 Because we did not detect significant TCF4 occupancy at the MYC 3′ WRE in quiescent cells or cells in S phase, the underlying mechanisms accounting for MYC repression through this element were unknown at that time.
In the present study, we hypothesized that TCF3 functions as a repressor of MYC expression in CRC cells, and that an exchange of TCF3 with TCF4/β-catenin complexes accompanies activation of MYC expression. In asynchronously growing cells, depletion of TCF3 stimulated TCF4/β-catenin binding to the MYC 3′ WRE. When CRC cells and normal intestinal epithelial cells were treated with lithium to activate downstream Wnt/β-catenin signaling, an exchange of TCF3 with TCF4/β-catenin complexes at the MYC 3′ WRE accompanied the increase in MYC expression. Finally, in quiescent CRC cells cultured in serum-deprived media, TCF3 complexes bound the MYC 3′ WRE to repress MYC expression. When these cells were stimulated with media-containing serum, an exchange of TCF3 with TCF4/β-catenin accompanied the increase of MYC expression. As cells progressed to S phase, TCF3 replaced TCF4/β-catenin complexes at this WRE to repress MYC expression. Thus, for the first time, these findings indicate that a dynamic interplay of TCF family members controls MYC expression in CRC cells.
Results
TCF3 is a transcriptional repressor in CRC cells
Depending on the target gene and cell type analyzed, TCF3 has been shown to function either as an activator or repressor of gene expression.Citation31 To study the function of TCF3 in the HCT116 human CRC cell line, we generated 5 independent lentiviruses containing shRNAs that targeted non-overlapping regions of the TCF3 transcript. We infected HCT116 cells with these lentiviruses and 3 days after transduction, RNAs were isolated, cDNAs were synthesized, and TCF3 levels were assessed using quantitative PCR (qPCR). Cells expressing TCF3 shRNA1 or TCF3 shRNA2, contained a 90% or greater reduction in TCF3 transcripts relative to levels seen in control cells that were transduced with lentiviruses expressing a scrambled shRNA control sequence (). Western blot analysis demonstrated that this reduction was also seen at the protein level (). TCF4 protein levels were stable in these cells, indicating that the TCF3 shRNAs do not target TCF4 (). In addition, we analyzed β-catenin levels in purified cytoplasmic and nuclear fractions isolated from TCF3-depleted cells. Reduction of TCF3 levels did not affect β-catenin levels in either compartment or in whole cell extracts ().
Figure 1. TCF3 functions as a transcriptional repressor. (A) qPCR analysis of cDNAs synthesized from RNAs isolated from HCT116 cells 3 days after they were infected with lentiviruses expressing a scrambled sequence (Ctrl.) or 2 independent shRNAs targeting TCF3. TCF3 levels are normalized to GAPDH. (B) Western blot analysis of TCF3 and TCF4 protein levels in HCT116 cells expressing the indicated shRNAs. Tubulin served as a loading control. (C) Western blot analysis of β-catenin and TCF3 levels in whole cell [W], cytoplasmic [C], and nuclear [N] lysates prepared from HCT116 cells transduced with lentiviruses expressing control or TCF3-shRNA2. The blots were reprobed with tubulin antibodies or histone H3 antibodies to analyze the purity of the cytoplasmic and nuclear extracts, respectively. (D) Luciferase reporter assays conducted in HCT116 cells transduced with control or TCF3 shRNA2 lentiviruses. Two days after infection, the cells were transfected with the indicated luciferase reporter constructs along with a construct expressing Renilla luciferase to control for transfection efficiency. The data are normalized to levels detected in control cells. (E) As in D except cells were transfected with pCMV-Tag2B-TCF3 or vector alone as a control. Inset, Western blot analysis showing levels of TCF3 expression in transfected cells. All experiments were repeated at least 3 times and error bars are ± SEM.
![Figure 1. TCF3 functions as a transcriptional repressor. (A) qPCR analysis of cDNAs synthesized from RNAs isolated from HCT116 cells 3 days after they were infected with lentiviruses expressing a scrambled sequence (Ctrl.) or 2 independent shRNAs targeting TCF3. TCF3 levels are normalized to GAPDH. (B) Western blot analysis of TCF3 and TCF4 protein levels in HCT116 cells expressing the indicated shRNAs. Tubulin served as a loading control. (C) Western blot analysis of β-catenin and TCF3 levels in whole cell [W], cytoplasmic [C], and nuclear [N] lysates prepared from HCT116 cells transduced with lentiviruses expressing control or TCF3-shRNA2. The blots were reprobed with tubulin antibodies or histone H3 antibodies to analyze the purity of the cytoplasmic and nuclear extracts, respectively. (D) Luciferase reporter assays conducted in HCT116 cells transduced with control or TCF3 shRNA2 lentiviruses. Two days after infection, the cells were transfected with the indicated luciferase reporter constructs along with a construct expressing Renilla luciferase to control for transfection efficiency. The data are normalized to levels detected in control cells. (E) As in D except cells were transfected with pCMV-Tag2B-TCF3 or vector alone as a control. Inset, Western blot analysis showing levels of TCF3 expression in transfected cells. All experiments were repeated at least 3 times and error bars are ± SEM.](/cms/asset/cd2e741b-a094-4c5f-8417-5c9859562f5f/kccy_a_980643_f0001_b.gif)
To determine how TCF3 regulated Wnt/β-catenin-dependent transcription, we first conducted luciferase reporter assays. In comparison to cells expressing the control shRNA sequence, knocking down TCF3 activated luciferase expression driven from the TOPflash reporter (). TOPflash contains 6 TCF consensus binding sites inserted upstream of the thymidine kinase minimal promoter that drives luciferase. Luciferase levels driven from the control FOPflash reporter, containing mutations in the TCF binding sites, were not affected by TCF3 depletion (). To confirm these findings on a more physiologic target, we conducted assays using a luciferase reporter driven by the MYC 3′ WRE. Citation28 This construct contains the MYC 3′ WRE inserted downstream from the luciferase gene, and TCF3 knock-down activated expression from this reporter in transiently transfected cells (). TCF3 knock-down did not affect luciferase expression from the MYC 3′ WRE (mut) construct that contains mutations in the 2 TCF binding sites within the 3′ WRE ().Citation28 In converse experiments, transfecting cells with a mammalian expression plasmid harboring TCF3 cDNA decreased luciferase expression from TOPflash and the MYC 3′ WRE-luciferase construct, while not influencing expression from the TCF-mutant controls (). These findings indicate that TCF3 functions as a transcriptional repressor in HCT116 cells.
TCF3 directly represses MYC gene expression
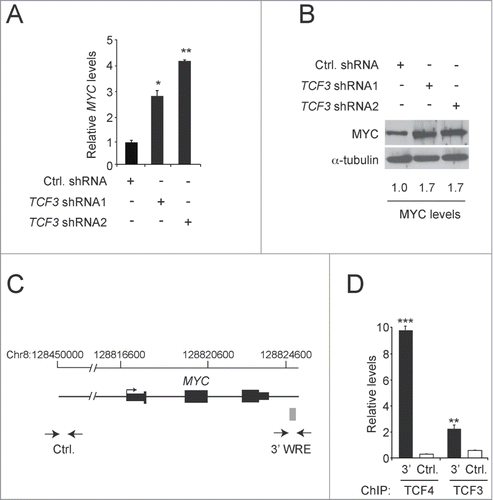
The results from the luciferase experiments suggested that TCF3 might directly bind the MYC 3′ WRE to repress MYC gene expression in HCT116 cells. To investigate this possibility, we first measured MYC transcript levels in cells transduced with lentiviruses expressing control or TCF3-specific shRNAs. Relative to control cells, RT-qPCR analysis indicated that cells expressing TCF3 shRNA1 or TCF3 shRNA2 contained 3- and 4-fold higher levels of MYC transcript levels, respectively (). We found using Western blot analysis that TCF3-depletion also increased MYC protein levels 1.7-fold (). To determine whether TCF3 occupied the MYC 3′ WRE at the MYC gene locus, we conducted chromatin immunoprecipitation (ChIP) assays. We used primers designed against this element, and a control sequence that maps 290-kb upstream from the MYC transcription start site, to amplify precipitated DNAs in real-time qPCR reactions (). In addition, we evaluated TCF4 as a positive control, which we have previously shown to bind the MYC 3′ WRE in these cells. Citation28,Citation30 We detected TCF3 binding to the MYC 3′ WRE, although levels of occupancy were lower than that of TCF4 (). Together, these findings support the hypothesis that MYC gene expression is negatively regulated, in part, through direct binding of TCF3 to a downstream regulatory element.
Figure 2. TCF3 represses MYC gene expression. (A) qPCR analysis of cDNAs prepared from HCT116 cells expressing control or TCF3-specific shRNAs. The values presented are normalized to GAPDH levels. (B) Western blot analysis of MYC protein levels in HCT116 cells expressing control or TCF3-specific shRNAs. The blots were reprobed with anti-α-tubulin antibodies to control for equal loading. (C) Diagram of the MYC gene locus. Exons and UTRs are depicted by black rectangles with an arrow marking the major transcription start site. The gray box marks the position of the MYC 3′ WRE. Opposed arrows indicate the positions of the PCR primers used for the chromatin immunoprecipitation (ChIP) assays depicted in D. The control region maps approximately 290-kb upstream from the MYC transcription start site. (D) qPCR analysis of DNA fragments precipitated using TCF4- or TCF3-specific antibodies in ChIP assays conducted in HCT116 cells. All experiments were repeated at least 3 times and error bars are ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001).
TCF3 and TCF4 compete for binding to the MYC 3′ WRE
We next tested whether altering levels of TCF3 affected TCF4 binding to the MYC 3′ WRE. Using ChIP assays, we found that TCF3 depletion enhanced TCF4 binding to this element (). We detected no difference in background levels of binding to the control sequence (). Because β-catenin binds TCF4 to activate transcription driven from WREs,Citation27,28,30,32,33 we asked whether TCF3 depletion also stimulated β-catenin binding to the MYC 3′ WRE. Indeed, ChIP assays with β-catenin-specific antibodies detected higher levels of β-catenin binding to this element when TCF3 was knocked down (). Given that TCF3 knock-down does not affect TCF4 protein levels () and that depleting TCF3 increases MYC gene expression (), these results suggest that in the absence of TCF3, the MYC 3′ WRE is more readily available to bind TCF4/β-catenin complexes to activate MYC expression.
Figure 3. TCF3 and TCF4 compete for binding to the MYC 3′ WRE. (A) qPCR analysis of DNA fragments precipitated using anti-TCF4 antibodies in ChIP assays conducted from HCT116 cells 3 days after they were infected with lentiviruses expressing control or TCF3-specific shRNAs. (B) As in A, except ChIP assays were conducted with anti-β-catenin antibodies. (C) Western blot analysis of TCF4 protein levels in HCT116 cells 3 days after they were infected with lentiviruses expressing a scrambled sequence (Ctrl.) or 4 independent shRNAs that targeted TCF4. Tubulin served as a loading control.(D) qPCR analysis of DNA fragments precipitated using anti-TCF3 antibodies conducted from HCT116 cells expressing control or TCF4-specific shRNAs. All experiments were repeated at least 3 times and error bars are ± SEM (*P < 0.05, **P < 0.01).
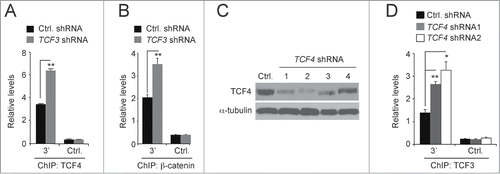
To test whether TCF4 depletion likewise stimulates TCF3 binding, we generated lentiviral particles expressing 4 independent shRNA sequences that targeted TCF4 transcripts. Western blot analysis indicated that 3 days after infection, TCF4 shRNA constructs 1 and 2 were most effective in reducing TCF4 protein levels in transduced HCT116 cells (). We then conducted ChIP assays with TCF3-specific antibodies and found that TCF4 depletion stimulated TCF3 binding to the MYC 3′ WRE (). Together, these findings suggest that TCF3 and TCF4/β-catenin transcription complexes compete for binding to this downstream MYC WRE.
Activation of the downstream Wnt/β-catenin signaling pathway induces a TCF3/TCF4 exchange
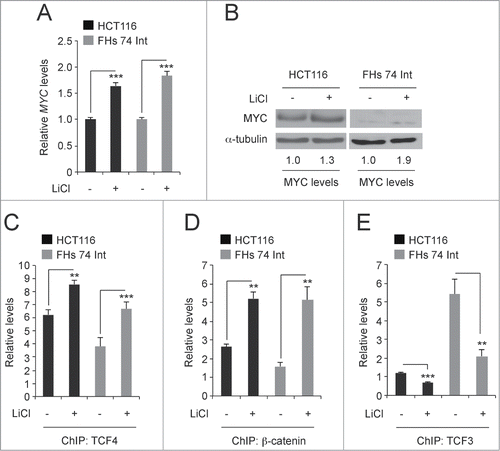
HCT116 cells have one mutant allele of CTNNB1 that contains a deletion in the codon for serine 45.Citation34 This lesion impairs proteasomal degradation of the pool of β-catenin encoded by this allele. The other CTNNB1 allele is wild-type and levels of β-catenin encoded by this allele are turned over by the APC/GSK3β destruction complex. Others and we have shown that inhibiting the activity of GSK3β in these cells using lithium chloride (LiCl), stabilizes β-catenin levels and further activates Wnt/β-catenin signaling. Citation26,33,35,36 FHs 74 Int is a cell line derived from normal human intestinal epithelia. Citation37 Treatment of HCT116 and FHs 74 Int cells with 25 mM LiCl for 6 hours increased MYC transcript and protein levels relative to those seen in control cells (). Using ChIP assays, we found that LiCl treatment increased TCF4/β-catenin binding to the MYC 3′ WRE and decreased TCF3 binding to this element (). Thus, lithium treatment induces an exchange of TCF3 with TCF4 transcription factors at the downstream enhancer to increase MYC gene expression.
Figure 4. Lithium induces a TCF3/TCF4 exchange at the MYC 3′ WRE. (A) qPCR analysis of MYC expression in cDNAs prepared from HCT116 and FHs 74 Int cells that were untreated or treated with 25 mM LiCl for 6 hours. MYC levels are normalized to GAPDH. (B) Western blot analysis of MYC protein levels in control and LiCl-treated cells. (C) qPCR analysis of DNA fragments precipitated using anti-TCF4 antibodies in ChIP assays conducted in cells treated with LiCl as indicated. Shown are levels of TCF4 binding at the MYC 3′ WRE. (D) As in C except anti-β-catenin antibodies were used in the ChIP assays. (E) As in C except anti-TCF3 antibodies were used in the ChIP assays. All experiments were repeated at least 3 times and error bars are ± SEM (**P < 0.01, ***P < 0.001).
A dynamic exchange of TCF3 and TCF4/β-catenin transcription complexes at the MYC 3′ WRE controls MYC expression during the cell cycle
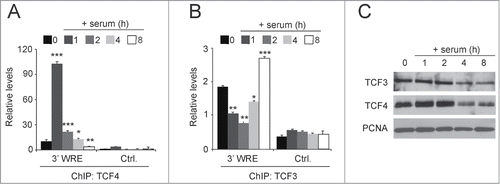
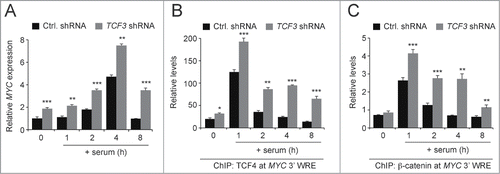
As an immediate early gene, MYC expression is silenced in quiescent cells and is subsequently induced as cells are stimulated to enter the G1 phase of the cell cycle.Citation38 When HCT116 cells are grown to confluence and deprived of serum for 48 hours, the cells enter G0/G1 and silence MYC expression.Citation28,39 When these cells are treated with media containing serum, they reenter the cell cycle and MYC expression is induced. In our prior study, we found MYC expression was induced after 2 hours in serum, and its maximal expression was seen at 4 hours of serum treatment.Citation28 At the 8 hour time point, when the cells fully transitioned into S phase, MYC expression returned to levels that were slightly above those seen in quiescent cells. We used these synchronized cells to test whether there was an exchange of TCF3 with TCF4 at times when MYC was repressed or actively transcribed. Confirming our published results, using ChIP assays we detected very little TCF4 binding to the MYC 3′ WRE in serum-starved cells and high levels of TCF4 binding as cells transition into G1 (0 versus 1h, respectively, ). Citation28 Likewise, TCF4 binding to the MYC 3′ WRE decreased as cells progressed through G1 when treated with serum for one, 2 and 4 hours and was absent when cells transitioned into S phase after 8 hours of serum treatment (). In contrast, TCF3 binding to the MYC 3′ WRE displayed the opposite pattern with highest levels in quiescent cells and cells in S phase and lowest levels in G1 cells (). A Western blot analysis indicated that the levels of nuclear TCF3 and TCF4 remained relatively constant during treatments, although levels were slightly reduced in cells at 4 and 8 hours of serum treatment (). Together, these findings, coupled with those from our previous study,Citation28 indicate that an exchange of TCF3 with TCF4/β-catenin complexes, and vice versa, governs MYC expression as cells transition from quiescence to G1 and from G1 to the S phase of the cell cycle.
Figure 5. Serum induces an exchange of TCF3 with TCF4 occupancy at the MYC 3′ WRE in synchronized cells. (A) HCT116 cells were synchronized in the cell cycle by culturing confluent cells in the absence of serum for 48 h. The cells were released into the cell cycle by culturing them in media-containing serum for one, 2, 4, and 8 hours. ChIP assays were conducted using anti-TCF4 antibodies in cells at each time point, with zero indicating starved cells, and qPCR assays were used to detect TCF4 binding to the MYC 3′ WRE (3′ WRE) and the control element (Ctrl.). (B) As in A, except ChIP assays were conducted with anti-TCF3 antibodies. (C) Western blot analysis of protein lysates prepared from isolated nuclei in starved cells (time 0) or cells treated for the indicated times with media-containing serum. Blots were probed with the indicated antibodies with PCNA serving as a loading control. All experiments were repeated at least 3 times and error bars are ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001).
To extend this analysis, we conducted similar assays in HCT116 cells that were transduced with lentiviruses harboring control or TCF3 shRNAs. Two days after delivery of the shRNAs, cells were deprived of serum for 48 hours and then released into the cell cycle with media-containing serum. Using qPCR analysis of cDNAs prepared from these cells, we found that TCF3 depletion increased MYC expression in quiescent cells and in cells as they entered the G1 and S phases of the cell cycle (). This increase in MYC expression was accompanied by an increase in TCF4 binding to the MYC 3′ WRE at each and every time point examined (). In addition, TCF3 knock-down increased β-catenin binding to the MYC 3′ WRE throughout the time course (). These findings suggest that in the absence of TCF3, TCF4/β-catenin complexes inappropriately activate MYC expression through the downstream WRE during G1 and S phase.
Figure 6. TCF3 knockdown alters proper regulation of MYC expression in synchronized cells. (A) qPCR analysis of MYC expression in cDNAs prepared from HCT116 cells expressing control or TCF3-specific shRNAs that were synchronized in the cell cycle. MYC transcripts were measured in quiescent cells (time 0), and in cells treated with media-containing serum for one, 2, 4, or 8 hours. At each time point, the data is normalized to GAPDH. (B) qPCR analysis of DNA fragments precipitated with anti-TCF4 antibodies in ChIP assays conducted in quiescent cells (time 0) or quiescent cells that were treated with media-containing serum for the number of hours indicated. TCF4 binding was assessed at the MYC 3′ WRE using oligonucleotides specific to the element in the qPCR reactions. (C) As in B, except ChIP assays were conducted with anti-β-catenin-specific antibodies. All experiments were repeated at least 3 times and error bars are ± SEM (*P < 0.05, **P < 0.01, **P < 0.001).
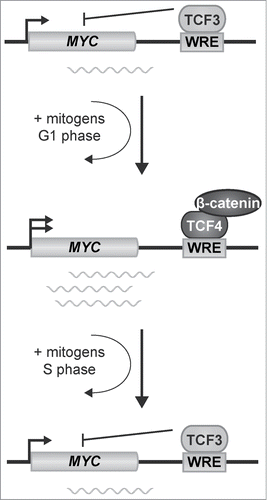
As summarized in our model, TCF3 binds the MYC 3′ WRE in quiescent cells to repress MYC expression (). Treating cells with media-containing serum induces an exchange of TCF3 with TCF4/β-catenin transcription complexes to activate expression of MYC. As the cells progress into S phase, TCF3 binding is restored at the MYC 3′ WRE to repress MYC gene expression. Thus a dynamic exchange of TCF3 and TCF4/β-catenin transcription factor complexes governs MYC expression during the cell cycle.
Figure 7. Model depicting the interplay between TCF3 and TCF4/β-catenin complexes in controlling MYC expression in CRC cells.
Discussion
Expression of the MYC proto-oncogene is deregulated in over 50% of all cancers.Citation40 In colorectal cancers, mutations in the Wnt/β-catenin signaling pathway drive oncogenic MYC expression through TCF/β-catenin transcription complexes bound to MYC WREs.Citation25,30,41-44 However, high levels of MYC induce an apoptotic program, which is disadvantageous to tumor growth.Citation45 Thus, a “just right” amount of MYC expression is required to promote colorectal carcinogenesis. Others and we have shown that TCF4/β-catenin complexes bound to proximal and distal MYC WREs induce MYC expression in human CRC cell lines.Citation27,28,30,46 In previous reports, we extensively characterized the MYC 3′ WRE and its role in regulating MYC expression in the human CRC cell line HCT116.Citation28,30 Using this WRE and these cells as a model in this report, we uncovered a critical role for the TCF3 transcription factor in repressing MYC expression. Therefore, our results suggest one mechanism for how oncogenic MYC levels are sustained without inducing apoptosis.
According to a transcription factor switch model, TCFs function as a platform that bind TLEs to repress target gene expression in the absence of a Wnt signal, and then bind β-catenin to activate target gene expression when the Wnt/β-catenin signaling pathway is engaged.Citation7 Our findings indicate that TCF3 represses MYC, and that under conditions where MYC expression is induced, TCF4/β-catenin complexes displace TCF3. We propose that TCF3 and TCF4 transcription factors compete to bind the MYC 3′ WRE; however, the highly conserved HMG DNA binding domain of TCF family members binds the consensus TCF recognition element with the same relative affinities.Citation47 Moreover, we have shown that these 2 factors are expressed to relatively equivalent levels in asynchronous cells and that nuclear levels of these factors are comparable in synchronous cells (). While our ChIP analysis suggests that TCF4 binds the MYC 3′ WRE more strongly than TCF3, differences in the antibodies used for precipitation do not allow a direct comparison of TCF occupancy by ChIP. One potential mechanism that might account for differential binding involves post-translational modification. TCF factors can be acetylated, phosphorylated and sumoylated.Citation47 Additional work is required to determine whether such post-translational modifications account for the differential occupancy of TCF3 and TCF4 at the MYC 3′ WRE.
Our model suggests that TCF3 represses MYC expression by blocking binding of TCF4/β-catenin complexes; however, the mechanistic details for how TCF3 represses transcription remain to be fully defined. We detect TCF3-binding to the MYC 3′ WRE in quiescent cells and cells in S-phase (). At these stages in the cell cycle, we previously showed that nucleosomes within this element contain deacetylated histones.Citation28 This finding suggests that TCF3 may be tethering an HDAC-containing complex to repress MYC. While the TLE/HDAC complex is an attractive candidate to fulfill this function, our attempts to detect this interaction using co-immunoprecipitation and ChIP assays have thus far been unsuccessful. It is therefore possible that TCF3 is interacting with CtBP or another, yet unknown HDAC complex to repress MYC expression. As an alternative, by simply blocking TCF4/β-catenin binding, TCF3 occupancy at this element may prevent aberrant MYC expression and this keeps the MYC gene locus poised to respond to the appropriate extracellular cues. In this scenario, the absence of β-catenin-associated histone acetyltransferase complexes, such as CBP, would explain the presence of deacetylated histones within the MYC 3′ WRE that we noted in our previous report.Citation28
Currently it is unclear how TCF3 is being removed from the MYC 3′ WRE. β-Catenin has been shown to interact with TCF3 in embryonic stem cells (ESCs) and target it for proteasomal degradation.Citation48 Our results in synchronized cells indicate that levels of TCF3 in the nucleus are unchanged when quiescent cells are stimulated to enter the cell cycle (), suggesting that β-catenin is not targeting TCF3 for degradation in CRC cells.Citation48 In addition, Chiaro et al. failed to detect a TCF3/β-catenin complex in HCT116 cells.Citation17 In another study, TCF3 was found to be a substrate of the homeodomain-interacting protein kinase (HIPK2).Citation49 When TCF3 was phosphorylated by HIPK2, it was displaced from a subset of WREs.Citation49 It will be interesting to test whether this HIPK2 pathway functions to remove TCF3 from the MYC 3′ WRE in CRCs.
The vast majority of colorectal cancers contain mutations in components of the Wnt/β-catenin pathway and these mutations cause de-regulated MYC expression. Therefore, it is imperative to understand how MYC is controlled at the transcriptional level in human CRC cell lines that harbor these mutations. TCF4/β-catenin complexes bound to distal and proximal WREs drive MYC expression in these cells.Citation28,30,41-44,46 In this report, we find that the TCF3 transcription factor plays a critical role in repressing MYC gene expression. Therefore, small molecules designed to promote TCF3 function as a transcriptional repressor could offer an effective therapeutic approach for treatment of individuals afflicted by colorectal cancer.
Materials and Methods
Cell lines
The HCT116 (ATCC, CCL-247) and HEK293FT (Invitrogen, R700-07) cell lines were maintained in DMEM and the FHs 74 Int cells (ATCC, CCL-241) were maintained in Hybri-Care Media (ATCC 46-x). The growth media was supplemented with 10% FBS, 50 units/ml penicillin, 2 mM Glutamax, and 0.1 mg/ml streptomycin and the cells were cultured at 37°C in 5% CO2. HEK293FT media was supplemented with 500 μg/ml neomycin and FHs 74 Int media was supplemented with 30 ng/ml epidermal growth factor (Peprotech, 315-09).
Plasmids
TOPflash and FOPflash luciferase reporters were purchased from Millipore (21-170 and 21-169, respectively). Generation of the MYC 3′ WRE- and MYC 3′ WRE (mut)-luciferase reporters was described previously. Citation28 The MYC 3′ WRE luciferase reporter contains the 615-bp MYC 3′ WRE inserted downstream from the luciferase gene in the pGL3 promoter vector (Promega, E1761). The MYC 3′ WRE (mut)-luciferase reporter contains mutations within the 2 consensus TCF recognition sites that have been shown to abolish TCF-dependent transcriptional activity. To generate the mammalian TCF3 expression vector, TCF3 was amplified from cDNAs prepared from HCT116 cells in standard PCR reactions with Phusion DNA polymerase (New England Biolabs). The primers used in this reaction were 5′-CGG AAT TCC CCC AGC TCG GCG GCG GG-3′ and 5′-CCC AAG CTT TTA GTG GGC AGA CTT GGT GAC C-3′, which contain 5′ EcoRI and HindIII sites, respectively. The amplified products were cloned as EcoRI-HindIII fragments into the pCMV-Tag2B vector, which provided an amino-terminal FLAG tag on TCF3 expressed from this plasmid (Agilent Technologies, 211172).
shRNA knockdown of TCF3 and TCF4
Lentiviral plasmids (pLKO.1) containing TCF3 and TCF4 shRNAs were obtained from Open Biosystems (GE Healthcare). The pLKO.1 plasmid harboring the scrambled sequence, 5′-CCT AAG GTT AAG TCG CCC TCG-3′ was previously described and this plasmid is also available from Addgene (plasmid 1864, deposited by D. Sabatini).Citation30 The following lists the sequences and clone identification numbers of the TCF3 shRNAs: TCF3 shRNA1; 5′-TTT CTT ACC ATA GTT GTC CCG-3′ (TRCN0000021704), TCF3 shRNA2; 5′-TTT ATT AGA CAA GTG TGC TGG-3′ (TRCN0000021705), TCF3 shRNA3; 5′-TTG TGC CAC TTT CTT CCA AGG-3′ (TRCN0000021706), TCF3 shRNA4; 5′-TAA TGG CTG CAC TTT CCT TCA-3′ (TRCN0000021707), and TCF3 shRNA5; 5′-ATT CCT GTC TTT GGA TCG ATC-3′ (TCRN0000021708). The following lists the sequences and clone identification numbers of the TCF4 shRNAs: TCF4 shRNA1; 5′-TTC TTT CCA TAG TTA TCC CGC-3′ (TRCN0000061893), TCF4 shRNA2; 5′-TTT CAT CTG GAG ATA GGT TCG-3′ (TRCN0000061894), TCF4 shRNA3; 5′-TTG CTG TAC GTG ATA AGA GGC-3′ (TRCN0000061896), and TCF4 shRNA4; 5′-TAA GAG GTT TCT TTA TGT GGG-3′ (TRCN0000061897). Lentiviruses were generated by transfecting 5 × 106 HEK293FT cells, seeded in a 10 cm dish, with 3.0 μg of the pLKO.1 shRNA plasmid, 3.0 μg each of pLP1 and pLP2 packaging plasmids and 3.0 μg of the pLP/VSVG envelope plasmid using Lipofectamine 2000 (Life Technologies, 11668019). Media containing the virus particles was harvested at 24 and 48 h after transfection and centrifuged at 1500 × g for 5 min at room temperature. The media was supplemented with 6.0 μg/ml hexadimethrine bromide and then added to HCT116 cells. Transcripts and proteins were analyzed 3 days after infection.
Cellular fractionation and western blot analysis
Approximately 5 × 106 HCT116 cells were fractionated into nuclear and cytoplasmic protein lysates as described previously.Citation33 Whole cell protein extracts were prepared using RIPA buffer and Western blot analysis was conducted as described previously.Citation41 The blots were probed with the following primary antibodies: anti-TCF3 (Cell Signaling, 2883, 1:1000), anti-α-tubulin (Sigma, T9026, 1:1000), anti-PCNA (Santa Cruz, sc-28250, 1:500), anti-TCF4 (Millipore, 05–511, 1:1000), anti-MYC (Santa Cruz, sc-764, 1:200) and anti-β-catenin (BD Transduction, 610154, 1:1000).
Luciferase reporter assays
Luciferase assays were conducted as described previously.Citation28 For the TCF3- knock-down experiments, 1.2 × 105 HCT116 cells were seeded in each well of a 24-well plate one day after they were infected with control or TCF3-specific shRNAs. The cells were seeded in quadruplicate for each experimental condition tested. After 24 h, the cells were transfected, using Lipofectamine 2000, with 100 ng of the firefly luciferase reporter plasmid, 2 ng pLRL-SV40 Renilla luciferase (Promega, E2231), and pBlueScript SK+ to obtain a 1μg final DNA concentration. For experiments involving overexpression of TCF3, the cells were seeded and transfected as above, except transfection complexes also contained 150 ng of pCMV-Tag2B vector or pCMV-Tag2B-TCF3 expression plasmids per well. Luciferase levels were measured 24 h after transfection using a firefly and Renilla luciferase assay kit (Biotium, 30005) and a GloMax 20/20 luminometer (Promega).
Reverse transcription and quantitative PCR (RT-qPCR)
RNA isolation, cDNA synthesis, and qPCR were performed as described except that cDNA was diluted 1:50 for qPCR. 26 The following primer sequences were used to detect expression of the indicated genes: TCF3; 5′-CT CGT CTC CCC CAT CGT CAA G-3′ and 5′-GGC TTC TTT TCC TCC TCC TTT TTC AC-3′, MYC; 5′-GCA AAC CTC CTC ACA GCC CAC-3′ and 5′-AAC TTG ACC CTC TTG GCA GCA-3′, and GAPDH; 5′-CCA GCA AGA GCA CAA GAG GAA GAG-3′ and 5′-CAA GGG GTC TAC ATG GCA ACT GTG-3′. Relative expression was measured with the 2ΔCT method using GAPDH as the reference transcript. Citation50
Chromatin immunoprecipitation (ChIP)
ChIP analysis was performed as previously describedCitation26 using 3 μg each of anti-TCF3 (Santa Cruz, sc-8635), anti-β-catenin (BD Transduction, 610154), and anti-TCF4 (Millipore, 05–511) antibodies. The precipitated DNA was amplified by qPCR using the primer set 5′-GCT CAG TCT TTG CCC CTT TGT GG-3′ and 5′-TAA CAC CTT CCC GAT TCC CAA GTG-3′ to interrogate the MYC 3′ WRE, or the primer set 5′-AAA AAC GGG GTC AGA AGT CAG GAA-3′ and 5′-AGG TAA AGA TTG GGG AAG CAG CAA-3′, which anneals to a control site 290-kb upstream from the MYC transcription start site that we previously demonstrated to lack TCF4 and β-catenin binding.Citation41 A standard curve that was generated with serial dilutions of purified input DNA was used as a reference to quantify the precipitated DNA to ensure that the ChIP signal was within the linear range of detection. Specifically, these reactions contained 50 ng, 10 ng, 2 ng, 0.4 ng, or 0.08 ng of sonicated and purified input DNA. The data is presented as relative levels obtained using the standard curve. For the TCF3 knock-down experiments, HCT116 cells were transduced with control or TCF3 shRNA lentiviral particles and ChIP was performed 48 hours after transduction.
Synchronizing HCT116 cells in the cell cycle
We used the serum-deprivation protocol that we previously described to synchronize the cells in the cell cycle.Citation28 Briefly, HCT116 cells were grown to confluence on a 10-cm dish and then cultured in media lacking serum for 48 hours. Media-containing serum was then added for one, 2, 4, or 8 hours. Cells were collected and processed for transcript analysis, Western blot analysis, and ChIP assays as described above. To synchronize TCF3 knockdown cells, serum was withdrawn from the media 48 h after infection with the lentiviruses harboring control or TCF3-specific shRNAs.
Statistics
Each experiment was repeated at least 3 times and statistical significance was calculated using Student's t-test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank all members of the Yochum lab for helpful comments during the course of this study.
Funding
This work was supported by NIH grant R01DK080805 (GSY) and funds provided by the Department of Biochemistry & Molecular Biology, Pennsylvania State University College of Medicine.
Related Research Data
References
- Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin 2014; 64:104-17; PMID:24639052; http://dx.doi.org/10.3322/caac.21220
- Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol 2011; 6:479-507; PMID:21090969; http://dx.doi.org/10.1146/annurev-pathol-011110-130235
- Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell 2012; 149:1192-205; PMID:22682243; http://dx.doi.org/10.1016/j.cell.2012.05.012
- MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17:9-26; PMID:19619488; http://dx.doi.org/10.1016/j.devcel.2009.06.016
- Cadigan KM. TCFs and Wnt/beta-catenin signaling: more than one way to throw the switch. Curr Top Dev Biol 2012; 98:1-34; PMID:22305157; http://dx.doi.org/10.1016/B978-0-12-386499-4.00001-X
- Mosimann C, Hausmann G, Basler K. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol 2009; 10:276-86; PMID:19305417; http://dx.doi.org/10.1038/nrm2654
- Cadigan KM, Waterman ML. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol 2012; 4; PMID:23024173
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997; 275:1784-7; PMID:9065401; http://dx.doi.org/10.1126/science.275.5307.1784
- Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, Clevers H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet 1998; 19:379-83; PMID:9697701; http://dx.doi.org/10.1038/1270
- Barker N, Huls G, Korinek V, Clevers H. Restricted high level expression of Tcf-4 protein in intestinal and mammary gland epithelium. Am J Pathol 1999; 154:29-35; PMID:9916915; http://dx.doi.org/10.1016/S0002-9440(10)65247-9
- van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002; 111:241-50; PMID:12408868; http://dx.doi.org/10.1016/S0092-8674(02)01014-0
- Angus-Hill ML, Elbert KM, Hidalgo J, Capecchi MR. T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc Natl Acad Sci U S A 2011; 108:4914-9; PMID:21383188; http://dx.doi.org/10.1073/pnas.1102300108
- Tang W, Dodge M, Gundapaneni D, Michnoff C, Roth M, Lum L. A genome-wide RNAi screen for Wnt/beta-catenin pathway components identifies unexpected roles for TCF transcription factors in cancer. Proc Natl Acad Sci U S A 2008; 105:9697-702; PMID:18621708; http://dx.doi.org/10.1073/pnas.0804709105
- Merrill BJ. Wnt pathway regulation of embryonic stem cell self-renewal. Cold Spring Harb Perspect Biol 2012; 4:a007971; PMID:22952393; http://dx.doi.org/10.1101/cshperspect.a007971
- Lien WH, Fuchs E. Wnt some lose some: transcriptional governance of stem cells by Wnt/beta-catenin signaling. Genes Dev 2014; 28:1517-32; PMID:25030692; http://dx.doi.org/10.1101/gad.244772.114
- van Es JH, Haegebarth A, Kujala P, Itzkovitz S, Koo BK, Boj SF, Korving J, van den Born M, van Oudenaarden A, Robine S, et al. A critical role for the Wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol Cell Biol 2012; 32:1918-27; PMID:22393260; http://dx.doi.org/10.1128/MCB.06288-11
- Chiaro C, Lazarova DL, Bordonaro M. Tcf3 and cell cycle factors contribute to butyrate resistance in colorectal cancer cells. Biochem Biophys Res Commun 2012; 428:121-6; PMID:23063976; http://dx.doi.org/10.1016/j.bbrc.2012.10.018
- Athineos D, Sansom OJ. Myc heterozygosity attenuates the phenotypes of APC deficiency in the small intestine. Oncogene 2010; 29:2585-90; PMID:20140021; http://dx.doi.org/10.1038/onc.2010.5
- Ignatenko NA, Holubec H, Besselsen DG, Blohm-Mangone KA, Padilla-Torres JL, Nagle RB, de Alboránç IM, Guillen-R JM, Gerner EW. Role of c-Myc in intestinal tumorigenesis of the ApcMin/+ mouse. Cancer Biol Ther 2006; 5:1658-64; PMID:17106247; http://dx.doi.org/10.4161/cbt.5.12.3376
- Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, Clarke AR. Myc deletion rescues Apc deficiency in the small intestine. Nature 2007; 446:676-9; PMID:17377531; http://dx.doi.org/10.1038/nature05674
- Wilkins JA, Sansom OJ. C-Myc is a critical mediator of the phenotypes of Apc loss in the intestine. Cancer Res 2008; 68:4963-6; PMID:18593890; http://dx.doi.org/10.1158/0008-5472.CAN-07-5558
- Yekkala K, Baudino TA. Inhibition of intestinal polyposis with reduced angiogenesis in ApcMin/+ mice due to decreases in c-Myc expression. Mol Cancer Res 2007; 5:1296-303; PMID:18171987; http://dx.doi.org/10.1158/1541-7786.MCR-07-0232
- Konsavage WM, Jr., Yochum GS. The myc 3' wnt-responsive element suppresses colonic tumorigenesis. Mol Cell Biol 2014; 34:1659-69; PMID:24567369; http://dx.doi.org/10.1128/MCB.00969-13
- Konsavage WM, Jr., Jin G, Yochum GS. The Myc 3' Wnt-responsive element regulates homeostasis and regeneration in the mouse intestinal tract. Mol Cell Biol 2012; 32:3891-902; PMID:22826434; http://dx.doi.org/10.1128/MCB.00548-12
- Sur IK, Hallikas O, Vaharautio A, Yan J, Turunen M, Enge M, Taipale M, Karhu A, Aaltonen LA, Taipale J. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science 2012; 338:1360-3; PMID:23118011; http://dx.doi.org/10.1126/science.1228606
- Bottomly D, Kyler SL, McWeeney SK, Yochum GS. Identification of {beta}-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res 2010; 38:5735-45; PMID:20460455; http://dx.doi.org/10.1093/nar/gkq363
- Yochum GS, McWeeney S, Rajaraman V, Cleland R, Peters S, Goodman RH. Serial analysis of chromatin occupancy identifies beta-catenin target genes in colorectal carcinoma cells. Proc Natl Acad Sci U S A 2007; 104:3324-9; PMID:17360646; http://dx.doi.org/10.1073/pnas.0611576104
- Yochum GS, Cleland R, Goodman RH. A genome-wide screen for beta-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol Cell Biol 2008; 28:7368-79; PMID:18852287; http://dx.doi.org/10.1128/MCB.00744-08
- Mautner J, Joos S, Werner T, Eick D, Bornkamm GW, Polack A. Identification of two enhancer elements downstream of the human c-myc gene. Nucleic Acids Res 1995; 23:72-80; PMID:7870592; http://dx.doi.org/10.1093/nar/23.1.72
- Yochum GS, Sherrick CM, Macpartlin M, Goodman RH. A beta-catenin/TCF-coordinated chromatin loop at MYC integrates 5′ and 3′ Wnt responsive enhancers. Proc Natl Acad Sci U S A 2010; 107:145-50; PMID:19966299; http://dx.doi.org/10.1073/pnas.0912294107
- Mao CD, Byers SW. Cell-context dependent TCF/LEF expression and function: alternative tales of repression, de-repression and activation potentials. Crit Rev Eukaryot Gene Expr 2011; 21:207-36; PMID:22111711; http://dx.doi.org/10.1615/CritRevEukarGeneExpr.v21.i3.10
- Konsavage WM, Jr., Kyler SL, Rennoll SA, Jin G, Yochum GS. Wnt/beta-catenin signaling regulates Yes-associated protein (YAP) gene expression in colorectal carcinoma cells. J Biol Chem 2012; 287:11730-9; PMID:22337891; http://dx.doi.org/10.1074/jbc.M111.327767
- Yochum GS, Cleland R, McWeeney S, Goodman RH. An antisense transcript induced by Wnt/beta-catenin signaling decreases E2F4. J Biol Chem 2007; 282:871-8; PMID:17121828; http://dx.doi.org/10.1074/jbc.M609391200
- Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci U S A 1997; 94:10330-4; PMID:9294210; http://dx.doi.org/10.1073/pnas.94.19.10330
- Deevi R, Fatehullah A, Jagan I, Nagaraju M, Bingham V, Campbell FC. PTEN regulates colorectal epithelial apoptosis through Cdc42 signalling. Br J Cancer 2011; 105:1313-21; PMID:21952626; http://dx.doi.org/10.1038/bjc.2011.384
- Kang DW, Min do S. Positive feedback regulation between phospholipase D and Wnt signaling promotes Wnt-driven anchorage-independent growth of colorectal cancer cells. PLoS One 2010; 5:e12109; PMID:20711340; http://dx.doi.org/10.1371/journal.pone.0012109
- Owens RB, Smith HS, Nelson-Rees WA, Springer EL. Epithelial cell cultures from normal and cancerous human tissues. J Natl Cancer Inst 1976; 56:843-9; PMID:176412
- Wierstra I, Alves J. The c-myc promoter: still MysterY and challenge. Adv Cancer Res 2008; 99:113-333; PMID:18037408; http://dx.doi.org/10.1016/S0065-230X(07)99004-1
- Toualbi K, Guller MC, Mauriz JL, Labalette C, Buendia MA, Mauviel A, Bernuau D. Physical and functional cooperation between AP-1 and beta-catenin for the regulation of TCF-dependent genes. Oncogene 2007; 26:3492-502; PMID:17146436; http://dx.doi.org/10.1038/sj.onc.1210133
- Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene 1999; 18:3004-16; PMID:10378696; http://dx.doi.org/10.1038/sj.onc.1202746
- Yochum GS. Multiple Wnt/beta-catenin responsive enhancers align with the MYC promoter through long-range chromatin loops. PLoS One 2011; 6:e18966; PMID:21533051; http://dx.doi.org/10.1371/journal.pone.0018966
- Tuupanen S, Turunen M, Lehtonen R, Hallikas O, Vanharanta S, Kivioja T, Björklund M, Wei G, Yan J, Niittymäki I, Mecklin JP, et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat Genet 2009; 41:885-90; PMID:19561604; http://dx.doi.org/10.1038/ng.406
- Wright JB, Brown SJ, Cole MD. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Mol Cell Biol 2010; 30:1411-20; PMID:20065031; http://dx.doi.org/10.1128/MCB.01384-09
- Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, Beckwith CA, Chan JA, Hills A, Davis M, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet 2009; 41:882-4; PMID:19561607; http://dx.doi.org/10.1038/ng.403
- Soucek L, Evan GI. The ups and downs of Myc biology. Curr Opin Genet Dev 2010; 20:91-5; PMID:19962879; http://dx.doi.org/10.1016/j.gde.2009.11.001
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science 1998; 281:1509-12; PMID:9727977; http://dx.doi.org/10.1126/science.281.5382.1509
- Arce L, Yokoyama NN, Waterman ML. Diversity of LEF/TCF action in development and disease. Oncogene 2006; 25:7492-504; PMID:17143293; http://dx.doi.org/10.1038/sj.onc.1210056
- Shy BR, Wu CI, Khramtsova GF, Zhang JY, Olopade OI, Goss KH, Merrill BJ. Regulation of Tcf7l1 DNA binding and protein stability as principal mechanisms of Wnt/beta-catenin signaling. Cell Rep 2013; 4:1-9; PMID:23810553; http://dx.doi.org/10.1016/j.celrep.2013.06.001
- Hikasa H, Sokol SY. Phosphorylation of TCF proteins by homeodomain-interacting protein kinase 2. J Biol Chem 2011; 286:12093-100; PMID:21285352; http://dx.doi.org/10.1074/jbc.M110.185280
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402-8; PMID:11846609; http://dx.doi.org/10.1006/meth.2001.1262